

Abstract
With only two different cell types, the haploid green alga Volvox represents the simplest multicellular model system. To facilitate genetic investigations in this organism, the occurrence of homologous recombination events was investigated with the intent of developing methods for gene replacement and gene disruption. First, homologous recombination between two plasmids was demonstrated by using overlapping nonfunctional fragments of a recombinant arylsulfatase gene (tubulin promoter/arylsulfatase gene). After bombardment of Volvox reproductive cells with DNA-coated gold microprojectiles, transformants expressing arylsulfatase constitutively were recovered, indicating the presence of the machinery for homologous recombination in Volvox. Second, a well characterized loss-of-function mutation in the nuclear nitrate reductase gene (nitA) with a single G → A nucleotide exchange in a 5′-splice site was chosen as a target for gene replacement. Gene replacement by homologous recombination was observed with a reasonably high frequency only if the replacement vector containing parts of the functional nitrate reductase gene contained only a few nucleotide exchanges. The ratio of homologous to random integration events ranged between 1:10 and 1:50, i.e., homologous recombination occurs frequently enough in Volvox to apply the powerful tool of gene disruption for functional studies of novel genes.
Green algae of the order Volvocales range in complexity from unicellular Chlamydomonas through colonial genera to multicellular organism in the genus Volvox. The unicellular members of this order have proven to be excellent model systems for the biochemical and genetic analysis of cellular processes like photosynthesis, phototaxis and motility, and cell wall biogenesis. The more advanced members of this group, like Volvox have developed a multicellular form of organization with a complete division of labor between somatic and reproductive cells; in Volvox carteri, about 2,000 cells are somatic and only 16 cells are reproductive. Therefore, the Volvocales represent an ideal model system to study the prerequisite for the transition from unicellularity to multicellularity. Recently, the development of nuclear transformation (1, 2) and the introduction of reporter genes (3, 4) for the unicellular as well as the multicellular members of the Volvocales have made it possible to apply the powerful techniques of molecular genetics. However, the important genetic technique of gene replacement and gene disruption by homologous recombination has not yet been established in Volvox. Only a few successful reports on homologous recombination events exist for Chlamydomonas reinhardtii (5–7).
Gene targeting by homologous recombination is a genetic tool that permits modification of cellular genes in a precise and predetermined fashion. This technique was initially used as a tool by yeast genetics because site-specific rather than random integration of homologous DNA is the preferred mode of integration in this organism (8, 9). Although homologous integration of transfected DNA into the genome was considered to be an extremely rare event in other organisms, techniques for this process have been established for several model systems (5, 10–16).
As found for higher eukaryotic genomes, nuclear transformation of Volvox has, thus far, only been characterized by illegitimate recombination events, resulting in ectopic integration of the introduced DNA. Any strategy for the detection of homologous recombination events in Volvox must take into account the technical impossibility of treating high numbers of Volvox algae at one time as well as the low level of transformation efficiency. To overcome these difficulties, experimental conditions should increase the frequency of homologous-to-random integration events. To that end, we drew on experience gained with unicellular Chlamydomonas, in which it was observed that bombardment with DNA-coated microprojectiles resulted in a much higher frequency of homologous recombination events than did another method of DNA delivery (5). In addition, we made only a very few changes in the nucleotide sequence of the targeting vector to avoid any possible hindrance to the process of homologous recombination.
In this paper, we present evidence for efficient homologous recombination between two introduced DNA fragments as well as for gene replacement events by homologous recombination at the nitrate reductase locus in the nuclear genome of V. carteri.
MATERIALS AND METHODS
Recipient Strain.
The Volvox strain 153–48, obtained from D. L. Kirk (Washington University, St. Louis), was used as the DNA recipient. Strain 153–48 is an F1-female progeny of HB11A, a female strain of V. carteri f. nagariensis that has been described (17). This strain with wild-type morphology inherited from HB11A an allele that carries a loss-of-function mutation of nitA, the structural gene encoding nitrate reductase (18, 19). The spontaneous reversion rate of this allele is less than 3 × 10−9 per reproductive cell (D. L. Kirk, personal communication).
Culture Conditions.
Synchronous Volvox recipients were grown in Volvox medium (20) at 28°C in an 8-h dark/16-h light (10,000 lx) cycle (21). The nonselective medium used was Volvox medium, supplemented with 1 mM NH4Cl; the selective medium was Volvox medium lacking NH4Cl and containing only nitrate as a nitrogen source.
Construction of the Chimeric β-Tubulin/Arylsulfatase Gene.
