

Abstract
The HLA class II-associated invariant chain (Ii)-derived peptide (CLIP) occupies the peptide binding groove during assembly in the endoplasmic reticulum, travels with HLA class II to endosomal compartments, and is subsequently released to allow binding of antigenic peptides. We investigated whether the exchange of CLIP with a known T helper epitope at the DNA level would lead to efficient loading of this helper epitope onto HLA class II. For this purpose, a versatile Ii-encoding expression vector was created in which CLIP can be replaced with a helper epitope of choice. Upon supertransfection of HLA-DR1-transfected 293 cells with an Ii vector encoding a known T helper epitope (HA307–319), predominantly length variants of this epitope were detected in association with the HLA-DR1 molecules of these cells. Moreover, this transfectant was efficiently recognized by a peptide-specific T helper clone (HA1.7). The results suggest that this type of Ii vector can be used to create potent class II+ cellular vaccines in which defined T cell epitopes are continuously synthesized.
T cells require short peptides presented in the context of HLA molecules for activation through the T cell receptor. Both HLA class II-restricted T helper cells and HLA class I-restricted cytotoxic T cells are important for the induction and maintenance of immune responses. To this purpose, professional antigen-presenting cells have the capacity to process antigens and present the resulting peptides in association with either HLA class I or HLA class II molecules (1, 2).
The efficient assembly of antigenic HLA class II/peptide complexes requires the presence of a molecular chaperone called the invariant chain (Ii, for review, see ref. 3). In the endoplasmic reticulum, the Ii associates with HLA class II, mainly through aa 81–104 of Ii, termed CLIP (4). This CLIP peptide, which is bound in the class II peptide binding groove, prevents loading of endoplasmic reticulum-resident peptides onto class II (5). Furthermore, signal sequences in the cytosolic tail of the Ii target the nonameric (αβIi)3 complex to endosomal peptide-loading compartments (6), where the Ii is subsequently proteolyzed. As CLIP is bound in the HLA peptide binding groove, it is protected from proteolysis.
The nonclassical HLA molecule, HLA-DM (7–9), and the low pH in these compartments (10) promote the dissociation of CLIP, allowing endosomally located peptides to bind the HLA class II peptide binding groove. The resultant HLA/peptide complexes are transported to the cell surface, allowing detection by T helper cells. In contrast to CLIP, several T helper epitopes have been shown to remain unaffected by the presence of HLA-DM at endosomal pH (7–9, 11). For instance, the influenza virus hemagglutinin (HA) 307–319 peptide (HA307–319) is not released from HLA-DR1 by affinity-purified DM (7, 11).
Peptide binding (12), as well as x-ray crystallography studies (13, 14), have demonstrated that CLIP binds to HLA class II in a fashion similar to well-described T helper epitopes. The main anchoring residues of CLIP bind in the HLA-DR peptide binding groove in pockets at relative positions P1 and P9, which are also used for the binding of helper peptides (12–14). In addition, both CLIP and the HA307–319 peptide form a nearly identical network of hydrogen bonds to conserved class II residues (13, 14). Given these similarities, the genetic exchange of CLIP for a T helper epitope might result in homogeneous loading of this epitope onto class II, which could be used to generate cells that constitutively synthesize and express immunogenic class II/peptide complexes.
To this end, HLA-DR1(HLA-DRA/DRB*0101)-transfected 293 cells were supertransfected with a Ii vector in which the CLIP sequence had been replaced by a sequence encoding the antigenic core of a known T helper epitope. This model peptide (HA307–319) is the immunodominant epitope of the influenza virus in HLA-DR1+ individuals (15). We find that this versatile invariant chain vector can be used to ensure the continuous, functional, and high density expression of this T helper epitope on DR1 positive cells.
MATERIALS AND METHODS
Cell Lines.
The T helper clone HA1.7, which is HLA-DR1 (HLA-DRA/DRB*0101) restricted and HA307–319 specific, was a generous gift of J. Lamb (15). The HLA-DR1-transfected human embryonic kidney 293 cell line (16) was a generous gift of J. Neefjes (Nederlands Kanker Instituut, Amsterdam). The HLA-DR1-homozygous Epstein–Barr virus (EBV)-transformed B cell line SA and the 293 transfectants were grown in Iscove’s medium supplemented with 8% fetal calf serum (FCS). The medium for the HLA-DR1+ 293 cells contained 250 μg/ml G418/neomycin, while the medium for the invariant chain supertransfected cells was supplemented with 150 μg/ml hygromycin. The HLA-DR-specific hybridoma B8.11.2 (17) was grown in protein free medium. Hybridoma supernatant was concentrated by dialysis and the antibodies were purified using a protein A column before use.
