

Abstract
Monoclonal antibodies (mAbs) that exert antitumor activity can do so by virtue of their effector function and/or their ability to signal growth arrest or cell death. In this study, we demonstrate that mAbs which have little or no signaling activity—i.e., anti-CD19, CD20, CD21, CD22 and Her-2—can become potent antitumor agents when they are converted into IgG–IgG homodimers. The homodimers exert antigrowth activity by signaling G0/G1 arrest or apoptosis, depending upon which cell surface molecule they bind. This activity is specific and, in the case of the anti-CD19 mAb, did not require an Fc portion. These results offer the possibility that homodimers of other tumor-reactive mAbs which have little antitumor activity as monomers might be potent, antitumor agents.
During the past two decades, a variety of monoclonal antibodies (mAbs) have been selected for clinical use based on their effector functions. Two examples are the epithelial cell-reactive mAb, 17.1A (1, 2), and the lymphoma reactive mAb, CAMPATH-1 (3, 4). In this regard, there is considerable experimental (5, 6), and some clinical (2, 4) evidence to indicate that effector functions play an important role in the antitumor activity of mAbs. Those mAbs that do not fix complement or mediate antibody-dependent culular cytotoxicity (ADCC) have been converted into useful ones by chimerization (7, 8), by generating switch variants (9–11), or by preparing cytotoxic immunoconjugates (12–14). Recently it has been shown that mAbs can exert antitumor activity in other ways—e.g., by inhibiting metastases (15), tumor cell–substrata interactions (16), or tumor cell extravasation (17). In addition, we (18–20) and others (21–27) have reported that some mAbs can signal growth arrest and/or apoptosis of tumor cells by acting as agonists (“negative signaling”). Indeed, in the case of B cell lymphoma, there is compelling evidence that both anti-idiotype (28, 29) and anti-CD19 mAbs (18, 19) exert their antitumor activities predominantly, if not exclusively, by signaling growth arrest and apoptosis. Other mAbs which also have signaling properties include anti-Fas (21), anti-CD40 (24), anti-class II major histocompatibility complex (23), anti-Her-2 (30), anti-Ley (26), and anti-IgM (20, 22, 25, 27). Furthermore, negative signaling can be optimized by hypercrosslinking with secondary antibodies or by using “cocktails” of primary antibodies (31).
In the case of anti-CD19, only a small percentage of mAbs can deliver growth-inhibiting signals to neoplastic B cells, and these require the addition of very large (i.e., hypersaturating) concentrations of antibody (19). Because of this peculiar behavior, we studied the physicochemical properties of one of these mAbs, HD37, in more detail and observed that it spontaneously formed homodimers of 300 kDa which constituted 20–30% of our purified antibody preparations. When these “natural” HD37 dimers were separated from the monomers, all the negative signaling activity could be attributed to the homodimers. This explained why such large amounts of the initial mAb were needed.
This finding led us to explore the possibility that chemically generated homodimers of HD37 and other mAbs that did not, as monomers, signal growth arrest very effectively, could be made into highly potent cytotoxic or growth-inhibiting mAbs by homodimerization.
METHODS
Cells.
Two human Burkitt lymphoma cell lines, Daudi and Ramos, were maintained in culture by serial passage in RPMI 1640 medium containing 25 mM Hepes, 10% heat-inactivated fetal calf serum (FCS), 100 units/ml penicillin, 100 μg/ml streptomycin (complete medium), and 100 mM glutamine. The cells were grown in a humidified atmosphere of 5% CO2 and air. Cell viability was determined by trypan blue exclusion. Cells from the breast cancer line, BT474, were maintained by serial passage in minimal essential medium (MEM) containing 10% heat-inactivated FCS, 2 mM l-glutamine, 100 nM nonessential amino acids, 1 mM sodium pyruvate, and 2% vitamins for MEM.
Preparation of the Anti-Her-2 mAb.
BALB/c mice were immunized with a recombinant form of the 641-amino acid extracellular domain of Her-2. Spleen cells from the immunized mice were harvested and fused with the myeloma cell line, SP2/0. The hybridomas were subcloned and assayed by ELISA for the ability to produce immunoglobulin. Antibody-containing supernatants from positive clones were tested by ELISA for reactivity against the Her-2 extracellular domain and by fluorescence-activating cell sorter (FACS) on a Her-2+ cell line, SKBr3. The antibody chosen for this study was designated HER-50.
mAbs.