The Volvox arylsulfatase gene was placed under the control of the Volvox β-tubulin promoter in Volvox by using genomic clones of Volvox β-tubulin (22) and Volvox arylsulfatase (23). Additional restriction sites were introduced by PCR to facilitate ligation of the parent DNAs. An EcoRV site was generated directly in front of the start codons both of the Volvox arylsulfatase gene and the Volvox β-tubulin gene. A KpnI site was introduced into the β-tubulin promoter region 493 bp upstream of the start codon. Then, a KpnI/EcoRV fragment bearing the 493-bp Volvox β-tubulin promoter region was ligated to a 10-kb EcoRV/SalI fragment containing the Volvox arylsulfatase gene with its 15 introns. The complete construct was confirmed by sequencing.
Construction of a Modified Nitrate Reductase Gene Fragment.
To introduce conservative nucleotide exchanges into the nitrate reductase gene as a marker, the gene splicing by overlap extension technique, or the recombinant PCR technique, was performed as described by Horton et al. (24). To construct the modified nitA fragment, the first PCR was performed on nitA plasmid pVcNR1 (2) with an 18-mer 5′ sense primer (5′-GTTCGGATCCCGGATCAA) and a 43-mer 3′ antisense primer (5′-ACAAACCTGTGATTTTGTTTGTAGGCAATAATGACGTCAGAGG) with only half of the primer completely matching; the other half contains mismatches as a marker. The second PCR was performed on the same template with a 42-mer 5′ sense primer (5′-TTGCCTACAAACAAAATCACAGGTTTGTCCTGTGCTATGTGT, in which the underlined nucleotides are complementary to the corresponding region of the antisense primer of the first PCR), and an 18-mer 3′ antisense primer (5′-GGTTGTCCATGAAGTGGT). The third PCR was carried out with both of these PCR products and only the flanking primers. PCRs were performed as described (4). The final construction of the nitA fragment was performed by standard techniques (25). All products produced by PCR and cloning were sequenced.
Stable Transformation.
Small gold beads (1 μm in diameter, Bio-Rad) were coated with the required targeting plasmids as described (2, 4, 26) and used as microprojectiles. Stable transformation of reproductive Volvox cells of strain 153–48 (carrying a loss-of-function mutation in the nitrate reductase gene) was performed by using a particle gun with flowing helium (Biolistic PDS-1000/He System, Bio-Rad) to bombard the cells with the DNA-coated microprojectiles (2, 4, 26). To select for nitrate reductase activity in transgenic Volvox, the bombarded cells were resuspended in Volvox medium with nitrate as the only nitrogen source (lacking NH4+). From the sixth day after transformation onward, bombarded cultures were observed stereomicroscopically, and each green alga was transferred into a microtiter well with fresh selective Volvox medium. Transformants were tested for restoration of chlorate sensitivity by cultivation in 8 mM potassium chlorate in Volvox medium (2).
Reverse Transcription (RT)-PCR.
RT-PCR was performed as described (4, 26). For transformants expressing the artificial tubulin/arylsulfatase gene, the antisense primer 5′-CAGTGGAATAGAAGCCG (arylsulfatase) and the sense primer 5′-ATAACAAGCGACCACTAC (tubulin) were used. For transformants with homologous recombination events at the nitrate reductase gene locus (nitA), the antisense primer 5′-GGTTGTCCATGAAGTGGT and the sense primer 5′-CCTGCACTCTCGTATGCG were used. Products of PCR amplification were ligated into the SmaI site of pUC18 and sequenced. Amplification, cloning, and sequencing of each RT-PCR fragment were repeated twice.
Genomic PCR.
Twenty Volvox spheroids were selected under the stereomicroscope and transferred into 10 μl of sterile lysis buffer (0.1 M NaOH/2.0 M NaCl/0.5% SDS). After 5 min at 95°C, 200 μl of 50 mM Tris⋅HCl (pH 7.5) was added immediately. The resulting lysate (2 μl) was used for PCR (in a total volume of 100 μl). PCR was performed by standard protocol. Products of PCR amplification were cloned and sequenced. Amplification, cloning, and sequencing of each PCR fragment was repeated twice.
Southern Blot Analysis.
Total genomic DNA was prepared from V. carteri by using the RapidPrep genomic DNA isolation kit from Pharmacia. Lysis of Volvox cells in extraction buffer was supported by adding glass beads (0.45–0.50 μm) and vortexing five times at high speed for 1 min. The tube was frozen and thawed between pulses. Restriction digests, gel electrophoresis, and Southern blot procedures followed standard techniques (25). Blots were probed with an ≈0.3-kb EcoRI/ApaI nitrate reductase (18) fragment (nucleotides 1175–1478 of nitA sequence as deposited in the GenBank database, accession no. X64136).