Plasmid DNA Constructs.
A cDNA coding for human p31/p33 Ii (a kind gift of J. Neefjes, ref. 18) was used to generate a cassette vector in which the CLIP-encoding sequence can be replaced with other sequences. The region upstream of CLIP was amplified by PCR using primers 5′-ACATTGGATCCTCCTTGGGGAGTGATGCAC-3′ and 5′-CTTGGAATTCCTTCGAAACAGGCTTGGGAGGCTTGGGAAG-3′, the downstream region was amplified with primers 5′-GAAGGAATTCCAAGCGCTGCCCATGGGAGCCCT-3′ and 5′-CAGGATCTCGAGGACCGCCTCTGCTGCTCT-3′. The products of these PCRs were blunted and phosphorylated, and subsequently ligated into the pIc20H vector. The correct sequence of the inserts of both constructs was confirmed through DNA sequencing. From these constructs the upstream region was isolated as a BamHI/EcoRI fragment, whereas the downstream region was isolated as an EcoRI/XhoI fragment. Both fragments were simultaneously ligated into pcDNAI/Amp, which had been cut with BamHI and XhoI. The resulting gene construct encodes a modified Ii, which carries unique SfuI and Eco47III cloning sites instead of the CLIP peptide encoding sequence. Into these sites double-stranded oligonucleotides, with sequences encoding the peptide of choice, were cloned. The sequences of these oligonucleotides were as follows: HA, 5′CGAAGTATGTCAAGCAAAACACCCTGAAACTACAAGC-3′/5′-GCTTGTAGTTTCAGGGTGTTTTGCTTGACATACTT-3′; CLIP, 5′-CGAAGATGCGCATGGCCACCCCGCTGCTGATGCAAGC-3′/5′-GCTTGCATCAGCAGCGGGGTGGCCATGCGCATCTT-3′; Mycobacterium tuberculosis HSP, 5′-CGAAGATTGCATACGACGAAGAGGCTCGACGTCAAGC-3′/5′-GCTTGACGTCGAGCCTCTTCGTCGTATGCAATCTT-3′. Again, the inserts were checked by DNA sequencing. The resulting plasmids were termed pHA, pCLIP, and pHSP, respectively.
Transfections.
Supertransfection of HLA-DR1+ 293 cells was performed as described (19). Briefly, 100,000 HLA-DR1-transfected 293 cells were seeded in 5-cm dishes the day before supertransfection. For supertransfection, 9 μg pHA, pCLIP, or pHSP, together with 1 μg hygromycin-resistance plasmid pTk hygro (20) (per dish) was precipitated with CaCl2. The precipitate was added to the cells. After 4 hr of incubation at 37°C and 5% CO2/95% air, the cells were shocked for 1.5 hr with 100% dimethyl sulfoxide. Hygromycin was added the next day at a final concentration of 150 μg/ml. Within 3 weeks, individual colonies were picked and expanded. Invariant chain positive transfectants were identified by intracellular fluorescence-activated cell sorter (FACS; Becton Dickinson) staining.
FACS Analysis.
For intracellular staining, cells were fixed in 4% paraformaldehyde for 5 min and then washed twice with PBS/10% FCS. The cells were then incubated with a 1,000-fold dilution of anti-Ii antibody Bu45 (Serotec) in PBS/10% FCS containing 0.2% Triton X-100 for 30 min at room temperature. They were then washed twice with PBS/10% FCS. Incubation with fluorescein isothiocyanate FITC-labeled goat anti-mouse IgG took place for another 30 min at room temperature. The cells were washed again and analyzed on a Becton Dickinson FACScan apparatus, using the lysisii software. For cell surface labeling, the Triton X-100 was omitted from the protocol.
Peptide Elution.