Mouse IgG1 mAbs specific for CD22 (RFB4), CD19 (HD37), CD20 (2H7), CD21 (B-ly4), and Her-2 (HER-50), and the purified isotype matched IgG1 of irrelevant specificity (3F12) were used. RFB4 and HD37 were prepared in our scale-up laboratory (32). 2H7 and B-ly4 were purchased from PharMingen. IgGs from 3F12 (control) and HER-50 were prepared in our laboratory by purification of hybridoma cell supernatants on a protein A-Sepharose column.
Preparation of Homodimers by Introducing a Thioether Bond.
Two heterobifunctional crosslinkers were used to dimerize the mAbs without using reducing reagents: SMCC [succinimidyl 4-(maleimidomethyl)cyclohexane-1-carboxylate] and SATA (N-succinimidyl S-acethylthio-acetate) (Pierce).
Derivatization with SMCC.
A total of 250 mg of mAb at 10 mg/ml in 0.05 M phosphate buffer containing 3 mM Na2-EDTA (PBE; pH 7.5) was mixed with 250 μl of SMCC (concentration of 10 mg/ml in dimethylformamide) for 1 hr at room temperature. The SMCC/IgG molar ratio was 4.5. After 1 hr, the protein was purified by chromatography on a Sephadex G-25 column (in the same buffer). The purified protein was concentrated to 10 mg/ml by Centriprep concentrators (Amicon).
Derivatization with SATA.
A total of 250 mg mAb at 10 mg/ml was mixed with 250 μl SATA (6.3 mg/ml in dimethylformamide) and incubated for 1 hr at room temperature (RT). The SATA/IgG molar ratio was 4.0. The excess SATA was removed by chromatography on a Sephadex G-25 column. The purified protein was concentrated to 10 mg/ml and was deacetylated with 0.15M hydroxylamine-HCl for 5 min at RT. Both derivatized proteins were mixed together and incubated at RT for 1–2 hr. Mixtures were analyzed by SDS/PAGE, and 20–25% of the IgG was in dimeric form (300 kDa). The preparation was dialyzed overnight (O.N.) vs. 0.05 M PBE at 4°C, filter sterilized, and further purified on a 600 × 21.5 mm Bio-Sil Sec.400 HPLC column (Bio-Rad) at flow rate of 2 ml/min. Generally, the dimer had a retention time of about 66 min, while the monomer had a retention time of about 74 min. The protein fractions purified by HPLC were concentrated to 5–10 mg/ml, filter sterilized on a 0.22-μm filter unit (sterile MILLEX-GV; Millipore), and analyzed by SDS/PAGE and HPLC.
Preparation of F(ab′)2 Homodimers.
HPLC-purified HD37 dimers were dialyzed O.N. against 0.1 M acetate buffer (pH 3.5–4.0) and digested with insoluble pepsin (Sigma) for 3 hr at 37°C. The dimer pepsin-digest was affinity purified on a SpA-Sepharose column. The material not bound to the column was further affinity purified on a protein G-Sepharose column. Sixty percent of this material was bound and eluted with 0.1 M acetic acid, then neutralized to pH 7.5, concentrated, and analyzed by analytical HPLC and SDS/PAGE gel to determine the molecular weight.
SDS/PAGE.
Proteins were analyzed under both reducing and nonreducing conditions by SDS/PAGE on 4–15% gels using a Phast System Separation Unit (Pharmacia). Protein bands were visualized by staining the gel with Coomassie blue in the same Phast System.
HPLC Analysis.
The molecular weights of the dimers were determined by analytical HPLC using a 600 × 7.5 mm Bio-Sil SEC-400 column (Bio-Rad) equilibrated in 0.05 M sodium phosphate buffer (pH 6.8). A standard protein mixture containing IgA (300 kDa) and IgG (150 kDa) was fractionated on the HPLC column to establish the retention time of known molecular weight standards.
[3H]Thymidine Assay.
