

Abstract
To develop a strategy that promotes efficient antiviral immunity, hybrid virus-like particles (VLP) were prepared by self-assembly of the modified porcine parvovirus VP2 capsid protein carrying a CD8+ T cell epitope from the lymphocytic choriomeningitis virus nucleoprotein. Immunization of mice with these hybrid pseudoparticles, without adjuvant, induced strong cytotoxic T lymphocyte (CTL) responses against both peptide-coated- or virus-infected-target cells. This CD8+ class I-restricted cytotoxic activity persisted in vivo for at least 9 months. Furthermore, the hybrid parvovirus-like particles were able to induce a complete protection of mice against a lethal lymphocytic choriomeningitis virus infection. To our knowledge, this study represents the first demonstration that hybrid nonreplicative VLP carrying a single viral CTL epitope can induce protection against a viral lethal challenge, in the absence of any adjuvant. These recombinant particles containing a single type of protein are easily produced by the baculovirus expression system and, therefore, represent a promising and safe strategy to induce strong CTL responses for the elimination of virus-infected cells.
CD8+ cytotoxic T lymphocytes (CTLs) play an important role in the elimination of cells infected by pathogens and in the regression of tumors. CTLs recognize antigen-derived peptides presented by major histocompatibility complex (MHC) class I molecules on the cell surface and are usually activated by peptides resulting from the processing of endogenous intracellular proteins (1). Because antigens have to gain access to the cytosol to enter the class I-restricted presentation pathway, exogenous soluble proteins are usually unable to stimulate CTL responses. Therefore, several strategies have been developed to deliver exogenous antigens into the cytosol. Protein or peptide antigens delivered in association with appropriate adjuvants [complete Freund’s adjuvant (2), incomplete Freund’s adjuvant (3), or saponin (4)], liposomes (5), ISCOMs (6), or in particulate form linked to latex microspheres (7) efficiently stimulate CTL responses. However, alum (aluminum salts) is still the only adjuvant currently licensed for use in human vaccines. Recombinant live vectors [such as attenuated virus, vaccinia virus (8), mengo virus (9)] or bacteria [bacillus Calmette–Guérin (10), Salmonella (11), or Listeria (12)] have also been shown to sensitize CTLs in vivo but are risk-prone. Recombinant canarypox virus expressing gp160 from HIV-1, which cannot replicate in mammalian species, was recently shown to induce CTL responses in humans but only in less than 40% of the volunteers (13). DNA vaccination may also represent a powerful strategy to activate CTL responses (14), but the safety of this method remains to be determined. Therefore, the development of a safe strategy to induce CTL responses with nonreplicating antigens is still an important prerequisite for the design of new efficient vaccines.
Recently, we have developed an antigen delivery system based on hybrid recombinant parvovirus like-particles [porcine parvovirus virus-like particles (PPV:VLP)] formed by the self-assembly of the VP2 capsid protein of PPV carrying a foreign epitope at its N terminus. We analyzed the capacity of these nonreplicative pseudoparticles, PPV:VLP, to prime in vivo class I-restricted cytotoxic responses. For this purpose, the CD8+ CTL epitope, residues 118–132 from the lymphocytic choriomeningitis virus (LCMV) nucleoprotein (15, 16), was inserted into the VP2 capsid protein of PPV [PPV:VLP-(LCMV)]. After expression in insect cells with the baculovirus vector system, the recombinant VP2 protein spontaneously self-assembles into VLPs with a morphology very similar to the native capsid. Recombinant PPV:VLP expressing the LCMV epitope were analyzed for their ability to stimulate in vivo specific cytotoxic responses and to protect mice against a lethal infection with the virus.
The present study demonstrates that chimeric PPV:VLP carrying a single LCMV CTL epitope induced a strong CD8+ class I-restricted CTL response that killed virus-infected cells. Moreover, in vivo immunization with these recombinant PPV:VLP-(LCMV) fully protected mice against lethal choriomeningitis and allowed complete viral clearance in the surviving mice.
METHODS
Mice, Virus, and Peptide.
Female BALB/c mice, 8–10 weeks old, were purchased from Iffa Credo (L’Arbresle, France). LCMV strain Arm/53b was kindly given by M. B. A. Oldstone and M. McChesney (Scripps Clinic, La Jolla, CA). The p118–132 synthetic peptide RPQASGVYMGNLTAQ corresponding to a H-2d-restricted CTL epitope from the LCMV nucleoprotein (15, 16) was synthesized by Neosystem (Strasbourg, France).
Construction of a Recombinant Baculovirus Expressing PPV:VLP-(LCMV).
