

Abstract
The molecular mechanisms responsible for the cellular uptake of copper in mammalian cells are unknown. We describe isolation of a human gene involved in this process by complementation of the yeast high-affinity copper uptake mutant, ctr1. Besides complementing ctr1 growth defect on nonfermentable media, the human gene also rescues iron transport and SOD1 defects in ctr1 yeast. Overexpression of the gene in yeast leads to vulnerability to the toxicity of copper overload. In addition, its expression in ctr1 yeast significantly increases the level of cellular copper, as demonstrated by atomic absorption. We propose this gene as a candidate for high-affinity copper uptake in humans and by analogy have named it hCTR1. The hCTR1 and yeast CTR1 predicted transmembrane proteins are 29% identical, but the human protein is substantially smaller in both the extracellular metal-binding and intracellular domains. An additional human gene similar to hCTR1, here named hCTR2, was identified in a database search. Both hCTR1 and hCTR2 are expressed in all human tissues examined, and both genes are located in 9q31/32. These studies, together with the previously recognized functional and sequence similarity between the Menkes/Wilson copper export proteins and CCC2 in yeast, demonstrate that similar copper homeostatic mechanisms are used in these evolutionarily divergent organisms.
Copper is an element essential for life. Because of its special redox properties, copper serves as a cofactor for proteins involved in a variety of biological reactions, such as photosynthesis and respiration (cytochrome c oxidase), free radical eradication (superoxide dismutase), connective tissue formation (lysyl oxidase), neurological development (dopamine β-hydroxylase), and iron homeostasis (ceruloplasmin). On the other hand, excessive copper is toxic or even lethal to the cell because of its ability to bind to some proteins, to interfere with homeostasis of other metals, and to generate hydroxyl radicals. Therefore, cells have developed sophisticated ways to maintain a critical balance between necessity and toxicity. The intake, export, and intracellular compartmentalization or buffering of copper is strictly regulated. In complex organisms such as mammals, this balance is achieved not only at the cellular level but also at the tissue and organismal level (reviewed in refs. 1–3).
The molecular mechanisms of copper homeostasis in mammals are starting to unfold, particularly in the area of copper export. The recent cloning of two related genes responsible for the human diseases Menkes syndrome and Wilson disease (4–9) have provided some insights into copper metabolism in mammals. Both genes encode transmembrane P-type ATPases that are required for copper export. The gene defective in Menkes syndrome is expressed in most tissues except the liver. In Menkes disease, the symptoms involving the central nervous system, hair, and connective tissues result from a global copper deficiency secondary to the lack of intestinal absorption of copper. In contrast, the gene responsible for Wilson disease is expressed predominantly in the liver, and as a result mutations lead to excessive copper accumulation in liver and eventually to hepatic cirrhosis.
However, the molecular basis for copper uptake in mammalian cells remains a mystery. It was suggested that one avenue for uptake is through ceruloplasmin, the most abundant copper-containing protein in the circulation, which might deliver copper to multiple tissues through a ceruloplasmin receptor (10–13). Later studies on aceruloplasminemic patients indicated that these patients have defects in iron homeostasis, whereas copper metabolism is normal (14–16). This indicates that ceruloplasmin is at least not necessary for normal copper transport.
The yeast Saccharomyces cerevisiae can serve as a model system to study copper homeostasis. Like mammalian cells, yeast cells use a copper-transporting P-type ATPase, a product of the CCC2 gene, to sequester copper into the secretory pathway (17). The CCC2 protein is both functionally and structurally similar to the gene products defective in Menkes syndrome and Wilson disease described above. Although no mammalian copper uptake gene has been isolated, in yeast three copper uptake genes, CTR1, CTR2, and CTR3, have been identified (18–20). CTR1 and CTR3 are high-affinity copper transport proteins, and mutations in both CTR1 and CTR3 are required to eliminate high-affinity copper uptake in yeast. In many laboratory strains, CTR3 function is interrupted by a Ty2 transposable element, and thus CTR1 alone becomes indispensable for high-affinity copper uptake for these laboratory strains (20). CTR2, whose sequence is similar to CTR1, appears to be a low-affinity copper uptake protein (19).
A probable Arabidopsis thaliana copper transporter, COPT1, was cloned recently by functional complementation of yeast high-affinity copper uptake mutant ctr1 (19). Here we describe the identification of a human gene by complementation of the same yeast mutant. We propose that this gene is responsible, at least in part, for high-affinity copper uptake in humans.
MATERIALS AND METHODS
Yeast Transformation and DNA Manipulation.
The human cDNA yeast expression library (21), made by cloning HeLa cDNA in the yeast pDB20 expression vector, was a generous gift from Leonard Guarente’s laboratory (Massachusetts Institute of Technology, Cambridge, MA). For complementation studies, the library was transformed into a ctr1 strain (MATa ura3 lys2 ade2 trp1 his3 leu2 Δctr1::LEU2, by using the lithium acetate method (22). Primary transformants recovered from SD-ura (synthetic defined medium with glucose as carbon source and uracil omitted) were then plated on yeast extract/peptone/glycerol or ethanol (YPG/E).