Peptide elution was performed as described (21). Briefly, 250*10 6 cells were lysed in 0.5% Nonidet P-40. The lysate was precleared with Sepharose CL-4B beads (Pharmacia). HLA-DR molecules were precipitated from the precleared lysate with MoAb B8.11.2 coupled to CNBr-activated Sepharose CL-4B beads. These beads were then washed extensively, before elution with 10% acetic acid. Subsequently, the peptides were separated from the HLA-DR α and HLA-DR β chains using Amicon filters with a cut-off value of 10 kDa. The peptide-containing low molecular weight fraction was concentrated by lyophilization. This material was fractionated by reverse-phase HPLC on a C2C18 SC2.1/10 SMART column (Pharmacia). HPLC fractions were analyzed by mass spectrometry on a LASERMAT (Finnigan-MAT, San Jose, CA), and were subjected to Edman degradation on a Hewlett–Packard G1005A sequencer.
Proliferation Assays.
T cells (20,000) were added to each well of a 96-well plate containing mitomycin C-treated (Sigma–Aldrich) stimulator cells (800, unless indicated otherwise), which had been pre-incubated with peptide for 3 hr when indicated. [3H]Thymidine was added (0.5 μCi/well; 1 Ci = 37 GBq) on day 3. After overnight incubation, thymidine incorporation was measured. Peptides were synthesized by standard 9-fluorenylmethoxycarbonyl chemistry on a multiple peptide synthesizer (AMS 422, Abimed Analyes-Technik, Langenfeld, Germany). Their identity was confirmed by mass spectrometry.
RESULTS AND DISCUSSION
Generation of Ii Vectors.
An invariant chain (Ii) vector was created in which the core CLIP sequence KMRMATPLLMQA can be exchanged with a sequence of choice (Fig. 1). For this purpose, a CLIP-cassette that is flanked by unique restriction sites was generated by site-directed mutagenesis (see Materials and Methods). To prevent improper folding of the recombinant Ii, we limited the modification to the sequence spanning the peptide binding groove, beginning in the P1 pocket and ending in the P9 pocket (MRMATPLLM). As a model epitope, the core of the influenza virus HA epitope HA307–319 (YVKQNTLKL) was chosen (vector pHA). Control vectors (Fig. 1) contained wild-type (wt) CLIP (pCLIP) and the HLA-DR3 (HLA-DRA/DRB*0301)-restricted Mycobacterium tuberculosis HSP5–13 epitope (pHSP).
Figure 1.
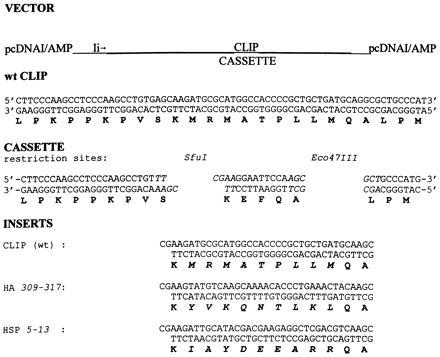
Invariant chain constructs. The cassette-containing Ii was cloned into the BamHI/XhoI-digested pcDNAI/amp vector. The cassette is flanked by unique restriction sites SfuI/Eco47III. One-letter codes for the amino acids are given in boldface type. Inserts CLIP, HA, and HSP were generated by annealing the encoding oligonucleotides. Inserts were ligated into the SfuI/Eco47III-digested cassette plasmid, resulting in plasmids pCLIP (wt p31/33 Ii), pHA, and pHSP.
FACS Analysis.
After supertransfection of HLA-DR1+ 293 cells with either the vector encoding the core of the HA peptide (pHA), pCLIP, or pHSP, single colonies were picked and expanded. Transfectants were subsequently screened for invariant chain and HLA-DR-expression by FACS analysis. Transfectants 293-pHA, 293-pHSP, and 293-pCLIP expressed high levels of Ii, whereas the control transfectant 293-Tk which had been transfected with the hygromycin resistance plasmid only, was negative for intracellular Ii (Fig. 2). All cell types were positive for HLA-DR on the cell surface (Fig. 2).
Figure 2.
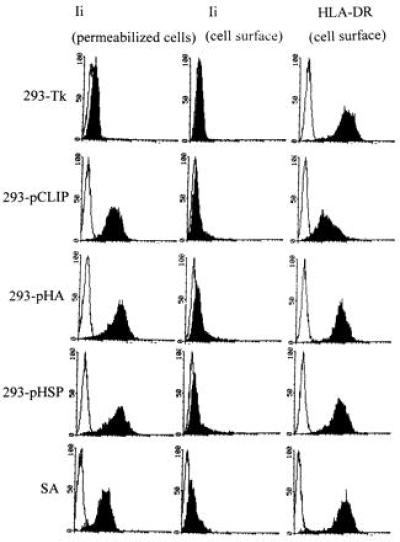
FACS analysis of the transfectants. Transfectants 293-pHA, 293-pCLIP, and 293-pHSP and control transfectant 293-Tk were screened for intracellular invariant chain expression (first column), as well as for cell surface expression of HLA-DR (third column) and invariant chain (second column). For reference, the EBV-transformed B cell line SA was also analyzed. Open areas indicate background staining.