Oligonucleotide 5′-TCGAGATGCGACCACAAGCTTCAGGAGTATACATGGGAAACCTAACAGCACAAC-3′ and its complementary were designed to encode the LCMV epitope of residues 118–132 (LCMV 118–132 epitope) plus an initiation codon and two flanking XhoI sites. The oligonucleotides were obtained from MedProbe (Oslo), phosphorylated with T4 polynucleotide kinase, annealed at 70°C for 15 min, ligated into XhoI-digested pPPV29 mod (which contains the PPV VP2 gene), and transformed into Escherichia coli D115 cells. Recombinants containing the LCMV insert were sequenced by dideoxynucleotide procedures to determine the orientation and integrity of the inserted sequences. The recombinant clone containing the LCMV 118–132 epitope in the proper orientation was named pPPV29 mod/LCMV. The chimeric VP2 sequence was released from pPPV29 mod/LCMV by BamHI digestion and subcloned into the unique restriction site BamHI from the baculovirus transfer vector pAcYM1. Recombinant clones were analyzed by restriction mapping. The recombinant clone was called pAcYM1/LCMV/ppv29 mod.
To generate the recombinant baculoviruses, a mixture of 2 μg of purified transfer vector DNA plus 500 ng of parental baculovirus DNA AcRP23lacZ+ was added to Spodoptera frugiperda clone 9 (Sf9) insect cells in the presence of the transfection reagent N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP; Boehringer Mannheim). Recombinant baculoviruses were prepared by standard procedures and plaque-purified until no more blue plaques (wild-type) could be detected. Then, high-titer viral stocks of the recombinant baculovirus AcNPV.PPV-LCMV (>108 viruses) were prepared.
Characterization and Purification of Recombinant PPV:VLP-(LCMV).
Sf9 cells were infected with AcNPV.PPV-LCMV at a multiplicity of infection of 1 plaque-forming unit (pfu) per cell. Cellular extracts were harvested at 3 days after infection. Protein dissociation buffer was added to each cell extract, the mixtures were heated at 100°C for 5 min and loaded on SDS/9% polyacrylamide gels. The gels were stained with Coomassie blue or transferred to nitrocellulose membranes for Western blot analysis of PPV VP2 and LCMV 118–132 epitope. The membranes were incubated with rabbit serum containing anti-PPV or mouse serum containing anti-LCMV p118–132 for 2 h at room temperature. After washing, bound antibodies were detected by peroxidase-conjugated protein A with 4-chloronaphthol as substrate until development of color.
For PPV:VLP purification, cells were harvested 72 h after infection, washed with PBS, and lysed by osmotic shock with 25 mM bicarbonate solution at 4°C. PPV:VLP were then purified by salt precipitation with 20% ammonium sulfate. The precipitate was recovered by centrifugation at 12,000 × g for 20 min, resuspended in PBS, and dialyzed overnight against PBS. The identity and properties of the particles were confirmed by SDS/PAGE, immunoblotting, and electron microscopy. Characterization of PPV:VLP obtained by CsCl sedimentation analysis and electron microscopy revealed properties identical to native PPV virions.
Mice Immunization and in Vivo Depletion of CD4+ or CD8+ Cells.
Purified VLPs were diluted in PBS. Mice (two to five mice per group) were injected i.p. in the absence of adjuvant with 10 μg of control or recombinant PPV:VLP or with 105 pfu of LCMV (17). In some experiments, mice were injected i.p. with anti-CD4 (GK1.5) or anti-CD8 (H35.17.2) mAbs prepared from ascitic fluids as described (18). They received 300 μg of mAbs at each injection as detailed in figures.
In Vitro Cytotoxic Assay.
After immunization of mice, spleens were surgically removed and spleen cells were in vitro-stimulated with 1 μM p118–132 peptide and tested for their cytotoxic activity in a 51Cr release assay, as described (19). 51Cr-labeled target cells, P815 (H-2d), EL4 (H-2b), RDM4 (H-2k), or 1T22 (H-2q), were pulse-labeled with 50 μM p118–132, and the J774 macrophage-like cell line was infected with LCMV. The released radioactivity was measured in the supernatants. The percentage of specific lysis was calculated as 100 × (experimental release − spontaneous release)/(maximum release − spontaneous release). Maximum release was generated by adding 1 M HCl (P815 assay) or 1% Triton X-405 (J774 assay) to target cells, and spontaneous release was obtained with target cells incubated without effector cells. In all experiments, the spontaneous release from the various targets did not exceed 20%.
Virus Protection Experiment.
LCMV (101.7 pfu) were inoculated intracerebrally (30 μl) to perform protection experiments. Death and survival were recorded during 21 days after the viral challenge and clearance of the virus was checked by anti-LCMV antigen-capture ELISA on mice kidneys. Purified anti-LCMV guinea pig IgG was used as the capture antibody, and purified anti-LCMV mouse IgG was used to detect captured LCMV. Alkaline phosphatase-linked anti-mouse IgG antibodies were then added to detect bound anti-LCMV mouse antibodies.