RESULTS
Homologous Recombination in Volvox.
The aim of our initial experiments was to search for an indication that the machinery for homologous recombination exists in reproductive cells of Volvox. A plasmid-based test system was used to detect the occurrence of homologous recombination between a pair of plasmids encoding supplementing parts of a reporter gene. The reporter gene used was the Volvox arylsulfatase gene (4, 23). To distinguish the plasmid-encoded arylsulfatase from the endogenous arylsulfatase, which is only expressed under conditions of sulfur starvation, the plasmid-encoded arylsulfatase gene was placed under the control of the constitutive β-tubulin-promoter (22) of Volvox (Fig. 1A; plasmid 1). First, Volvox was transformed with this intact artificial gene construct to check its function; as expected, transformants showed arylsulfatase activity in sulfur-sufficient medium and the presence and the function of the chimeric gene was proven by genomic PCR and RT-PCR.
Figure 1.
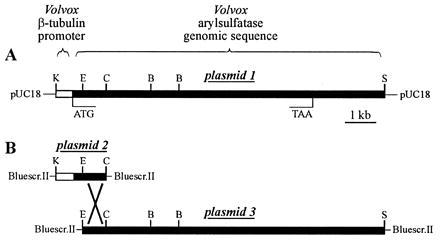
(A) Structure of the tubulin/arylsulfatase chimeric gene, harbored in plasmid 1, containing the Volvox β-tubulin promoter region and the Volvox arylsulfatase coding region. (B) Plasmids 2 and 3 carry parts of the original plasmid 1. Plasmids 2 and 3 share a 776-bp overlap of arylsulfatase genomic sequence. B, BamHI; C, ClaI; E, EcoRI; K, KpnI; S, SalI.
Then, two plasmids were constructed, each carrying only a fragment of the chimeric tubulin/arylsulfatase gene (Fig. 1B; plasmids 2 and 3). Transformation of Volvox with either of these plasmids alone (plus selectable marker plasmid) did not yield any transformant constitutively expressing the arylsulfatase gene, confirming that the deletions had inactivated the artificial tubulin/arylsulfatase gene. Then, Volvox was transformed with both (circular) plasmids, that share a 776-bp region of identity but harbor a deletion at the 5′ or 3′ end. In addition, a selectable transformation marker (the complete nitA gene) was supplied by a third plasmid because co-transformation frequency in V. carteri is high (40–80%) (2). Transformation with both arylsulfatase plasmids (plus selectable marker plasmid) resulted in a significant number of transformants expressing the arylsulfatase gene in sulfur-sufficient medium. Six out of 20 (or 30%) transformants exhibited this phenotype. These results indicated that a functional chimeric tubulin/arylsulfatase gene had resulted from homologous recombination between the introduced nonfunctional gene fragments.
The RT-PCR technique was used to verify the existence of recombinant mRNA in transgenic Volvox clones constitutively expressing the arylsulfatase gene. For this purpose, RNA was extracted from 20 Volvox spheroids, reverse transcribed, and subsequently amplified by PCR. Oligonucleotide primers that matched sequences up- or downstream from the overlap region of the plasmids and, in particular, that allowed the amplification of the fusion region of the chimeric β-tubulin/arylsulfatase gene construct were selected. The sense primer anneals to a sequence on the tubulin part present on the original plasmid 2, and the antisense primer anneals to a sequence on the arylsulfatase part present on the original plasmid 3 (Fig. 1B). A correct RT-PCR product could be expected only if a homologous recombination event, i.e., a single crossover, took place. In this case, a 663-bp cDNA was predicted from the sequence data and the known intron–exon boundaries of the parent genes. As documented in Fig. 2A, the RT-PCR yielded the expected chimeric cDNA fragment in all six Volvox clones (named HRA-1 to HRA-6) constitutively expressing the arylsulfatase gene. RT-PCRs with wild-type algae or the parent nitA− strain 153–48 failed to produce this product. The corresponding DNA sequence analysis of these hybrid cDNA fragments (663 bp) is shown in Fig. 2B. The five introns originally present within this stretch of chimeric DNA were excised by the splicing machinery of Volvox exactly as the parent arylsulfatase gene was spliced. Thus, homologous recombination, i.e., a single crossover, between identical regions of both plasmids took place, resulting in the functional reconstitution of the interrupted artificial tubulin/arylsulfatase gene. Homologous recombination between introduced circular DNA molecules was observed to occur with a high frequency (about 1:3) during transformation of Volvox.