Deletion and mutational analysis of hCTR1 was achieved by PCR. To facilitate cloning, PCR primers are designed with either HindIII or NotI site overhangs. To minimize mutation introduced by PCR, amplification was done with Pfu polymerase for about 10 cycles on plasmid template. Amplification products were purified with Qiagen PCR purification columns and then digested with appropriate enzymes before cloning in pDB20. Accuracy of all PCR-derived constructs was confirmed by sequencing.
Sequencing and Sequence Analysis.
DNA sequencing was performed by both manual and automated sequencing by using the Sanger dideoxy method. blast, tblastx, and tblastn (23) were used to search the databases. The method of Kyte and Doolittle (37) in dna strider program was used for hydrophobicity analysis. Alignment was done by using the University of Wisconsin Genetics Computer Group package.
SOD1 Assay.
Yeast was grown in SD-ura liquid media, and cells were broken in lysis buffer (25 mM Tris⋅Cl, pH 7.5/150 mM NaCl/1 mM EDTA/1 mM phenylmethylsulfonyl fluoride) by vortexing with glass beads. The protein concentration of the lysate was determined by the Bio-Rad DC protein assay with use of BSA as standard. SOD1 activity was assayed by a nitro blue tetrazolium test in polyacrylamide gel (24). Briefly, 10 μg of protein was loaded in each lane. The gel was soaked with gentle shaking in the dark for 10 min, in coloring stain [100 ml of 0.036 M potassium phosphate buffer, pH 7.8/20 ml of 0.5 M EDTA, pH 8.0/1.13 mg of riboflavin (Sigma)/16.4 mg of nitro blue tetrazolium (Sigma)/175 μl of N,N,N′,N′-tetramethylethylenediamine], and then developed on a light box. SOD1 activity is shown as a colorless band in the blue background. The gel was dried and scanned, and the SOD1 colorless band was inverted to black with adobe photoshop program. SOD1 activity was distinguished by its sensitivity to potassium cyanide at pH 9 (25).
Atomic Absorption Spectrophotometry.
The copper concentration of yeast lysates was determined by using a Perkin–Elmer 2380 Atomic Absorption Spectrophotometer with PE Pure copper (Perkin–Elmer) as standard. hCTR1-UTR(F), hCTR1-UTR(R), and pDB20 transformants were all grown in SD-ura medium (Bio 101). Protein was obtained and concentration was determined as described for the SOD1 assay.
Copper and Iron Sensitivity Assay.
Yeast grown to OD 1.0 was used for a 10-fold serial dilution. A 5-μl volume was applied to each spot. Copper-rich plates were made by adding 1 M CuSO4 to SD-ura media to a final concentration of 900 μM. Iron-limiting plates were made by adding 0.2 M bathophenanthrolinedisulfonic acid disodium salt (Fluka) to SD-ura media to a final concentration of 50 μM.
Northern Blot Analysis and Human cDNA Library Screen.
A CLONTECH multiple-human-tissue blot was probed with the hCTR1 cDNA, stripped, and then reprobed with hCTR2. Both probes were made from the whole coding region. Both hybridization and washing were done under stringent conditions (26). Randomly primed fibroblast (Stratagene) and oligo(dT)-primed liver and placenta libraries (a gift from Jeff Edman, University of California, San Francisco) were screened with hCTR1 and hCTR2 probes.
RESULTS
Isolation of a Human cDNA by Complementation of the Yeast Copper Uptake Mutant, ctr1.
To clone the human CTR1 homologue by functional complementation, we transformed a yeast ctr1 strain with a human cDNA expression library (21) and selected for growth on uracil-deficient plates. About 2 million primary transformants were obtained, and these colonies were collected and pooled. Because yeast ctr1 cannot grow in nonfermenting media because of intracellular copper deficiency, as described previously, a portion of the transformants pool was then plated on YPG or YPE plates for selection of a complementing human cDNA. After 7–10 days of growth, larger colonies were picked and analyzed. Plasmids from 50 individual colonies were purified and retransformed into the ctr1 strain. Five of these 50 clones were able to grow on YPG plates (Fig. 1A). The remaining 45 seemed to be false positives because the original growth phenotype in the YPG(E) plate was not conferred by the harbored human cDNAs; these may have been yeast suppressor mutations. The transformed strain grows rather slowly on YPG compared with the wild-type CTR1 strain, indicating that the recovered cDNA clones complement yeast ctr1 inefficiently, even with high copy number and driven by a strong promoter. As will be shown later, this inefficiency results from a property of the 5′ untranslated region (UTR) of this human clone in the yeast vector.
Figure 1.