RESULTS
Preparation of Recombinant PPV:VLP Carrying a LCMV CD8+ T Cell Epitope.
The baculovirus vector system was used for the expression of the chimeric PPV:VLP containing the LCMV epitope. After electrophoretic analysis and Coomassie blue-staining, a visible extra band of 67 kDa was detected in the extracts of Sf9 cells infected with the recombinant baculovirus AcNPV.PPV-LCMV (Fig. 1a). The size observed corresponds to the expected size for the chimeric VP2 protein carrying the LCMV epitope. The identity of this protein was confirmed by immunoblotting. Specific antisera against the PPV VP2 or LCMV 118–132 epitope gave a clear positive reaction with the 67-kDa protein (Fig. 1b). The chimeric VP2 protein was expressed as a cytoplasmic soluble protein that was easily purified by osmotic shock lysis and salt precipitation. Electron microscopy analysis of purified PPV:VLP-(LCMV) confirmed its 25-nm particle structure (Fig. 1c).
Figure 1.

Analysis of the expression in insect cells of the recombinant PPV:VLP carrying the 118–132 LCMV CD8+ epitope. (a) Coomassie blue-stained SDS/polyacrylamide gels showing the different steps of PPV:VLP-(LCMV) purification. Lanes: M, molecular mass markers; 1, mock-infected Sf9 cells; 2, crude extract from infected cells; 3, supernatant from lysed cells; 4, pellet from 20% ammonium sulfate precipitation. (b) Immunoblotting analysis of PPV:VLP-(LCMV) with anti-PPV rabbit antiserum (lane 1) and anti-LCMV 118–132 peptide mouse antiserum (lane 2). (c) Electron microscopy analysis of PPV:VLP-(LCMV) particles negatively stained with 2% uranyl acetate.
In Vivo Induction of Peptide-Specific Cytotoxic Responses by PPV:VLP Carrying a LCMV CD8+ Epitope.
PPV:VLPs carrying the H-2d CTL epitope of residues 118–132 from LCMV nucleoprotein (15, 16) were analyzed for their capacity to stimulate in vivo cytotoxic responses against the inserted epitope. BALB/c (H-2d) mice were injected i.p. once or twice with 10 μg of either control or recombinant PPV:VLP, in the absence of any adjuvant (Fig. 2). Fourteen or 7 days later, respectively, immune splenocytes were in vitro-stimulated with the priming 118–132 peptide (p118–132) for 5 days. The cytotoxic activity of these effector cells was then tested on p118–132-coated P815 (H-2d) target cells. As illustrated in Fig. 2, a strong peptide-specific cytotoxic response was induced after in vivo priming with the hybrid PPV:VLP-(LCMV), which reached a similar level after one or two injections, demonstrating the strong immunogenicity of these hybrid VLPs in the absence of any adjuvant. Moreover, a strong CTL response was also observed after injection of PPV:VLP-(LCMV) in the presence of 1 mg of alum (Fig. 3A). The optimal dose of PPV:VLP-(LCMV) to induce a cytotoxic response was found to be 10 μg, although a dose as low as 2 μg still stimulated a cytotoxic response (data not shown). In vitro-stimulated spleen cells from PPV:VLP-(LCMV)-immunized mice did not kill P815 target cells incubated only with medium, demonstrating the specificity of the cytotoxic response. Furthermore, splenocytes from mice injected with PBS or with control PPV:VLP did not kill p118–132-coated P815 target cells after in vitro stimulation with p118–132 (data not shown and Fig. 2). Therefore, the induction of p118–132-specific cytotoxic response requires in vivo priming by recombinant PPV:VLP carrying the CTL epitope. Thus, these results indicate that these recombinant VLP stimulated high level of CD8+ CTL precursors specific for the LCMV epitope.
Figure 2.

In vivo induction of CTL responses by recombinant PPV:VLP carrying the 118–132 LCMV CD8+ epitope. BALB/c mice were immunized i.p. on day 0 (A) or on days 0 and 21 (B) with 10 μg of control PPV:VLP or PPV:VLP-(LCMV). Fourteen (A) or 7 (B) days later, spleen cells were in vitro-stimulated for 5 days with the p118–132 peptide and irradiated syngeneic spleen cells. Cytotoxic activity of the effector cells was measured on 51Cr-labeled P815 target cells pulse-labeled with the p118–132 peptide (•) or incubated with medium alone (▴). Data represent the mean of percent specific lysis from duplicate samples.
Figure 3.