Figure 2.
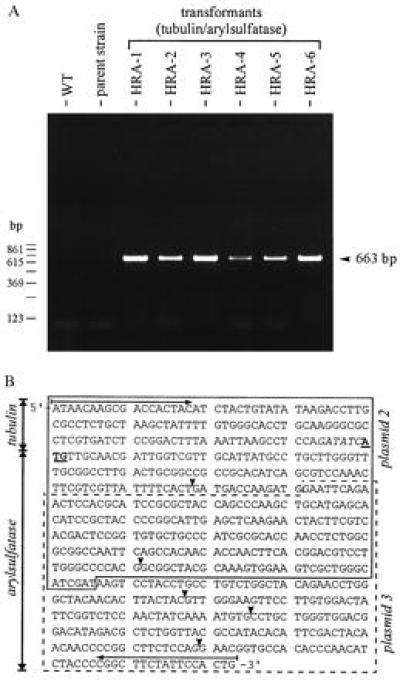
(A) RT and subsequent PCR amplification of the chimeric mRNA. RNA was extracted from 20 Volvox spheroids of parent strain 153–48, wild-type algae (WT), or the six transformants constitutively expressing the arylsulfatase gene (HRA-1 through HRA-6). Sizes of PCR products (663 bp) were determined by using a 123-bp ladder as size marker and by DNA sequencing. (B) Sequence of cDNA (663 bp) obtained from RT and subsequent PCR amplification of the overlap region of the tubulin/arylsulfatase gene from Volvox clones HRA-1 through HRA-6. All clones resulted from transformation experiments using a mixture of plasmids 2 and 3 (and nitA as a selection marker). The sequence stretch contributed by plasmid 2 is boxed with a solid line; the contribution of plasmid 3 is boxed with a dashed line. The translation initiation site (ATG) is highlighted (shown underlined and in boldface type). The artificial EcoRV restriction site in front of the translation initiation site, shown in italic type, separates the β-tubulin sequence from the arylsulfatase sequence. Introns were spliced correctly. The positions of the introns within this cDNA-fragment are indicated by solid arrowheads. PCR primers used are indicated by horizontal arrows.
Chimeric cDNA was detectable in transgenic Volvox clones even after 6 months, i.e., after about 90 generations. Because nonreplicating plasmids were used, the product of homologous recombination between both plasmids must have been integrated into the Volvox genome, most probably by illegitimate recombination.
Gene Replacement by Homologous Recombination.
The above experiments indicated that the machinery for homologous recombination exists in reproductive cells of Volvox. Subsequent experiments investigated the possibility of a targeted gene replacement in the Volvox genome.
Our first attempts to achieve and detect homologous recombination in the Volvox genome followed standard procedures, i.e., a replacement vector in which the coding region of the target gene (Volvox arylsulfatase gene; refs. 4 and 23) had been interrupted by inserting a marker gene (neomycin phosphotransferase gene; ref. 27) or by deleting a part (0.3 kb) of the arylsulfatase gene was constructed. All transformants obtained with these constructs turned out to be the result of illegitimate recombination events (data not shown).
Therefore, a different approach was initiated. A nuclear single copy gene was selected as a target in which minor changes, such as exchange of a few nucleotide positions, would lead to a selectable phenotype. The defective nitrate reductase gene in a well characterized nitA− mutant of Volvox was chosen as an ideal target. The stable nitA loss-of-function mutation in the nitA− Volvox strain 153–81 has been shown to reside in a G → A transition of the first nucleotide within the 5′-splice site of the nitA intron 2, and, consequently, all splicing products are only nonfunctional mRNA variants (19). Because strain 153–81 does not show wild-type morphology because of additional mutations (regA and gls), the nitA− strain 153–48 with wild-type morphology was selected for all experiments. In strain 153–48, the spontaneous reversion rate of the defective nitA allele is less than 3 × 10−9 per reproductive cell (D. L. Kirk, personal communication). Because both strains, 153–81 and 153–48, have a common ancestor, the presence of the same G → A transition in the nitA gene of strain 153–48 was verified by genomic PCR, followed by DNA sequencing. Next, we tried to repair this defective gene by homologous recombination. The targeting vector carried a G nucleotide at the corresponding position, and another four nucleotides were exchanged within the coding region (exon 2) of the targeting plasmid. These four exchanges did not affect the amino acid sequence, but they allowed the distinction between a homologous recombination event and a spontaneous point mutation. In addition, the 5′ region and the first exon of the nitA gene were removed on the targeting vector to prevent vector integrations by illegitimate recombination from producing the nitA+ phenotype. The nitA fragment on the targeting plasmid starts within intron 1 (≈0.5 kb upstream of the five nucleotide exchanges) and ends ≈0.4 kb downstream of the polyadenylylation signal (5.4 kb downstream of the five nucleotide exchanges). The complete targeting vector is shown in Fig. 3A (plasmid 4). The differences in sequence between parent strain 153–48 and the targeting plasmid are shown in Fig. 3B.