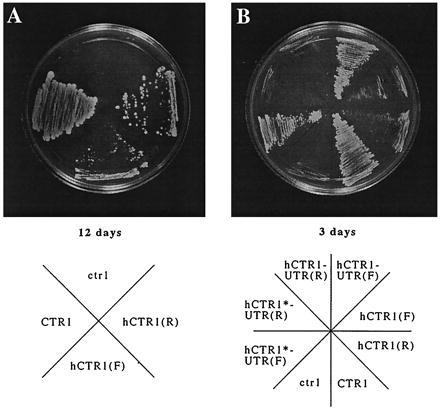
Complementation of yeast ctr1 growth on YPG medium with human hCTR1 cDNA. (A) hCTR1 expression complements ctr1 growth defect on YPG medium. Both sense and antisense hCTR1 cloned in vector pDB20 have the complementing ability. The plate was photographed after 12 days of growth at 30°C. The antisense complements slightly better, as shown by slightly larger colonies. (B) Growth of different hCTR1 transformants on YPG medium compared with ctr1 and CTR1 strains. The photograph was taken after 3 days of growth. Notice that there is only minor growth for either hCTR1(F) or hCTR(R). The growth of hCTR1-UTR(F) transformant is nearly as robust as the wild-type CTR1. (See Table 1 for the nomenclature of constructs and the results.)
Sequence Analysis of the Cloned cDNA.
Sequencing of the plasmids from the five positive clones indicated that they are from the same original clone because they have exactly the same length (1.7 kb), the same sequence, and the same orientation. Translation in three reading frames of the potential transcript from the adh promoter (Padh) to the adh terminator (Tadh) revealed no reading frame longer than 100 aa. However, translation of all six reading frames indicated one potential product of 190 aa in the unexpected orientation, from Tadh to Padh.
In addition to the significance of the derived amino acid sequence (described below), several lines of evidence suggested that this reverse reading frame is authentic. First, there are several mammalian expressed sequence tags (ESTs) that have their 5′-to-3′ direction corresponding to the isolated human cDNA from the Tadh-to-Padh direction (see later in Results). Second, a search (July 1996) against the STS database indicated that the cDNA portion adjacent to Tadh is associated with a human CpG island (accession no. Z65139) and, therefore, is presumably adjacent to the site of transcription initiation. Third, using the putative coding region to screen a cDNA library, we recovered several cDNAs with poly(A)+ tails, all of which are located at the cDNA end adjacent to Padh. Finally, reading from the Tadh-to-Padh direction, the first ATG of the ORF appears to be the authentic translational start site because the sequence is in good agreement with the Kozak consensus sequence AXXATGG and because there is an in-frame stop codon shortly upstream. Thus we conclude that the authentic human transcript corresponds to the transcript from the Tadh-to-Padh direction in the isolated clones.
Comparative analysis of the amino acid sequence derived from the ORF indicates that the human cDNA encodes a protein similar to CTR1 and to COPT1, an Arabidopsis copper transporter also isolated by functional complementation of yeast ctr1 (19) (Fig. 2A). All three proteins are predicted to have three transmembrane domains by hydrophobicity analysis. Interestingly, hCTR1 (190 aa), like its closer relative COPT1 (169 aa), is significantly smaller than its yeast counterpart CTR1 (406 aa), mostly because the carboxyl terminus is truncated. At the amino terminus, hCTR1 and COPT1 lack significant primary sequence identity with CTR1, but they do share some common features. All three proteins are rich in methionine and serine at the amino termini, but hCTR1 is additionally abundant in histidines. The methionine-rich feature is observed in some bacterial copper-transport proteins, including the Enterococcus hirae P-type ATPase CopB and Pseudomonas syringae CopA and CopB (27, 28). The amino termini of two putative Escherichia coli heavy metal transporters are also abundant with histidines (29). As both methionine and histidine are common ligands for copper and as the amino-terminal domains of the CTR1 family of proteins are predicted to lie on the extracellular surface, these domains are likely to serve as scavengers for copper preceding copper uptake.
Figure 2.

Sequence comparison between hCTR1 and some other copper transporters. (A) Sequence comparison of hCTR1, Arabidopsis COPT1, and yeast CTR1. (B) Sequence comparison of hCTR2, hCTR2X (a variant of hCTR2), and yeast CTR2. The cores of putative transmembrane-spanning regions are boxed. With hCTR1 protein as the query, a blastp search revealed only CTR2 as a significant homologous sequence with a score of 98 and a possibility 1.5 × 10−9. Gaps have to be introduced to notice the homology between hCTR1, COPT1, and CTR1. A tblastn search revealed several hCTR2 ESTs, of which the highest scores are for H60113 (score, 117; possibility, 6.4 × 10−8) and W31703 (score, 118; possibility, 8.1 × 10−8).
Analysis of the Effects of the UTRs on Expression in the Yeast Vector.
Because the cDNA isolated by complementation was actually cloned in the reverse orientation and because growth of the complemented ctr1 yeast on glycerol was far less robust than that of wild-type yeast (Fig. 1), we assumed that reorientation of the cDNA to the forward direction would result in a more efficient complementation. This hypothesis turned out to be incorrect as complementation with the forward clone was actually no better, and in fact slightly worse, than that with the reverse clone, as shown by growth at 12 days (Fig. 1A). As shown in Fig. 1B, significant sizes of colonies for wild-type CTR1 yeast were observed about 2–3 days later, whereas there was very limited growth for full-length hCTR1 transformants in either orientation.