Immunization of mice with PPV:VLP-(LCMV) induces a long-lasting memory CD8+ CTL activity. (A) Mice were immunized i.p. on days 0 and 21 with 10 μg of PPV:VLP-(LCMV) in PBS (□) or with alum (▪). Spleens were removed 7 days later. (B) Mice were treated on days −1, 0, +1, and 9 with anti-CD4 (•) or anti-CD8 (▵) mAbs or with PBS (□) and were injected on day 0 with 10 μg PPV:VLP-(LCMV). Control mice received 10 μg of control PPV:VLP (⧫). Spleens were removed on day 14. (C) Mice were immunized i.p. on days 0 and 21 with 10 μg of either PPV:VLP (hatched bars) or PPV:VLP-(LCMV) (solid bars). Spleens were removed at various times after the second PPV:VLP injection as indicated. CTL activity was determined after in vitro stimulation of splenocytes as described in Fig. 2. Lysis of uncoated target cells was less than 10% and is not shown. Data represent the mean of percent specific lysis from duplicates (SD < 5%). Data in C were obtained at a 100:1 effector/target (E/T) ratio.
The Cytotoxic Activity Induced by Recombinant PPV:VLP Is Mediated by MHC Class I-Restricted CD8+ T Lymphocytes.
To characterize the effector cells stimulated by recombinant PPV:VLP, the cytotoxic response was analyzed with various target cells expressing different MHC class I molecules, P815 (H-2d), EL4 (H-2b), RDM4 (H-2k), or 1T22 (H-2q). BALB/c mice (H-2d) received a single injection of PPV:VLP-(LCMV) without adjuvant. The splenic effector cells were able to kill the syngeneic target cells, P815, coated with p118–132 but did not exhibit any cytotoxic activity toward p118–132-coated target cells expressing other MHC class I haplotypes (Table 1).
Table 1.
Cytotoxic responses induced in vivo by recombinant PPV:VLPs are restricted by MHC class I molecules
| Target cells | % specific 51Cr release
|
||||
|---|---|---|---|---|---|
| Peptide-coated target cells
|
Control target cells
|
||||
| PPV:VLP-(LCMV) | PPV:VLP | PPV:VLP-(LCMV) | PPV:VLP | ||
| P815 | (H-2d) | 84.5 | 14.3 | 8.3 | 0 |
| EL4 | (H-2b) | 0 | 0 | 0 | 0.5 |
| RDM4 | (H-2k) | 0 | 0 | 0 | 0 |
| 1T22 | (H-2q) | 8.2 | 4.5 | 8.8 | 9.5 |
Mice were immunized i.p. on day 0 with 10 μg of PPV:VLP or 10 μg of PPV:VLP-(LCMV). After 14 days, immune spleen cells were in vitro-stimulated for 5 days with the p118-132 peptide and syngeneic spleen cells. Cytotoxic activity of the effector cells was measured on 51Cr-labeled target cells pulse-labeled with the p118-132 peptide or incubated with medium alone, at a 30:1 effector/target ratio. Data represent mean values of duplicate samples (SD < 10%).
To determine the phenotype of cells responsible for the cytolytic response induced by PPV:VLP-(LCMV), mice were depleted of CD4+ or CD8+ T lymphocytes during immunization. The efficiency of CD4+ or CD8+ T cell depletion was controlled by FACScan analysis of spleen cells and was >99% (data not shown). The peptide-specific cytotoxic response induced by recombinant PPV:VLP carrying the LCMV epitope was not affected by in vivo CD4+ T cell depletion, whereas anti-CD8 treatment of mice totally abolished the cytotoxic response (Fig. 3B). Therefore, these results demonstrated that the CTL response stimulated by PPV:VLP-(LCMV) was mediated by MHC class I-restricted CD8+ T lymphocytes. Moreover, these experiments indicate that recombinant PPV:VLP-(LCMV) did not require CD4+ T cells to stimulate a strong CTL response.
Recombinant PPV:VLP Induces a Long-Lasting Memory CTL Response.
To analyze the persistence of the CTL response stimulated by recombinant PPV:VLP carrying the 118–132 epitope, mice received two injections of PPV:VLP-(LCMV) in the absence of adjuvant. One week to 9 months later, their splenic cytolytic activity was tested against p118–132-coated P815 target cells. As illustrated in Fig. 3C, immunization with PPV:VLP-(LCMV) induced a very efficient and long-lasting CTL response that persisted at least 9 months after the last injection. This study also provided evidence that recombinant PPV:VLP were not toxic for mice, because these animals did not develop any side effects or loss of weight during these 9 months.
In Vivo Induction of Virus-Specific CTLs by Immunization with Recombinant PPV:VLP.
To assess the capacity of effector cells from PPV:VLP-(LCMV)-immunized mice to recognize the CTL epitope presented by virus-infected cells, their cytotoxic activity was tested against LCMV-infected J774 target cells. This experiment showed that splenocytes from PPV:VLP-(LCMV)-primed mice were able to very efficiently kill LCMV-infected target cells after in vitro stimulation with p118–132 (Fig. 4). These effector cells did not lyse uninfected J774 target cells, thus, showing the specificity of the CTL response. Injection of BALB/c mice with control PPV:VLP or with PBS did not induce any LCMV-specific cytotoxic activity, whereas mice inoculated i.p. with LCMV developed a strong CTL response, as reported (20, 21).