Figure 3.
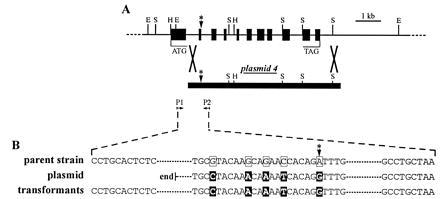
(A) Physical map and intron–exon structure of the Volvox nitrate reductase gene (18) (GenBank accession no. X64136). Plasmid 4 carries a 5′-deleted part of the original nitrate reductase gene with five base pair exchanges with respect to the parent gene, one of them repairing the ruined intron–exon splice site (which is marked by an asterisk). The primers used in genomic PCR are indicated (P1 and P2). P1 matches only the genomic sequence and not the plasmid sequence. For clarity, only the insert of the circular plasmid 4 is shown. (B) Part of the genomic sequence of the nitrate reductase gene in the original Volvox strain 153–48 (parent strain), part of the sequence of the 5′-deleted nitrate reductase plasmid 4 (plasmid), and part of the genomic sequence in the recombinant Volvox strains HRN-1 and HRN-2 (transformants), showing a homologous recombination event. E, EcoRI; H, HindIII; S, SalI.
Volvox strain 153–48 was transformed by bombardment with gold microparticles coated with the targeting plasmid in its circular form. Microparticles were accelerated by the use of a particle gun (28). Seven nitA+ transformants were recovered (HRN-1 through HRN-7). The number of transformations performed to collect these seven clones usually results in 60–300 transformants (with illegitimate recombinations) if a complete nitA gene is used. Therefore, in contrast to illegitimate recombination, which occurs with a frequency of ≈2.5 × 10−5 per reproductive cell (2), frequency of homologous recombination ranges between ≈2.5 × 10−6 and ≈5 × 10−7 per reproductive cell.
The seven nitA+ transformants were examined by genomic PCR. One of the oligonucleotide primers (Fig. 3A, primer P1) used for PCR anneals to a nitA sequence upstream of the 5′ end of the nitA fragment used for transformation. This strategy prevents the amplification of illegitimately integrated plasmids and allows only the amplification of nitA DNA from the nitA locus. A 867-bp PCR fragment was predicted from the nitA locus with or without the event of homologous recombination. For all seven transformants, genomic PCR yielded this expected 867-bp PCR fragment. Each of the PCR products were cloned into pUC18 vector and sequenced. In all seven transformants, the sequence of the 5′ half of the PCR fragment, which cannot be derived from the targeting vector, was the same as the sequence from the intact nitA gene. Furthermore, in all seven transformants, the mutation of the 5′-splice site of nitA intron 2 was corrected, i.e., it contained a G nucleotide instead of the A nucleotide present in the parent strain. Most importantly, in two (clones HRN-1 and HRN-2) of these seven transformants, the four marker mutations were found in the nitA gene exactly where they were introduced into the replacement vector (Fig. 3B, transformants). Another two transformants exhibited again the A → G transition in the splice site as well as one (clone HRN-3) or two (clone HRN-7) marker mutations next to this site. The remaining, more distantly located marker mutations did not show up. The remaining three transformants exhibited only the essential A → G transition within the splice site.
The RT-PCR technique was used to verify the existence of the recombinant mRNA and to prove the correct splicing of intron 2. Again, one of the oligonucleotide primers used for RT-PCR anneals to a sequence upstream of the 5′ end of the nitA fragment in the replacement vector to prevent the amplification of products resulting from illegitimately integrated plasmids. A 438-bp cDNA fragment was predicted for RT-PCR if homologous recombination took place and if intron 2 of the nitA gene was spliced correctly. RT-PCR yielded the expected 438-bp PCR fragment in all of the transformants (Fig. 4A). The DNA sequence analysis of the cDNA fragment of transformants HRN-1 and HRN-2 is shown in Fig. 4B. Again, the four marker mutations were found in the nitA cDNA of transformants HRN-1 and HRN-2, and the introns (introns 1 and 2) originally present within this stretch of DNA were excised by the splicing machinery of Volvox exactly as the wild-type nitA gene was spliced. Thus, gene replacement by homologous recombination restored a functional nitA gene containing four marker mutations.