These complications prompted further investigation of the effects on expression of the UTRs, as summarized in Table 1. Constructs that removed essentially all of the 3′ UTR (about 1 kb) of hCTR1 resulted in the same behavior as that for the original cDNAs, i.e., both orientations complement and the reverse orientation works slightly better. However, additional removal of the 5′ UTR (about 150 bp) caused a surprising change: the construct with insert in the reverse orientation no longer complements ctr1, but the construct with the insert in the forward orientation does. In fact, the forward 5′-UTR-less construct complements much more efficiently than the full-length original hCTR1 in either orientation and restores growth comparable to that of the wild-type strain on a YPG plate. These experiments also confirm that complementation is indeed due to the predicted hCTR1 protein that lies in the remaining 600 bp. To completely eliminate the possibility that the one short reading frame in the opposite orientation plays any role in ctr1 complementation, we introduced a stop codon near the beginning of its ORF (indicated by an asterisk in Fig. 1B). This mutation also leads to a conservative substitution of serine for threonine at position 186 of hCTR1. The resulting construct still complements as well as hCTR1-UTR, demonstrating that the opposite reading frame does not play any role in the complementation. The result also indicates that the threonine-to-serine change does not affect hCTR1 activity. Based on these results, we conclude that hCTR1 is the protein that effects the complementation of the yeast ctr1 mutation. We thus used the UTR-less hCTR1 in all following studies involving expression of the hCTR1 gene.
Table 1.
Effects of the UTRs on hCTR1 expression in yeast
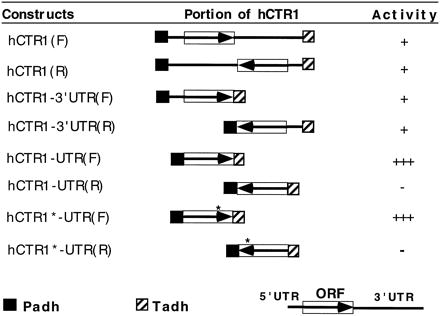
The box indicates the hCTR1 ORF, and the arrow denotes the ORF orientation. 5′ and 3′ UTRs are shown as lines adjacent to the ORF. ∗, A stop codon introduced to the shorter reading frame in the antisense orientation of hCTR1. −, +, and +++, No, weak, and strong activity, respectively, of the constructs to complement ctr1 defect of growth on YPG plates.
Multiple Defects of ctr1 Mutation Complemented by hCTR1 Expression.
The inability to use a nonfermentable carbon source is just one aspect of ctr1 mutation. It has been shown that the ctr1 strain is less sensitive to the killing by high level of copper and that overexpression of CTR1 renders the transformed strain more sensitive (30). We find a similar situation for hCTR1: overexpression of hCTR1 in either CTR1 or ctr1 background makes them more vulnerable to copper overload compared with vector-alone-derived strains (Fig. 3A). Another defect in ctr1 strain is its inability to take up iron with high affinity because of defect in copper incorporation into FET3, an iron oxidase-reductase (18, 31). This high-affinity iron uptake defect is also rescued by hCTR1 expression, as shown in Fig. 3B. The control strain with vector alone cannot grow in SD-ura media in the presence of 50 μM bathophenanthrolinedisulfonic acid disodium salt, whereas the strains with hCTR1 expression grow robustly. Expression of antisense (reverse orientation) UTR-less hCTR1 has no effect as compared with the control (vector alone) in these assays.
Figure 3.
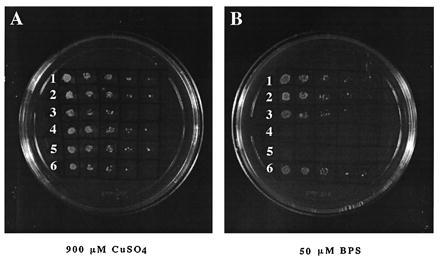
Increased sensitivity to copper overload and rescue of high-affinity iron transport defect of ctr1 strain by hCTR1 expression. (A) hCTR1 expression makes yeast more sensitive to the toxicity of copper overload. Rows: 1 and 4, hCTR1-UTR(R); 2 and 5, pDB20; 3 and 6, hCTR1-UTR(F). The transformed hCTR1 strains and ctr1 mutant strains are grown in rows 1–3 and 4–6, respectively. Spotted from left to right are 10-fold serially diluted yeast cultures. (B) hCTR1 rescues iron transport defect of ctr1 strain. ctr1 yeast cannot grow in 50 μM bathophenanthrolinedisulfonic acid disodium salt, whereas hCTR1 expression rescues this defect. Strains and plasmids in rows 1 to 6 are as in A.