The antiproliferative activity of different mAb monomers and dimers on Daudi cells was determined using a [3H]thymidine incorporation assay (33). Daudi cells at 5 × 104 cells/100 μl in RPMI 1640 medium containing 10% FCS, glutamine, and antibiotics were distributed into triplicate wells of 96-well microtiter plates containing 100 μl of antibodies (monomer or dimer) diluted in the same medium at concentrations ranging from 10−8 to 10−6 M. The plates were incubated for 24–48 hr at 37°C in 5% CO2 and pulsed for 4 hr with 1 μCi [3H]thymidine (Amersham; 1 Ci = 37 MBq). Wells were harvested on a Titertek cell harvester (Flow Laboratories), and the radioactivity retained on Skatron filters was determined in a liquid scintillation spectrometer. BT474 cells at a concentration of 104 cells/100 μl in MEM containing 10% heat-inactivated FCS, 2 mM l-glutamine, 100 nM nonessential amino acids, 1 mM sodium pyruvate, and 2% vitamins were plated into triplicate wells of 96-well microtiter plates and allowed to adhere overnight. The cells were then treated with 100 μl antibodies diluted in the same medium in varying concentrations. The plates were incubated for 72 hr at 37°C in 5% CO2 and pulsed for 6 hr with 1 μCi [3H]thymidine (Amersham). The percent reduction in [3H]thymidine incorporation, as compared with untreated controls, was used to quantitate killing. Nine wells of untreated cells were included in each experimental plate as controls.
Measurement of the Dissociation Rate.
IgG and F(ab′)2 fragments were radioiodinated with Na125I to a specific activity of ≈1 μCi/μg using the IODOGEN reagent (Pierce) (34), and dissociation rates were determined as described (35). Daudi cells (1 × 107/ml) were incubated in a 15-ml tissue culture tube with a concentration of 125I-labeled IgG1 (either monomer or dimer, in a volume of ≈4 ml of complete RPMI 1640 medium at 4°C) that occupied at least half of the binding sites at equilibrium. This amount was sufficient for 10 duplicate time points. A duplicate reaction mixture containing a 200-fold molar excess of unlabeled antibody was also prepared. The reaction mixtures were incubated at 4°C with slow shaking on a platform shaker until equilibrium was achieved (as determined from the association experiments 2 hr was sufficient). To prevent internalization, the cells were maintained at 4°C. After a 2-hr incubation the cells were centrifuged in a tabletop centrifuge for 5 min at 1000 × g. To initiate the dissociation of antibody from the cell surface, cells were washed at 4°C with cold complete RPMI 1640 medium, resuspended in 4 ml complete medium, and incubated on ice for the final step. At various time points (0, 1, 4, 8, 24, 48, 72, 96, 120 hr) duplicate aliquots of the cell suspension were removed and centrifuged through binding-oil columns, and the specific cell-bound radioactivity was determined. The dissociation rate (Kd) was measured by plotting ln(B) versus time where B represents bound cpm. The time point at which 50% of the protein bound to the cells was dissociated was calculated from the curve.
Internalization Experiments.
Daudi cells (1 × 106/ml) were treated on ice for 1 hr with 125I-labeled mAb (monomer or dimer) at 1 μg/ml (specific activity, 0.25 μCi/μg). This concentration of antibody was sufficient to saturate all available binding sites on Daudi cells (about 105 IgG molecules bound per cell). Then the cells were washed free of excess ligand with complete RPMI 1640 medium at 4°C, resuspended at the initial concentration, and returned to 37°C for different periods of time (2, 4, 8 and 24 hr), and then assayed for the radioactivity in the supernatant, surface membranes, and intracellular compartments as described (36). Samples of 106 cells were pelleted by centrifugation at 4°C, supernatants were aspirated, and the radioactivity in both the pellet and supernatant were measured. Surface-bound antibody was then removed by a 10 min incubation in RPMI complete medium acidified with 1 M HCl to pH 2.5. Then cells were sedimented and the supernatant and sediment were measured. After stripping off the surface 125I-labeled IgG, the radioactivity in the cell pellet represented the internalized (“acid-resistant”) radiolabeled antibody. All supernatant fractions collected were then analyzed by trichloroacetic acid precipitation. The fraction of antibody bound to cell membranes and that internalized into the cells were compared.
Cell Cycle Analysis.