Figure 4.
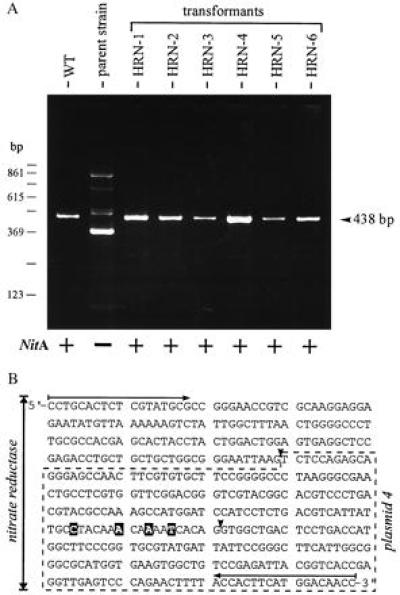
(A) RT and subsequent PCR amplification of the (modified) nitrate reductase mRNA. RNA was extracted from 20 Volvox spheroids of parent strain 153–48, wild-type algae (WT), or transformants HRN-1 through HRN-6 (RT-PCR of clone HRN-7 is not shown). As expected, RT-PCR with parent strain 153–48 yielded no correct product (19). Expression of nitrate reductase (NitA) is indicated by a plus or a minus. Sizes of PCR products (438 bp) were determined by using a 123-bp ladder as a size marker and by DNA sequencing. (B) Sequence of cDNA (438 bp), corresponding to nucleotides 1400–1837 of nitA sequence (18), obtained from RT and subsequent PCR amplification of the modified nitrate reductase gene harbored in Volvox clones HRN-1 and HRN-2, showing a homologous recombination event. Clones HRN-1 and HRN-2 resulted from transformation with plasmid 4. The sequence harbored in plasmid 4 is boxed (dashed line). The artificial base exchanges, with regard to the parent strain, are highlighted. Both introns (solid arrowheads) within this part of the cDNA were spliced correctly; in parent strain 153–48, splicing of the second intron is affected by mutation (G → A transition). The PCR primers used are indicated by horizontal arrows.
The mode of integration of the targeting vector at the nitA locus was investigated in Southern blot experiments. Integration by a single crossover would result in the integration of one copy of the replacement vector at the nitA locus and thus would result in a duplication of the nitA sequences. As a consequence, the surroundings of the original nitA gene would be changed. A double crossover or, alternatively, a recombination event between the duplicated regions generated by a single crossover would restore the original surrounding of the nitA gene. A probe (≈0.3 kb) covering the nitA gene region upstream of the 5′ end of the nitA sequence present in the replacement vector was used for the Southern blot experiments. This probe excludes signals resulting from sites of illegitimate plasmid integration. Both a SalI and a EcoRI digest (see the physical map in Fig. 3) of genomic DNA extracted from transformants HRN-1, HRN-2, and HRN-3 were probed, and the results are shown in Fig. 5. All transformants exhibited the same hybridization patterns as those found for the parent Volvox strain, 153–48 (Fig. 5), i.e., the homologous recombination event did not affect the immediate surroundings at either the 5′ end or the 3′ end of the nitA gene.
Figure 5.
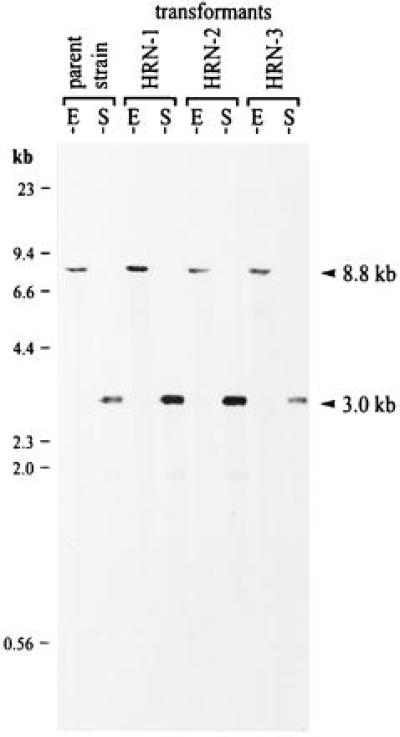
Southern blots of genomic DNA from the parent Volvox strain, 153–48, and from transformants HRN-1, HRN-2, and HRN-3. The sizes of DNA standards are indicated on the left, and hybridizing bands (arrowheads) are indicated on the right. DNA was digested with EcoRI or SalI, and the blot was probed with an ≈0.3-kb EcoRI/ApaI nitrate reductase fragment. The probe covers the nitA gene region upstream of the 5′ end of the nitA sequence present in the replacement vector used for the Southern blot experiments and, therefore, excludes signals resulting from sites of illegitimate plasmid integration. An 8.8-kb (EcoRI) fragment and a 3.0-kb (SalI) fragment were predicted if the homologous recombination event did not affect the immediate surroundings at the 5′ end or at the 3′ end of the nitA gene. E, EcoRI; S, SalI.