It is also well established that ctr1 yeast is SOD1-deficient in the absence of surplus copper (18). To determine whether hCTR1 can rescue the SOD1 defect observed in ctr1 yeast, we tested SOD1 activity in UTR-less hCTR1(F)-derived, vector-alone-derived, and UTR-less hCTR(R)-derived yeast ctr1 strains. The UTR-less hCTR1(F)-derived strain has obvious SOD activity, whereas the other two have no detectable level of SOD activity (Fig. 4A). This antioxidant activity is inhibited by cyanide, indicating that it is from SOD1 (data not shown). The SOD1 defect in ctr1 strain is presumably a result of low levels of cellular copper. By using atomic absorption spectrophotometry, we were able to show that expression of hCTR1 does increase the cellular copper level in the ctr1 background strain (Fig. 4B). This increase of the cellular copper level presumably results in restoration of SOD1 activity in the ctr1 mutant strain. As expected, there is no effect of hCTR1 on growth on the CTR1 strain compared with the controls (Fig. 3B, rows 1–3).
Figure 4.
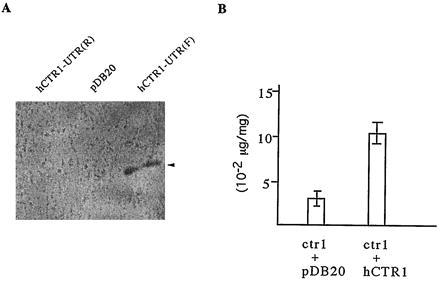
Rescue of SOD1 defect and increase of cellular copper concentration in ctr1 yeast by hCTR1 expression. (A) SOD1 activity is observed in hCTR1 expression strain. The control strains with vector alone or antisense hCTR1 have no detectable SOD1 activity. hCTR1-UTR(R), pDB20, and hCTR1-UTR(F) represent transformed ctr1 yeast strains with antisense UTR-less hCTR1, vector alone, and sense UTR-less hCTR1, respectively. The arrowhead indicates the SOD1 activity band. (B) hCTR1 expression in the ctr1 strain increases its cellular copper concentration. The y axis denotes the copper amount per milligram of protein. The numbers are means of duplicate experiments. hCTR1-UTR(F) was used for hCTR1 expression in ctr1.
A Homologous Human Gene, hCTR2.
hCTR1 was used for a tblastn search (July 1996) against the EST database and a set of overlapping human ESTs that could code for a similar protein were found. Using this information, we obtained the full-length coding region of the related gene by screening cDNA libraries and have named it hCTR2. Though the majority of the hCTR2 cDNAs had identical sequences, one alternatively spliced cDNA (designated hCTR2X in Fig. 2B) was also obtained. The rare, alternative transcript has one extra exon immediately after the hCTR2 start codon, ATG, creating a premature translational stop; however, immediately after the stop codon, another ATG could be used to produce a product that is 56 aa longer but is in-frame with hCTR2.
Although the topology and length of hCTR2 are similar to that of hCTR1, the lack of obvious metal-binding motifs and the lower abundance of histidine and methionine residues suggested to us that perhaps hCTR2 is more analogous to the yeast protein CTR2. CTR2 was proposed to work as a low-affinity copper uptake protein based on two pieces of evidence. First, overexpression of CTR2 make yeast more sensitive to copper overload, and second, the ctr2 mutant strain is more resistant to copper toxicity than the wild-type strain (19). To address the function of hCTR2 in terms of copper transportation, we overexpressed hCTR2 in yeast and tested the resulting strain in a copper-replete plate. Compared with the strain transformed with vector alone, we saw no obvious difference in terms of resistance to the toxicity of high copper levels. Expression of the whole minor form cDNA hCTR2X (including the preceding short reading frame) in yeast gave the same result. Thus we cannot make any conclusions about the function of hCTR2 based on these experiments.
We also noticed two groups of highly similar rodent ESTs (accession nos. AA107389, AA116457, AA124593, D28666, H33015) in the database. They are probably mouse and rat CTR1 homologues because translation of the partially sequenced regions yields predicted products that share more than 90% identity to hCTR1.
Tissue Distribution of Expression of hCTR1 and hCTR2.
By Northern blot analysis of human tissues, both hCTR1 and hCTR2 were found to be expressed in all organs and tissues examined, though at an uneven level (Fig. 5). Liver exhibits the highest level of expression, whereas expression in the brain and skeletal muscle is relatively low.
Figure 5.
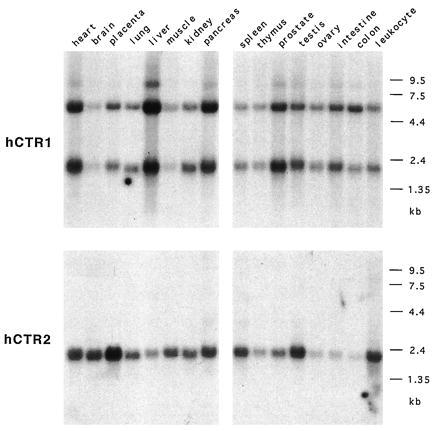
Expression of hCTR1 and hCTR2 by Northern blot analysis. A pair of multiple-tissue Northern blots (CLONTECH) were probed with hCTR1 and hCTR2 coding regions. Approximately 2 (1.5–2.5) μg of mRNA was loaded in each lane.
hCTR1 has two major transcripts of approximately 2 and 5.5 kb and a less abundant transcript of about 8.5 kb. The ratios of these transcripts appear to be relatively constant in all organs/tissues, suggesting that they are under the same transcriptional control (and posttranscriptional control if there is any). The cDNA we obtained in the complementation studies corresponds to the 2-kb isoform. The hCTR1 coding region was used to screen oligo(dT)-primed and randomly primed cDNA libraries in an effort to find any alternatively spliced cDNA forms corresponding to the 5.5-kb mRNA, but none were recovered. We did not use the 3′ UTR to screen the cDNA library because it contains a CA repeat and an Alu repeat.