Peptide Elution of the pHA Transfectant.
To investigate the efficiency of peptide loading in these transfectants, peptides were eluted from immunoprecipitated HLA-DR molecules from the 293-pHA transfectant. The peptide fraction was subsequently separated by reverse-phase HPLC. The absorption profile at 214 nm showed several distinct peaks (Fig. 3). The corresponding fractions were analyzed by mass spectrometry and sequenced by Edman degradation. The five largest peaks all contained length variants of the vector-encoded peptide HA-CLIP (Table 1). Two minor peaks were shown to contain nonvector-encoded peptides. As a control, the peptides identified in fractions 50, 80, and 87 were synthesized and shown to coelute with the original material upon reverse-phase HPLC separation (data not shown).
Figure 3.
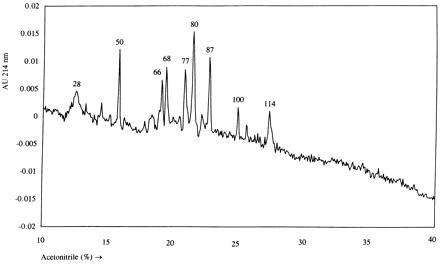
Reverse-phase HPLC analysis of HLA-DR1-eluted peptides isolated from transfectant 293-pHA. During a linear gradient of 10–40% of acetonitrile in water containing 0.1% trifluoroacetic acid, 180 fractions were collected. Fraction numbers corresponding to peaks with an absorption of >0.005 absorption unit (AU) at 214 nm are given. The peaks at fractions 28 and 77 are ghost peaks, also present in the reverse-phase HPLC profile of eluates from mock-precipitations (data not shown).
Table 1.
Peptide sequences from the 293-pHA HLA-DR peptide elution experiment
| Fraction | Mass (MH+, Da) | Amino acid sequence |
|---|---|---|
| 50 | 2,168 | KPPKPVSKYVKQNTLKLQA |
| 66 | 2,282 | KPPKPVSKYVKQNTLKLQAL |
| 68 | 2,380 | KPPKPVSKYVKQNTLKLQALP |
| 80 | 2,588 | LPKPPKPVSKYVKQNTLKLQALP |
| 87 | 2,719 | LPKPPKPVSKYVKQNTLKLQALPM |
| 100 | ND | RETNLDSL |
| 114 | ND | ND |
Masses were determined by mass spectrometry and sequences by Edman degradation. The combination of mass and N-terminal sequence allowed completion of the sequences. In fraction 100, no mass was detected (ND, none detected), but a vimentin-derived peptide, starting at residue 423, could be partially sequenced. Neither mass nor sequence was found in fraction 114, although the presence of peptide material could be established by Edman degradation.
In conclusion, a large proportion of the HLA-DR molecules on the transfectant 293-pHA is filled with length variants of the vector-encoded HA-CLIP peptide (Table 1), demonstrating a high efficiency of peptide loading by this Ii vector. These length variants are identical to naturally occurring CLIP peptides in HLA-DR1 (22), which suggests that processing of Ii variants occurs similarly to the degradation of wt Ii.
Functional Analysis of the Transfectant.
The transfectant 293-pHA was tested for its capacity to stimulate T cell clone HA1.7. This T helper clone recognizes HA307–319 in the context of HLA-DR1. For comparison, control transfectants 293-Tk, 293-pCLIP, and 293-pHSP, as well as the HLA-DR1-homozygous EBV-transformed B cell line SA, were also tested (Fig. 4A). When loaded with 1 μg/ml of the 24-mer HA-CLIP peptide (LPKPPKPVSKYVKQNTLKLQALPM), all cell types stimulated the proliferation of HA1.7 equally well. In contrast, in the absence of peptide, only 293-pHA was able to stimulate T helper clone HA1.7.
Figure 4.