Cells were simultaneously examined for viability and cell cycle status by flow cytometric analysis using the DNA-binding dyes 7-amino actinomycin D (7-ADD) and Hoechst 33342 (both from Molecular Probes) (37). Daudi cells (1 × 106) were incubated for 24 hr at 37°C either with media (control) or with different mAbs, washed twice with 10% FCS-containing RPMI 1640 medium, and the cell pellet was treated with 50 μl of 400 μM 7-ADD and incubated on ice for 30 min (7-ADD acting as a vital dye). Cells were then fixed (1.0 ml of 0.5% paraformaldehyde in PBS), and simultaneously permeabilized and stained with the Hoechst dye (220 μl of Hoechst at 10 μg/ml in 5% Tween-20) overnight at 4°C. After filtration through a 50-μm nylon mesh, samples were analyzed on a dual laser/pulse processor-equipped FACStar (Becton Dickinson) (105 cells/analysis). After gating on single, viable cells (viable cells are 7-ADD-negative and aggregates were excluded using an area-versus-width plot of the Hoechst signal) the percent of cells in each stage of the cell cycle was determined using the paint-a-gate plus data analysis program (Becton Dickinson Immunocytometry Systems). BT474 cells (1 × 105) were plated out in 1 ml medium and allowed to adhere overnight at 37°C. The cells were then treated with medium alone or with 50 μl medium containing 10 μg of antibody. Cells were incubated with antibody for 2 hr and then were trypsinized, washed twice in cold PBS, and stained simultaneously with fluorescein isothiocyanate-Annexin V and propidium iodide for 15 min at 25°C. The samples were then analyzed for surface expression of phosphatidyl serine and propidium iodide exclusion on a FACScan (Becton Dickinson).
Apoptosis Assays.
Treated or untreated Daudi cells (2–5 × 106) were collected by centrifugation and lysed in 0.2–0.5 ml hypotonic buffer (5 mM Tris⋅HCl, pH 7.4/5 mM Na2-EDTA/0.5% Triton X-100). The lysates were centrifuged and the supernatants were deproteinated as described (19). The DNA extracts were analyzed on a 2% agarose gel containing 0.0001% ethidium bromide (for apoptosis). BT474 cells (1 × 105) in 1 ml MEM containing 10% heat-inactivated FCS, 2 mM l-glutamine, 100 nM nonessential amino acids, 1 mM sodium pyruvate, and 2% vitamins were plated into duplicate wells of 24-well microtiter plates and allowed to adhere overnight. Cells were treated with 50 μg/ml antibody or 20 ng/ml tumor necrosis factor α (positive control) for 1, 2, or 4 hr and harvested following trypsinization in 0.5% trypsin-EDTA solution. BT474 cells were treated with Annexin V-fluorescein isothiocyanate and propidium iodide for 15 min and analyzed for the presence of phosphatidyl serine on their surface by FACScan. Cells permeable to propidium iodide were excluded from the analysis. Annexin V positive cells were taken as apoptotic cells.
Pharmacokinetic Studies.
Pharmacokinetic analyses were carried out in BALB/c mice using a method described elsewhere (38). Data analysis was carried out using a noncompartmental model (39) and a pkcal program (40). The half lives were calculated using 0 to 24-hr (α-phase) and 24- to 96-hr (β-phase) intervals of time following injection.
Severe Combined Immunodeficient (SCID)/Daudi Mice.
Female C.B-17 Scid/Scid mice originated from M. Bosma’s colony at the Fox Chase Cancer Center (Philadelphia) and were bred at the Carolinas Medical Center. Six- to 10 week-old female mice were inoculated i.v. with 5 × 106 Daudi cells in 0.1 ml RPMI 1640 medium. Groups of 5–7 mice were used for the follow up; control (untreated mice), mice treated with doxorubicin, mice treated with HD37 dimer ± doxorubicin. Mice were monitored daily and sacrificed at the onset of hind leg paralysis, a clinical symptom which precedes death (41). Comparison of survival curves was carried out using log-rank and Wilcoxon tests (42, 43). The median survival time of mice was calculated by the log-rank test at the 5% significance level.
RESULTS AND DISCUSSION
Characterization of Homodimers.