DISCUSSION
The targeted introduction of in vitro manipulated DNA to homologous sites in the genome provides a powerful tool for the study of physiological processes. Gene knockout or targeted gene disruption techniques provide important information about the biological function of unknown genes. Any organism allowing the application of these techniques fulfills an important prerequisite for serving as a model system. The green algae of the order Volvocales offer useful candidates for model organisms because this order includes organisms that demonstrate as clearly as a diagram the different stages of the evolutionary transition from unicellularity to multicellularity. Targeted gene replacement has recently been shown to be applicable to the unicellular member of this order, Chlamydomonas (5). The detection of targeted gene replacement in the most advanced member of the Volvocales makes this group of organisms an attractive model for molecular genetic and biochemical studies.
With a few exceptions, such as yeast, targeted gene replacement by homologous recombination is a rare event and requires highly selective screening procedures for detection; these procedures are available for a number of animal and plant cell systems (5, 10–16). The ratio of homologous to nonhomologous recombination events ranges between 10−2 and 10−5 in animal cells and between 10−4 and 10−5 in plant cells (10–16). In the unicellular green alga Chlamydomonas, the frequency ranged between 1:24 and 1:1000 (5). As a consequence, large numbers of transformants are required for the detection of homologous recombination events. This is not a serious problem if one is dealing with small unicellular algal cells such as Chlamydomonas. In this case, more than 107 cells can be handled per shot by using the particle gun for transformation. In contrast, a reproductive cell of Volvox with a mean diameter of 80 μm is a huge cell compared with Chlamydomonas, and at best 104 cells can be handled per shot. With a transformation rate of about 2.5 × 10−5 per reproductive cell, the detection of homologous recombination events becomes a serious problem. Indeed, we were unable to detect homologous recombination events using standard selection techniques such as the insertion of a selectable marker gene within the target gene sequence. Minimizing the extent of sequence modification to only a few nucleotide exchanges (a total of five in our experiments) within the targeting sequence increased the ratio of homologous to nonhomologous recombination events up to 1:10, thus greatly facilitating the detection of gene replacement events.
Genetic recombination involves the exchange of DNA strands between two duplex DNA partners. The Holliday junction, the branch point between two duplex DNAs that have exchanged a pair of strands, is thought to be an important intermediate in homologous recombination (29). Movement of the Holliday junction by branch migration can increase the amount of genetic information exchanged between homologues. But a single base mismatch, insertion, or deletion is sufficient to pose in vitro a substantial barrier to spontaneous branch migration (30). In our experiments, three of our gain-of-function transformants exhibited only the essential A → G nucleotide exchange, two transformants exhibited in addition either only one or two out of the four nucleotide exchanges serving as a marker, and two transformants exhibited all five nucleotide exchanges. Because of the low number of processed cells, it is highly unlikely that the first group of transformants arose by a spontaneous mutation event (spontaneous reversion rate < 3 × 10−9). Therefore, our data indicate that, even in vivo, single mismatches may pose a barrier to the movement of the Holliday junction.
This paper has shown that gene replacement by homologous recombination occurs frequently enough in Volvox to apply the powerful tool of gene disruption. A general and routine system for knocking out nuclear genes of the haploid multicellular Volvox by homologous recombination is an important goal in the further development of this organism as a model system for molecular genetic analyses.
Acknowledgments
We wish to thank C. Friederich for expert technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 521).