T cell stimulating capacity of the 293-pHA transfectant in comparison with control transfectants 293-Tk, 293-pCLIP, 293-pHSP, and EBV-transformed B cell line SA. (A) Stimulation of HA1.7 by stimulator cells in the presence or absence of 1 μg/ml of peptide. (B) HA1.7 proliferation induced by transfectant 293-pHA and control cells in the presence of various doses of peptide. (C) Stimulator cell titration. Control cells have been pulsed with the indicated amount of peptide for 3 hr. The cpm values are medians of triplicates (SEM < 5%). The data are representative of three separate experiments.
To investigate the efficiency of the interaction between a peptide-reactive T helper cell and this type of transfectant, two types of experiments were performed. In the first experiment, peptide HA-CLIP was titrated from 10 μg/ml to 100 pg/ml, and added to various stimulator cells (Fig. 4B). The response of HA1.7 to 293-pHA could not be increased by the addition of extra peptide. In all cases, the response to 293-pHA was at least as high as the response to peptide-loaded stimulator cells. Therefore, supertransfection of the HLA-DR1+ 293 cells with Ii vector pHA results in optimal presentation of the vector-encoded T helper epitope on the cell surface.
In the second type of experiment, stimulator cells, either peptide-pulsed or not, were titrated from 25.000 to 32 cells per well, and tested for stimulation of T cell clone HA1.7. Transfectant 293-pHA was compared with the EBV-transformed B cell line SA and the control transfectant 293-Tk, which were pulsed with 10 or 500 μg/ml HA-CLIP peptide for 3 hr. Only 160 293-pHA cells were needed to induce T cell proliferation, whereas approximately 10-fold more peptide-pulsed cells were necessary to induce a similar proliferative response. Taken together, these data show that the 293-pHA transfectant is a specific and efficient stimulator of T helper clone HA1.7.
These results show that, in a HLA-DM and Ii-negative cell line, a CLIP substitute-containing vector can be used to efficiently load a specific peptide onto HLA class II for presentation to T cells. Since it has been shown that HLA-DM is unable to enhance the dissociation of T helper epitope HA307–319 from HLA-DR1 or the dissociation of HSP3–13 from HLA-DR3 at endosomal pH (11), it is likely that in HLA-DM-positive cells the CLIP substitute-containing vector can be used to load T helper peptides of choice with similar efficiency. However, there will be an inverse correlation between the sensitivity of the chosen peptide to HLA-DM-mediated dissociation and the loading efficiency. In the presence of wt Ii, the efficiency of peptide loading will depend on the expression level of the recombinant Ii. The expression level of recombinant Ii that was reached in this set of experiments was as high as the endogenous expression of wt Ii in an EBV-transformed B cell line (Fig. 2). Thus, competition for binding to class II molecules between the wt Ii and the transfected recombinant Ii can be expected and is likely to result in reasonable loading of the helper peptide of choice in Ii positive cells as well.
Efficient and stable steady-state loading of HLA class II with peptides of choice may be a useful tool for the development of effective cellular vaccines. Exogenous loading of synthetic peptides onto HLA class II results in antigenic complexes with a limited life span, which may be antigenically distinct from those generated by intracellular processing (23). Fusion genes using the Ii- or LAMP-1 sorting signals have been used to target protein antigen to the class II processing pathway (24, 25), but this does not result in the homogeneous loading of T helper epitopes as obtained by replacement of CLIP. An alternative approach has been to place the peptide at the C terminus of Ii behind a cathepsin D cleavage site (26), which results in efficient generation of the epitope of choice in the major histocompatibility complex class II compartment. To achieve homogeneous loading of a peptide onto class II, others have used constructs in which the peptide was covalently linked to class II by a flexible linker (27). In contrast, our approach makes use of the natural assembly pathway for class II/Ii complexes to obtain homogeneous loading of T helper epitopes on DR positive cells. It will be of interest to investigate if such cells can be used to modulate immune responses in vitro and in vivo. This will be the subject of further studies.
Acknowledgments
We thank Dr. T. H. M. Ottenhoff, Dr. B. O. Roep, Dr. J.-W. Drijfhout, and Dr. E. Zanelli for carefully reviewing the manuscript. We are also indebted to Dr. J. Neefjes for helpful discussions and for kindly providing the HLA-DR1-transfected 293 cells, as well as the Ii-encoding cDNA. We are grateful to Willemien Benckhuizen and Jan-Wouter Drijfhout for peptide synthesis. J.v.B., F.V., and F.K. were supported by Netherlands Organization for Scientific Research Grant 030-93-001.