Figure 4.
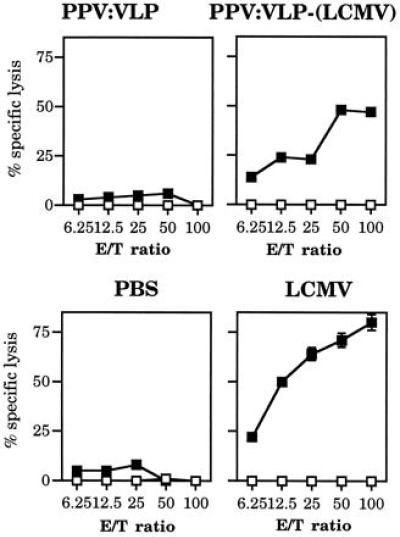
In vivo immunization with PPV:VLP-(LCMV) stimulates a strong virus-specific CTL response. BALB/c mice were injected i.p. on days 0 and 21, with 10 μg of PPV:VLP, 10 μg of PPV:VLP-(LCMV), PBS, or 105 pfu of LCMV. Seven days later, splenocytes were in vitro-stimulated and cytotoxic activity of the effector cells was measured on 51Cr-labeled J774 target cells infected with LCMV (▪) or incubated with medium (□). Data represent the mean of percent specific lysis from triplicate samples.
Recombinant PPV:VLP Protects Mice Against a Lethal LCMV Challenge.
To analyze the capacity of hybrid PPV:VLP-(LCMV) to protect mice against lethal viral infection, BALB/c mice were immunized twice, on days 0 and 21 with 10 μg of control or hybrid PPV. Seven or 49 days later, they were injected intracerebrally (i.c.) with 101.7 pfu of LCMV Armstrong strain, which induces a lethal acute choriomeningitis in normal mice (20, 21). Mice immunized with recombinant PPV:VLP-(LCMV) were fully protected against a lethal virus challenge performed on day 28 (Fig. 5A). As expected, control PPV:VLP- or PBS-immunized mice developed a fatal choriomeningitis in the 8 days after the i.c. viral inoculation, whereas mice previously injected i.p. with LCMV were protected against an i.c. challenge. An effective protection (60%) was also observed after challenge of PPV:VLP-(LCMV)-immunized mice with 103.5 pfu of LCMV (data not shown). In addition, mice immunized with PPV:VLP-(LCMV) were also protected against a 101.7-pfu viral challenge performed on day 70 (Fig. 5B), indicating that recombinant PPV:VLP induced a long-term protective immunity against LCMV. Twenty-one days after LCMV challenge, no virus (titer < 1:20) was detected by antigen-capture ELISA in the kidneys of surviving mice immunized with PPV:VLP-(LCMV) or with LCMV, indicating a complete viral clearance (data not shown). Moreover, all surviving mice developed high levels of LCMV-specific IgG (data not shown).
Figure 5.

CD8+ T cells induced by recombinant PPV:VLP-(LCMV) fully protect mice against a lethal LCMV challenge. BALB/c mice were injected i.p. on days 0 and 21 with 10 μg of PPV:VLP, 10 μg of PPV:VLP-(LCMV), PBS, or 105 pfu of LCMV. Two groups were also treated on days −1, 0, +1, 7, 14, and 20 with anti-CD4 or anti-CD8 mAbs. After 7 (A) or 49 (B) days, mice were challenged i.c. with 101.7 pfu of LCMV. Survival was recorded for 21-days period. The percent survival and the number of surviving mice/the number of total mice (∗) are the cumulative results of four experiments. ND, not determined.
We investigated the role of CD4+ and CD8+ T cells in the hybrid PPV:VLP-induced protection of mice against the virulent virus. Mice were in vivo-depleted of CD4+ or CD8+ T cells before and during immunization with PPV:VLP-(LCMV) and were then i.c. challenged at day 28. Whereas only 1 of 11 CD8+-depleted mice survived the LCMV challenge, CD4+ depletion did not modify the survival of immunized animals (Fig. 5A). These results demonstrate that (i) CD4+ are not required to induce protective immunity against LCMV and (ii) immunization of mice with hybrid PPV:VLP carrying a single viral CTL epitope induced a strong CD8+ CTL response, which conferred full protection against a lethal viral challenge.