ABBREVIATION
- RT
reverse transcription
References
- 1.Kindle K. Proc Natl Acad Sci USA. 1990;87:1228–1232. doi: 10.1073/pnas.87.3.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiedlmeier B, Schmitt R, Müller W, Kirk M M, Gruber H, Mages W, Kirk D L. Proc Natl Acad Sci USA. 1994;91:5080–5084. doi: 10.1073/pnas.91.11.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies J P, Weeks D P, Grossman A R. Nucleic Acids Res. 1992;20:2959–2965. doi: 10.1093/nar/20.12.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallmann A, Sumper M. Proc Natl Acad Sci USA. 1994;91:11562–11566. doi: 10.1073/pnas.91.24.11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sodeinde O A, Kindle K L. Proc Natl Acad Sci USA. 1993;90:9199–9203. doi: 10.1073/pnas.90.19.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gumpel N J, Rochaix J D, Purton S. Curr Genet. 1994;26:438–442. doi: 10.1007/BF00309931. [DOI] [PubMed] [Google Scholar]
- 7.Nelson J A, Lefebvre P A. Mol Cell Biol. 1995;15:5762–5769. doi: 10.1128/mcb.15.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinnen A, Hicks J B, Fink G R. Proc Natl Acad Sci USA. 1978;75:1929–1933. doi: 10.1073/pnas.75.4.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orr-Weaver T L, Szostak J W, Rothstein R S. Proc Natl Acad Sci USA. 1981;78:6354–6358. doi: 10.1073/pnas.78.10.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smithies O, Gregg R G, Boggs S S, Koralewski M A, Kucherlapati R S. Nature (London) 1985;317:230–234. doi: 10.1038/317230a0. [DOI] [PubMed] [Google Scholar]
- 11.Doetschman T, Gregg R G, Maeda N, Hooper M L, Melton D W, Thompson S, Smithies O. Nature (London) 1987;330:576–578. doi: 10.1038/330576a0. [DOI] [PubMed] [Google Scholar]
- 12.Thomas K R, Capecchi M R. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 13.Mansour S L, Thomas K R, Capecchi M R. Nature (London) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 14.Eid J, Sollner-Webb B. Proc Natl Acad Sci USA. 1991;88:2118–2121. doi: 10.1073/pnas.88.6.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaertig J, Thatcher T H, Gu L, Gorovsky M A. Proc Natl Acad Sci USA. 1994;91:4549–4553. doi: 10.1073/pnas.91.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee K Y, Lund P, Lowe K, Dunsmuir P. Plant Cell. 1990;2:415–425. doi: 10.1105/tpc.2.5.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams C R, Stamer K A, Miller J K, McNally J G, Kirk M M, Kirk D L. Curr Genet. 1990;18:141–153. doi: 10.1007/BF00312602. [DOI] [PubMed] [Google Scholar]
- 18.Gruber H, Goetinck S D, Kirk D L, Schmitt R. Gene. 1992;120:75–83. doi: 10.1016/0378-1119(92)90011-d. [DOI] [PubMed] [Google Scholar]
- 19.Gruber H, Kirzinger S H, Schmitt R. Plant Mol Biol. 1996;31:1–12. doi: 10.1007/BF00020601. [DOI] [PubMed] [Google Scholar]
- 20.Provasoli L, Pintner I J. In: The Ecology of Algae, Special Publication No. 2, Pymatuning Laboratory of Field Biology. Tyron C A, Hartman R T, editors. Pittsburgh: Univ. Pittsburgh Press; 1959. pp. 84–96. [Google Scholar]
- 21.Starr R C. Arch Protistenk. 1969;111:204–222. [Google Scholar]
- 22.Harper J F, Mages W. Mol Gen Genet. 1988;213:315–324. doi: 10.1007/BF00339597. [DOI] [PubMed] [Google Scholar]
- 23.Hallmann A, Sumper M. Eur J Biochem. 1994;221:143–150. doi: 10.1111/j.1432-1033.1994.tb18723.x. [DOI] [PubMed] [Google Scholar]
- 24.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 26.Hallmann A, Sumper M. Proc Natl Acad Sci USA. 1996;93:669–673. doi: 10.1073/pnas.93.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraley R T, Rogers S G, Horsch R B, Sanders P R, Flick J S, Adams S P, Bittner M L, Brand L A, Fink C L, Fry J S, Galluppi G R, Goldberg S B, Hoffmann N L, Woo S C. Proc Natl Acad Sci USA. 1983;80:4803–4806. doi: 10.1073/pnas.80.15.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeuchi Y, Dotson M, Keen N T. Plant Mol Biol. 1992;18:835–839. doi: 10.1007/BF00020031. [DOI] [PubMed] [Google Scholar]
- 29.Holliday R. Genet Res. 1964;5:282–304. [Google Scholar]
- 30.Panyutin I G, Hsieh P. J Mol Biol. 1993;230:413–424. doi: 10.1006/jmbi.1993.1159. [DOI] [PubMed] [Google Scholar]