ABBREVIATIONS
- CLIP
class II-associated invariant chain (Ii)-derived peptides
- HA
hemagglutinin
- HSP
heat shock protein
- FACS
fluorescence-activated cell sorter
- wt
wild type
- EBV
Epstein–Barr virus
References
- 1.Jondal M, Schirmbeck R, Reimann J. Immunity. 1996;5:295–302. doi: 10.1016/s1074-7613(00)80255-1. [DOI] [PubMed] [Google Scholar]
- 2.Norbury C C, Hewlett L J, Prescott A R, Shastri N, Watts C. Immunity. 1995;3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 3.Cresswell P. Cell. 1996;84:505–507. doi: 10.1016/s0092-8674(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 4.Freisewinkel I M, Schenck K, Koch N. Proc Natl Acad Sci USA. 1993;90:9703–9706. doi: 10.1073/pnas.90.20.9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busch R, Cloutier I, Sekaly R-P, Haemmerling G. EMBO J. 1996;15:418–428. [PMC free article] [PubMed] [Google Scholar]
- 6.Arneson L S, Miller J. J Cell Biol. 1995;129:1217–1228. doi: 10.1083/jcb.129.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherman M A, Weber D, Jensen P E. Immunity. 1995;3:197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 8.Denzin L K, Cresswell P. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 9.Sloan V S, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller D M. Nature (London) 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 10.Kropshofer H, Vogt A B, Stern L J, Haemmerling G J. Science. 1995;270:1357–1359. doi: 10.1126/science.270.5240.1357. [DOI] [PubMed] [Google Scholar]
- 11.Weber D A, Evavold B D, Jensen P E. Science. 1996;274:618–620. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- 12.Geluk A, van Meijgaarden K E, Drijfhout J-W, Ottenhoff T H M. Mol Immunol. 1995;32:975–981. doi: 10.1016/0161-5890(95)00058-m. [DOI] [PubMed] [Google Scholar]
- 13.Stern L J, Brown J H, Jardetzky T S, Gorga J C, Urban R G, Strominger J L, Wiley D C. Nature (London) 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh P, Amaya M, Mellins E, Wiley D C. Nature (London) 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 15.Lamb J R, Eckels D D, Lake P, Woody J N, Green N. Nature (London) 1982;300:66–69. doi: 10.1038/300066a0. [DOI] [PubMed] [Google Scholar]
- 16.Calafat J, Nijenhuis M, Janssen H, Tulp A, Dusseljee S, Wubbolts R, Neefjes J. J Cell Biol. 1994;126:967–977. doi: 10.1083/jcb.126.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rebai N, Malissen B, Pierres M, Accolla R S, Corte G, Mawas C. Eur J Immunol. 1983;13:106–111. doi: 10.1002/eji.1830130205. [DOI] [PubMed] [Google Scholar]
- 18.Claesson L, Larhammar D, Rask L, Peterson P A. Proc Natl Acad Sci USA. 1983;80:7395–7399. doi: 10.1073/pnas.80.24.7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham F L, van der Eb A J. Virology. 1977;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 20.Kast W M, Offringa R, Peters P J, Voordouw A C, Meloen R H, van der Eb A J, Melief C J. Cell. 1989;59:603–614. doi: 10.1016/0092-8674(89)90006-8. [DOI] [PubMed] [Google Scholar]
- 21.Verreck F A W, Elferink D, Vermeulen C J, Amons R, Breedveld F, Vries R R P, Koning F. Tissue Antigens. 1995;45:270–275. doi: 10.1111/j.1399-0039.1995.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 22.Chicz R M, Urban R G, Corga J C, Vignali D A A, Lane W S, Strominger J L. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viner N J, Nelson C A, Deck B, Unanue E R. J Immunol. 1996;156:2365–2368. [PubMed] [Google Scholar]
- 24.Wu T-C, Guarnieri F G, Stavely-O’Carrol K F, Viscidi R P, Levitsky H I, Hedrick L, Cho K R, August J T, Pardoll D. Proc Natl Acad Sci USA. 1995;92:11671–11675. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanderson S, Frauwirth K, Shastri N. Proc Natl Acad Sci USA. 1995;92:7217–7221. doi: 10.1073/pnas.92.16.7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano N, Rooke R, Benoist C, Mathis D. Science. 1997;275:678–683. doi: 10.1126/science.275.5300.678. [DOI] [PubMed] [Google Scholar]
- 27.Ignatowicz L, Kappler J, Marrack P. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]