DISCUSSION
Several live vectors were previously shown to induce a protective anti-LCMV immune response. Attenuated Listeria monocytogenes carrying the whole LCMV nucleoprotein or a nucleoprotein epitope were able to protect mice against a lethal viral challenge (12, 22). Other live attenuated viral vectors such as vaccinia virus (23, 24), mengo virus (9) or influenza virus (25) expressing LCMV proteins or epitopes also stimulate protective immune responses. However, the majority of these live vectors are potentially risk-prone.
To our knowledge, the present study demonstrates for the first time that nonreplicative parvovirus-like particles carrying a single viral CTL epitope are able to induce complete protection of mice against a lethal viral infection through the induction of virus-specific MHC class I-restricted CD8+ CTLs. These CTLs kill both peptide-coated target cells and virus-infected cells, indicating that the effector cells recognize naturally processed epitope from the infectious virus. This strong in vivo CTL response induced by PPV-like particles formed by the hybrid VP2 protein carrying the LCMV 118–132 CTL epitope does not require adjuvant and persists during months after immunization. Moreover, the ability of these hybrid PPV:VLP-(LCMV) to induce CTL responses and to protect mice against viral infection does not require CD4+ helper T cell function. These results are in accordance with results obtained with LCMV (26, 27).
Because they provide a safe antigen delivery system, various particulate carrier systems using viral proteins have been widely analyzed for their immunogenicity. The chimeric particles made by self-assembly of hepatitis B surface antigen-containing HIV-1 determinants (28) or HIV Gag particles (29) have been shown to stimulate specific CTL responses. VLPs made with the yeast retrotransposon Ty protein carrying HIV epitopes or influenza virus NP epitopes also induced CTL responses when injected to mice in the absence of adjuvant (30). However, so far, none of these VLPs was shown to induce protective immunity in a lethal infection model. Moreover, a phase I study showed that p17/p24-Ty-VLP was unable to induce antigen-specific CTL responses in healthy subjects (31).
Interestingly, Ty:VLP or hepatitis B surface antigen particles are unable to induce CTL response when administered to mice in the presence of adjuvant such as complete Freund’s adjuvant, incomplete Freund’s adjuvant, or alum (30, 32). In contrast, mice immunized with PPV:VLP-(LCMV) mixed with alum developed a CTL response similar to those induced after injection of PPV:VLP-(LCMV) in PBS. The reasons for such a discrepancy are unclear. However, it was recently suggested that alum suppresses the capacity of Ty:VLP to induce CTLs because the intact particulate structure of hybrid Ty:VLP is strictly required for CTL induction and is compromised by emulsification or precipitation with adjuvant (33). It should therefore be proposed that PPV:VLPs are either more stable than other VLPs or follow a different antigen-presentation pathway.
Several hypothesis may explain the strong potential of chimeric PPV:VLP to induce CD8+ CTL response. They may gain access to the cytosol after fusion between lipidic structure and cellular membrane, as shown for liposomes (5, 34). However, since PPV:VLPs are made by self-assembly of a single protein and do not carry any lipid, this mechanism is unlikely. The high immunogenicity of PPV:VLP is, therefore, more probably due to its 25-nm particulate form that can favor its optimal delivery to the class I antigen presentation pathway. Indeed, it was demonstrated that native 22-nm hepatitis B surface antigen particles displayed high-efficiency induction of class I-restricted CTLs in vivo (35). Ovalbumin linked to 1-μm synthetic latex particles also stimulated CTL responses in vitro and in vivo by an alternative novel pathway to present MHC class I-restricted peptides (7, 36). Thus, particulate PPV:VLPs can probably enter this novel cytosolic class I antigen presentation pathway after endocytic uptake by macrophages and/or dendritic cells. These pseudoparticles may also be taken up by macropinocytosis in macrophages, providing access for VLPs to the cytosol of antigen-presenting cells (37).
The internalization of hybrid PPV:VLP could also be increased by binding to a PPV-specific receptor expressed by the virus target cells, as reported, for instance, for the CD46 receptor of the measles virus, implicated in virus MHC class II-restricted presentation (38). Although the PPV receptor is yet to be characterized, one receptor of human parvovirus B19 has been identified on erythrocytes and at a lower level on endothelial cells, and placenta, fetal liver, and heart cells (39). Moreover, it has been demonstrated that some parvoviruses such as canine parvovirus and minute virus of mice use carbohydrates for cellular attachment (40). These data suggest that glycan-binding properties of parvoviruses may favor a nonspecific quick uptake and processing of these PPV:VLP by antigen-presenting cells. The detailed process carried out by PPV:VLP to enter the cytosolic class I presentation pathway is currently under investigation.