For hCTR2, a single major 2-kb transcript is observed, corresponding to the longest cDNA we have obtained. hCTR2 is most abundantly expressed in the placenta. We saw markedly reduced expression of hCTR2 in the liver in contrast to hCTR1. Expression of hCTR2 in the small intestine, colon mucosal lining, and peripheral blood leukocyte is also very low.
Localization of hCTR1 and hCTR2 to 9q31/32.
Examination of the STS and EST databases showed that both hCTR1 and hCTR2 have been mapped to 9q31/32. The 3′ UTR of hCTR1 contains a CA repeat marker (D9S262) that was placed in human 9q31/32. ESTs corresponding to hCTR2 were placed by the Massachusetts Institute of Technology Human Genome Project near markers also in 9q31/32. To explore the possibility that CTR1 and hCTR2 are adjacent genes, we tested whether they are colocalized in the same yeast artificial chromosome. Yeast artificial chromosome clones 945D1 and 738F10 (Research Genetics) were reported to contain the D9S262 maker. We confirmed that hCTR1 is indeed located in these two yeast artificial chromosomes by DNA Southern blotting but did not detect hCTR2 in these two yeast artificial chromosomes, suggesting that hCTR1 and hCTR2 are not adjacent genes.
DISCUSSION
Taking advantage of the inability of ctr1 mutant yeast to grow on a nonfermentable carbon source, we isolated a human cDNA that codes for a putative copper transporter. We propose that hCTR1 is a human high-affinity copper uptake gene for the following reasons. First, hCTR1 can complement the yeast ctr1 mutation, which is deficient for high-affinity copper uptake. We have shown that hCTR1 can rescue multiple aspects of defect in ctr1 mutant: the inability to use nonfermentable carbon source, the iron transport defect, and the SOD1 deficiency. Second, hCTR1 expression increases yeast cellular copper concentration (as shown by atomic absorption) and makes yeast more vulnerable to copper overload. Third, hCTR1 and CTR1 are predicted to have similar transmembrane topology in addition to their sequence similarity, suggesting that complementation occurs at the same physiological level (copper transportation) instead of as, for instance, a downstream suppressor. Furthermore, hCTR1 has a copper binding domain that it shares with some bacterial copper transporters, suggesting that hCTR1 is directly involved in copper homeostasis.
Our result attests to the remarkable conservation of proteins between yeast and humans. Indeed, it has been found that a quarter of cloned human disease genes have highly homologous sequences in yeast, and many human genes have been shown to function in yeast (ref. 32; also refer to http://www.ncbi.nlm.nih.gov/XREFdb/). Nevertheless, we still found it intriguing that hCTR1 and Arabidopsis COPT1, two proteins from two evolutionarily distant organisms, both function very well in yeast despite a complete lack of the region homologous to the carboxyl terminus of CTR1. This region has recently been shown to be involved in endocytosis of CTR1 in the presence of excess copper (33). The same authors showed that neither endocytosis nor the bulk of the region is necessary for copper transportation. It is thus expected that even in the presence of excess copper neither COPT1 protein nor hCTR1 will endocytose as authentic CTR1 does. The complementation of the ctr1 mutation by hCTR1 supports previous findings that yeast can be used to study metabolism of copper, and possibly other metals, in mammalian cells.
Our studies show that the 5′ UTR of hCTR1 has an intriguing effect on the expression of this gene in yeast. By trimming UTRs of hCTR1, we show that the predicted hCTR1 coding region is the only reading frame that can complement ctr1 mutation and it only complements in the correct orientation. The 5′ UTR, though only about 150 bp long, somehow leads to inefficient translation in yeast. It appears that the 5′ UTR of hCTR1 may also have some promoter activity in yeast because hCTR1 can function in either orientation and removal of 5′ UTR disrupts the ability to complement in the reverse orientation. The promoter activity in the 5′ UTR may somehow be inhibited by the Padh in the forward orientation. This may be the reason that in the presence of the 5′ UTR, the reverse orientation clone works slightly better than the forward one.
Although hCTR1 can complement multiple defects of ctr1 yeast, neither hCTR2 or its minor, alternatively spliced form hCTR2X, is functional in the yeast assay we performed. It is important to note however, that the copper-resistance assay is not very robust. In addition, we did not make any efforts to optimize cDNA expression as we did with hCTR1. Thus, in our view, speculation that hCTR2 could be the mammalian counterpart of yeast CTR2 remains a possibility.