Homodimerization should yield tetravalent antibody molecules that can crosslink their target molecules more efficiently. We therefore prepared homodimers of HD37 anti-CD19 and three other lymphoma-reactive mAbs: RFB4 anti-CD22, 2H7 anti-CD20, and B-ly4 anti-CD21; one breast tumor-reactive mAb, HER-50 anti-Her-2; and an isotype-matched (control) IgG1 (3F12). All homodimers were prepared using two heterobifunctional crosslinkers to introduce a thioether bond between the two IgG molecules. The homodimers were purified by preparative HPLC. As shown in Fig. 1, the typical 300-kDa homodimers were ≈90% pure as determined by SDS/PAGE and by analytical HPLC (data not shown).
Figure 1.
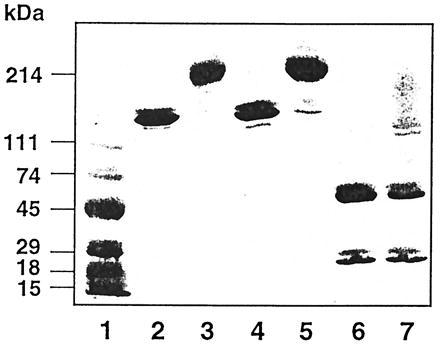
SDS/PAGE of different mAbs under nonreducing (lanes 2–5) and reducing (lanes 6 and 7) conditions. Lanes: 1, molecular weight markers; 2, RFB4 monomer; 3, RFB4 dimer; 4, HD37 monomer; 5, HD37 dimer; 6, HD37 monomer, reduced; 7, HD37 dimer reduced.
The in Vitro Antigrowth Activity of Homodimers.
The activities of the CD19, CD20, CD21, and CD22-reactive homodimers were tested on antigen-expressing Daudi and Ramos cells and the anti-Her-2 homodimers were tested on the human breast carcinoma cells, BT474. As shown in Fig. 2, the homodimers had significant antigrowth activity on their target cells whereas the monomers showed no effect even at the highest concentrations tested. The B cell-specific homodimers did not affect antigen-negative T cell tumors (data not shown). Furthermore, at the same concentrations, the isotype-control homodimers did not inhibit the growth of target cells (Fig. 2). Thus, the antigrowth activity is specific.
Figure 2.
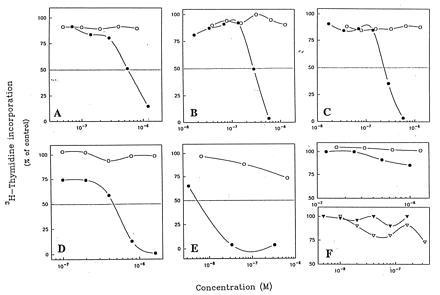
The effect of different mAbs monomers (○) or dimers (•) on [3H]thymidine incorporation in Daudi cells (A–D and F Upper) and BT474 cells (E and F Lower). (A) HD37 anti-CD19. (B) RFB4 anti-CD22. (C) 2H7 anti-CD20. (D) B-ly4 anti-CD21. (E) HER-50 anti-Her-2. (F) 3F12 (IgG1-isotype matched). These are representative results of three to seven experiments.
The Mechanisms by Which Homodimers Signal Cells.
The mechanisms by which the homodimers exerted their antigrowth activity were further explored by carrying out cell cycle analyses, as determined by DNA content using the FACS, and apoptosis, as determined by either the Annexin V assay (44) or by DNA laddering (19). As shown in Fig. 3A, HD37 homodimers but not monomers reduced the percentage of cells in S phase and arrested Daudi cells in G0/G1. As reported previously for HD37 (19), there was no evidence that dimeric HD37 induced apoptosis (data not shown). In contrast, the homodimers of the anti-Her-2 mAb induced significantly more apoptosis than the monomers (Fig. 3B). These results are in accord with a previous report which demonstrated that a dimeric vs. monomeric monoclonal IgA against the anti-Thy-1 antigen could more effectively induce apoptosis in cultured rat glomerular mesangial cells (45), supporting the concept that the degree of crosslinking and/or avidity of a mAb influences the extent of apoptosis. Thus, homodimers can induce or increase antigrowth activity in target cells in two ways depending upon which surface molecule they bind.
Figure 3.