Our results clearly demonstrate that hybrid PPV:VLPs provide a safe and efficient strategy to induce CTLs and to stimulate anti-viral immunity. These recombinant pseudoparticles represent a promising strategy to present several CTL epitopes from the same or different viral proteins or to deliver simultaneously helper CD4+ and cytotoxic CD8+ T cell epitopes to the immune system. Indeed, we (ref. 41 and unpublished observations) demonstrated that chimeric PPV:VLPs expressing a CD4+ T cell epitope from the VP1 protein of poliovirus type 1 or from the PreS2 region of hepatitis B virus stimulated CD4+ T cell proliferative responses and cytokine product.
In conclusion, to our knowledge, the present study represents the first demonstration that recombinant pseudoparticles formed by hybrid viral protein carrying a single viral CTL epitope can confer protection against a lethal viral infection, providing a novel and safe strategy to develop vaccine applications.
Acknowledgments
We thank J. Foulon, H. Tran, and A. Membrillera-Pizzaro for their excellent technical assistance. This work was carried out as a collaborative project between INGENASA and Institut Pasteur in BIOTECH projects (European Economic Community Biotechnology BI02-CT92-0290 and BI04-CT96-024). C.S. was supported by a fellowship from Association pour la Recherche sur le Cancer.
ABBREVIATIONS
- VLP
virus-like particle
- LCMV
lymphocytic choriomeningitis virus
- CTL
cytotoxic T lymphocyte
- MHC
major histocompatibility complex
- PPV
porcine parvovirus
- pfu
plaque-forming unit(s)
- i.c.
intracerebrally
References
- 1.Germain R N. Annu Rev Immunol. 1993;11:403–450. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 2.Ke Y, Li Y, Kapp J A. Eur J Immunol. 1995;25:549–553. doi: 10.1002/eji.1830250237. [DOI] [PubMed] [Google Scholar]
- 3.Schulz M, Zinkernagel R M, Hengartner H. Proc Natl Acad Sci USA. 1991;88:991–993. doi: 10.1073/pnas.88.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J Y, Gardner B H, Murphy C I, Seals J R, Kensil C R, Recchia J, Beltz G A, Newman G W, Newman M J. J Immunol. 1992;148:1519–1525. [PubMed] [Google Scholar]
- 5.Zhou F, Rouse B T, Huang L. J Immunol. 1992;149:1599–1604. [PubMed] [Google Scholar]
- 6.Takahashi H, Takeshita T, Morein B, Putney S, Germain R N, Berzofsky J A. Nature (London) 1990;344:873–875. doi: 10.1038/344873a0. [DOI] [PubMed] [Google Scholar]
- 7.Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock K L. Proc Natl Acad Sci USA. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irvine K R, McCabe B J, Rosenberg S A, Restifo N P. J Immunol. 1995;154:4651–4657. [PMC free article] [PubMed] [Google Scholar]
- 9.Altmeyer R, Girard M, van der Werf S, Mimic V, Seigneur L, Saron M F. J Virol. 1995;69:3193–3196. doi: 10.1093/benz/9780199773787.article.b00034516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter N, Lagranderie M, Gangloff S, Leclerc C, Gheorghiu M, Gicquel B. Vaccine. 1995;13:471–478. doi: 10.1016/0264-410x(94)00001-4. [DOI] [PubMed] [Google Scholar]
- 11.Harding C V, Pfeifer J D. Immunology. 1994;83:670–674. [PMC free article] [PubMed] [Google Scholar]
- 12.Gossens P L, Milon G, Cossart P, Saron M-F. Int Immunol. 1995;7:797–805. doi: 10.1093/intimm/7.5.797. [DOI] [PubMed] [Google Scholar]
- 13.Pialoux G, Excler J L, Rivière Y, Gonzalez-Canali G, Feuillie V, Coulaud P, Gluckman J C, Matthews T J, Meignier B, Kieny M P, Gonnet P, Diaz I, Méric C, Paoletti E, Tartaglia J, Salomon H, Plotkin S the Agis Group & Agence National de Recherche sur le SIDA. AIDS Res Hum Retroviruses. 1995;11:373–381. doi: 10.1089/aid.1995.11.373. [DOI] [PubMed] [Google Scholar]
- 14.Ulmer J B, Sadoff J C, Liu M A. Curr Opin Immunol. 1996;8:531–536. doi: 10.1016/s0952-7915(96)80042-2. [DOI] [PubMed] [Google Scholar]
- 15.Aichele P, Hengartner H, Zinkernagel R M, Schulz M. J Exp Med. 1990;171:1815–1820. doi: 10.1084/jem.171.5.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Motal U M A, Berg L, Jondal M. Eur J Immunol. 1992;22:3085–3090. doi: 10.1002/eji.1830221209. [DOI] [PubMed] [Google Scholar]