We noticed that expression of hCTR1 in yeast has a marginal effect in the copper-overload-sensitivity assay. A similar effect was observed in overexpression of COPT1 (19). One possible explanation is that the copper transport capacity of hCTR1 in yeast is not high, although affinity may be high. A second possibility is that the rate limiting factor here may not be hCTR1 but some other factors in the pathway, for instance, FRE1, which is negatively regulated by copper. Another explanation could be that hCTR1 may not be perfectly compatible with other yeast components in the copper transport pathway.
Before these studies, nothing was known about the molecular basis for copper entry into mammalian cells. In the blood, copper is bound to ceruloplasmin, albumin, possibly transcuprein, and small molecules, such as histidine. Although the majority of copper is bound to ceruloplasmin and albumin, neither of these two proteins seems to be necessary for normal copper absorption or transport because aceruloplasminia patients (who lack ceruloplasmin) and analbuminemic rats (with albumin deficiency) both have normal copper metabolism (14–16, 34).
Our results suggest that humans have at least one system for copper uptake homologous to that of yeast. We found that hCTR1 is expressed in all tissues examined, suggesting that all tissues possess the ability to absorb free copper or copper conjugated to small molecules. It would be interesting to ask whether hCTR1 is indispensable for normal copper uptake in human cells. Mammalian cells might have the ability to take up copper either in the free form (or in complex with small molecules like histidine) or in the retrieved from serum proteins such ceruloplasmin, albumin, or transcuprein. If multiple uptake pathways exist, then a copper uptake defect may be manifested only if more than one is disrupted. Also, it is intriguing that yeast has two copper high-affinity transport systems (CTR1 and CTR3) that functionally seem to be redundant. Given the complexity encountered in yeast, it is possible that additional free copper uptake genes are present in man. In our complementation screen of about 2 million clones, only hCTR1 was recovered. We have not eliminated the possibility that some other complementing cDNAs are in the library but were not retrieved or that potential cDNAs do not exist in the library in their full-length forms. On the other hand, we must also keep in mind that all the hCTR1 functional studies have been performed in yeast. Although less likely, we cannot rule out the possibility that hCTR1 could behave differently in human cells .
The positioning of two potential copper-transporting genes to a particular chromosomal location allows one to speculate on their involvement in inherited conditions that map to the position. Several human disorders map to 9q31/32, but two struck us as possible candidate disorders given their reported map position and their physiological basis. One, familial dysautonomia (Riley–Day syndrome), is a neuronal disorder that involves sensory and autonomic neuropathy. The other, Fukuyama muscular dystrophy, is a disease of the nervous system affecting the muscle, brain, and eyes. Preliminary investigation of patient materials suggests that neither disease is associated with a defect in the hCTR1 or hCTR2 gene. Sequence analysis of the coding regions of hCTR1 and hCTR2 cDNAs obtained by reverse transcription–PCR from familial dysautonomia cells revealed no abnormalities. Fukuyama muscular dystrophy has been mapped within a 100-kb region, and neither hCTR1 nor hCTR2 is located there (T. Toda, personal communication).
Aberrant copper transport has been implicated in only one human disease other than Menkes disease (or its variant occipital horn syndrome) and Wilson disease. The fatal, early onset disorder Indian childhood cirrhosis, which may have both genetic and environmental contributions (35, 36), is similar to Wilson disease in that copper accumulates in the liver and seems unlikely to be due to mutations in a ubiquitously expressed gene. Thus the involvement of hCTR1 or hCTR2 with an inherited human condition remains to be seen.
Acknowledgments
The authors thank Dr. L. Guarente for providing the HeLa cDNA yeast expression library, and Drs. A. Dancis, K. Kampfenkel, and M. Minet for providing yeast strains and plasmids. We also gratefully acknowledge Zack Ma for providing technical information about high efficiency yeast transformation, Elaine Carlson for SOD assay, Barbara Levinson for atomic absorption spectrophtometry, Martha Gunthorpe and Jeff Goldman for technical assistance, and Seymour Packman and members of our laboratory for helpful discussions.
ABBREVIATIONS
- SD-ura
synthetic defined medium with glucose as carbon source and uracil omitted
- YPG
yeast extract/peptone/glycerol
- UTR
untranslated region
- EST
expressed sequence tag
- Padh
adh promoter
- Tadh
adh terminator
Footnotes
References
- 1.Linder M C, Hazegh-Azam M. Am J Clin Nutr. 1996;63:797S–811S. doi: 10.1093/ajcn/63.5.797. [DOI] [PubMed] [Google Scholar]
- 2.Vulpe C D, Packman S. Annu Rev Nutr. 1995;15:293–322. doi: 10.1146/annurev.nu.15.070195.001453. [DOI] [PubMed] [Google Scholar]
- 3.Frieden E. Clin Physiol Biochem. 1986;4:11–19. [PubMed] [Google Scholar]
- 4.Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 5.Chelly J, Tumer Z, Tonnesen T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco A P. Nat Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 6.Mercer J F, Livingston J, Hall B, Paynter J A, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D, Clover T W. Nat Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi R E, Petrukhin K, Chernov I, Pellequer J L, Wasco, et al. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 8.Bull P C, Thomas G R, Rommens J M, Forbes J R, Cox D W. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi Y, Heiny M E, Gitlin J D. Biochem Biophys Res Commun. 1993;197:271–277. doi: 10.1006/bbrc.1993.2471. [DOI] [PubMed] [Google Scholar]
- 10.Marceau N, Aspin N. Biochim Biophys Acta. 1973;328:338–350. doi: 10.1016/0005-2795(73)90267-5. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh H S, Frieden E. Biochem Biophys Res Commun. 1975;67:1326–1331. doi: 10.1016/0006-291x(75)90172-2. [DOI] [PubMed] [Google Scholar]
- 12.Campbell C H, Brown R, Linder M C. Biochim Biophys Acta. 1981;678:27–38. doi: 10.1016/0304-4165(81)90044-1. [DOI] [PubMed] [Google Scholar]
- 13.Dameron C T, Harris E D. Biochem J. 1987;248:669–675. doi: 10.1042/bj2480669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris Z L, Takahashi Y, Miyajima H, Serizawa M, MacGillivray R T, Gitlin J D. Proc Natl Acad Sci USA. 1995;92:2539–2543. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida K, Furihata K, Takeda S, Nakamura A, Yamamoto K, Morita H, Hiyamuta S, Ikeda S, Shimizu N, Yanagisawa N. Nat Genet. 1995;9:267–272. doi: 10.1038/ng0395-267. [DOI] [PubMed] [Google Scholar]
- 16.Logan J I, Harveyson K B, Wisdom G B, Hughes A E, Archbold G P. Q J Med. 1994;87:663–670. [PubMed] [Google Scholar]
- 17.Yuan D S, Stearman R, Dancis A, Dunn T, Beeler T, Klausner R D. Proc Natl Acad Sci USA. 1995;92:2632–2636. doi: 10.1073/pnas.92.7.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dancis A, Yuan D S, Haile D, Askwith C, Eide D, Moehle C, Kaplan J, Klausner R D. Cell. 1994;76:393–402. doi: 10.1016/0092-8674(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 19.Kampfenkel K, Kushnir S, Babiychuk E, Inze D, Van Montagu M. J Biol Chem. 1995;270:28479–28486. doi: 10.1074/jbc.270.47.28479. [DOI] [PubMed] [Google Scholar]
- 20.Knight S A, Labbe S, Kwon L F, Kosman D J, Thiele D J. Genes Dev. 1996;10:1917–1929. doi: 10.1101/gad.10.15.1917. [DOI] [PubMed] [Google Scholar]
- 21.Becker D M, Fikes J D, Guarente L. Proc Natl Acad Sci USA. 1991;88:1968–1972. doi: 10.1073/pnas.88.5.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiestl R H, Gietz R D. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Beauchamp C, Fridovich I. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 25.Chang E C, Crawford B F, Hong Z, Bilinski T, Kosman D J. J Biol Chem. 1991;266:4417–4424. [PubMed] [Google Scholar]
- 26.Church G M, Gilbert W. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellano M A, Cooksey D A. J Bacteriol. 1988;170:2879–2883. doi: 10.1128/jb.170.6.2879-2883.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Odermatt A, Suter H, Krapf R, Solioz M. J Biol Chem. 1993;268:12775–12779. [PubMed] [Google Scholar]
- 29.Trenor C, III, Lin W, Andrews N C. Biochem Biophys Res Commun. 1994;205:1644–1650. doi: 10.1006/bbrc.1994.2856. [DOI] [PubMed] [Google Scholar]
- 30.Dancis A, Haile D, Yuan D S, Klausner R D. J Biol Chem. 1994;269:25660–25667. [PubMed] [Google Scholar]
- 31.Askwith C, Eide D, Van Ho A, Bernard P S, Li L, Davis-Kaplan S, Sipe D M, Kaplan J. Cell. 1994;76:403–410. doi: 10.1016/0092-8674(94)90346-8. [DOI] [PubMed] [Google Scholar]
- 32.Bassett D E, Jr, Boguski M S, Hieter P. Nature (London) 1996;379:589–590. doi: 10.1038/379589a0. [DOI] [PubMed] [Google Scholar]
- 33.Ooi C E, Rabinovich E, Dancis A, Bonifacino J S, Klausner R D. EMBO J. 1996;15:3515–3523. [PMC free article] [PubMed] [Google Scholar]
- 34.Vargas E J, Shoho A R, Linder M C. Am J Physiol. 1994;267:G259–G269. doi: 10.1152/ajpgi.1994.267.2.G259. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal S S, Lahori U C, Mehta S K, Smith D G, Bajpai P C. Hum Hered. 1979;29:82–89. doi: 10.1159/000153021. [DOI] [PubMed] [Google Scholar]
- 36.Sethi S, Grover S, Khodaskar M B. Ann Trop Paediatr. 1993;13:3–6. doi: 10.1080/02724936.1993.11747618. [DOI] [PubMed] [Google Scholar]
- 37.Kyte J, Doolittle R F. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]