- 17.Popescu M, Schaeffer H, Lehmann-Grübe F. J Virol. 1976;20:1–8. doi: 10.1128/jvi.20.1.1-8.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gengoux C, Leclerc C. Int Immunol. 1995;7:45–53. doi: 10.1093/intimm/7.1.45. [DOI] [PubMed] [Google Scholar]
- 19.Fayolle C, Sebo P, Ladant D, Ullmann A, Leclerc C. J Immunol. 1996;156:4697–4706. [PubMed] [Google Scholar]
- 20.Lehmann-Grübe F. Arch Virusforschung. 1964;14:344–350. doi: 10.1007/BF01555827. [DOI] [PubMed] [Google Scholar]
- 21.Kimmig W, Lehmann-Grübe F. J Gen Virol. 1979;45:703–710. doi: 10.1099/0022-1317-45-3-703. [DOI] [PubMed] [Google Scholar]
- 22.Slifka M K, Shen H, Matloubian M, Jensen E R, Miller J F, Ahmed R. J Virol. 1996;70:2902–2910. doi: 10.1128/jvi.70.5.2902-2910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klavinskis L S, Whitton J L, Oldstone M B A. J Virol. 1989;63:4311–4316. doi: 10.1128/jvi.63.10.4311-4316.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oldstone M B A, Tishon A, Eddleston M, de la Torre J C, McKee T, Whitton J L. J Virol. 1993;67:4372–4378. doi: 10.1128/jvi.67.7.4372-4378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castrucci M R, Hou S, Doherty P C, Kawaoka Y. J Virol. 1994;68:3486–3490. doi: 10.1128/jvi.68.6.3486-3490.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed R, Butler L D, Bhatti L. J Virol. 1988;62:2102–2106. doi: 10.1128/jvi.62.6.2102-2106.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahemtulla A, Fung-Leung W P, Schilham M W, Kündig T M, Sambhara S R, Narendran A, Arabian A, Wakeham A, Paige C J, Zinkernagel R M, Miller R G, Mak T W. Nature (London) 1991;353:180–184. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- 28.Michel M L, Mancini M, Schlienger K, Tiollais P. Res Virol. 1993;144:263–267. doi: 10.1016/s0923-2516(06)80038-5. [DOI] [PubMed] [Google Scholar]
- 29.Griffiths J C, Harris S J, Layton G T, Berrie E L, French T J, Burns N R, Adams S E, Kingsman A J. J Virol. 1993;67:3191–3198. doi: 10.1128/jvi.67.6.3191-3198.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Layton G T, Harris S J, Gearing A J H, Hill-Perkins M, Cole J S, Griffiths J C, Burns N R, Kingsman A J, Adams S E. J Immunol. 1993;151:1097–1107. [PubMed] [Google Scholar]
- 31.Weber J, Cheinsong-Popov R, Callow D, Adams S, Patou G, Hodgkin K, Martin S, Gotch F, Kingsman A. Vaccine. 1995;13:831–834. doi: 10.1016/0264-410x(94)00061-q. [DOI] [PubMed] [Google Scholar]
- 32.Schirmbeck R, Melber K, Mertens T, Reimann J. Eur J Immunol. 1994;24:1088–1096. doi: 10.1002/eji.1830240512. [DOI] [PubMed] [Google Scholar]
- 33.Harris S J, Woodrow S A, Gearing A J H, Adams S E, Kingsman A J, Layton G T. Vaccine. 1996;14:971–976. doi: 10.1016/0264-410x(96)00010-2. [DOI] [PubMed] [Google Scholar]
- 34.Nair S, Buiting A M J, Rouse R J D, van Rooijen N, Huang L, Rouse B T. Int Immunol. 1995;7:679–688. doi: 10.1093/intimm/7.4.679. [DOI] [PubMed] [Google Scholar]
- 35.Schirmbeck R, Böhm W, Melber K, Reimann J. J Immunol. 1995;155:4676–4684. [PubMed] [Google Scholar]
- 36.Kovacsovics-Bankowski M, Rock K L. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 37.Norbury C C, Hewlett L J, Prescott A R, Shastri N, Watts C. Immunity. 1995;3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 38.Gerlier D, Trescol-Biémont M-C, Varior-Krishnan G, Naniche D, Fugier-Vivier I, Rabourdin-Combe C. J Exp Med. 1994;179:353–358. doi: 10.1084/jem.179.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown K E, Hibbs J R, Gallinella G, Anderson S M, Lehman E D, McCarthy P, Young N S. N Engl J Med. 1994;330:1192–1196. doi: 10.1056/NEJM199404283301704. [DOI] [PubMed] [Google Scholar]
- 40.Chapman M S, Rossmann M G. Virology. 1993;194:491–508. doi: 10.1006/viro.1993.1288. [DOI] [PubMed] [Google Scholar]
- 41.Sedlik C, Sarraseca J, Rueda P, Leclerc C, Casal I. J Gen Virol. 1995;76:2361–2368. doi: 10.1099/0022-1317-76-9-2361. [DOI] [PubMed] [Google Scholar]