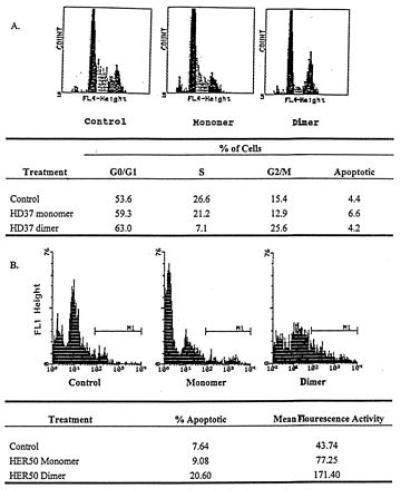
Cell cycle analysis (FACS) of Daudi or BT474 cells incubated with HD37 monomers or dimers vs. no treatment. (A) Daudi cells. This is a representative result of three experiments. (B) BT474 cells. This is a representative result of three experiments.
The Ka and Kd of Homodimers.
It has been reported that homodimers of some mAbs have a much slower dissociation rate from cells than monomers (46). We therefore determined whether the HD37 homodimers had a higher association rate (Ka), dissociation rate (Kd) or both, since these differences might be expected to affect not only their signaling properties but also their in vivo behavior. The results indicated that the Ka of the monomers and dimers were the same (data not shown) but, as shown in Fig. 4A, that the dissociation rate of the dimers was markedly slower, making their avidity much higher. In addition, the amount of dimer bound to the membrane and internalized into the target cells was higher for the dimer than the monomer (Fig. 4B). These results are entirely consistent with those of Wolff et al. (46), who previously reported similar changes in a breast tumor reactive mAb, ChiBR96, after dimerization. In the same report they also demonstrated that dimerization increased the effector function of ChiBR96 in vitro and in vivo (46). In the case of HD37, we excluded the possibility that the marked increase in antitumor activity observed following homodimerization was due to either an increase in effector function or more avid binding to FcRs on target cells by demonstrating that F(ab′)2 fragments of the homodimers were as active as the IgG homodimers in the cytotoxicity assay (Fig. 5). In addition, the HD37 IgG homodimers showed similar antigrowth activity on Ramos cells (data not shown), which lack FcγRIIs (47). These findings do not exclude an additional role for effector functions in vivo, although in our previous report we found that F(ab′)2 fragments of HD37 were highly therapeutic in SCID mice with Daudi tumors (19).
Figure 4.
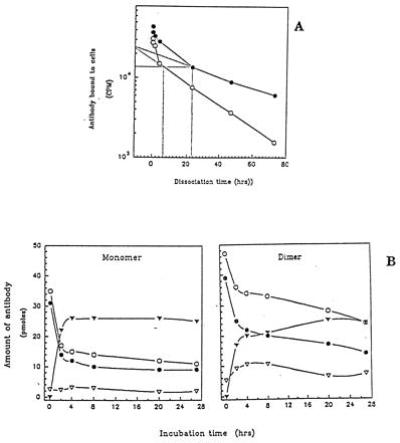
(A) The dissociation of HD37 monomers (○) and dimers (•) from Daudi cells. This is a representative result of two experiments. (B) The internalization of 125I-radiolabeled HD37 monomers and dimers into Daudi cells. ○, RBC-radioactivity bound to cell; •, RM-radioactivity bound to membrane; ▿, RI-radioactivity internalized; and ▾, RR-radioactivity released. This is a representative result of two experiments.
Figure 5.

The effect of HD37 on Daudi cells using a [3H]thymidine incorporation assay (19, 33) as monomers (○), IgG dimers (•), or F(ab′)2 dimers (▿). This is a representative result of three experiments. The difference between the curves representing the IgG and F(ab′)2 dimers is not statistically significant.
The in Vivo Antitumor Activity of Homodimers.
Based on the improved in vitro activity observed using the dimers, we next determined how the behavior of the homodimers and monomers compared in vivo. For this purpose, their half lives were first determined in BALB/c mice. While the T½α values were the same, the T½β values were different—i.e., 71.9 hr for dimers and 81.4 hr for monomers. These results are comparable to those reported using other mouse IgG1s (38). The antitumor activity of the HD37 monomers and dimers was then determined in SCID mice with the disseminated human Burkitt lymphoma, Daudi. As shown in Fig. 6 at the single therapeutic dose tested (1 mg/mouse) the dimers had significantly more therapeutic activity than the monomers. Furthermore, the antitumor activity of the dimers was further enhanced when the animals were injected with the chemotherapeutic drug, doxorubicin. Importantly, the homodimers completely prevented tumor growth in the organs where the majority of the Daudi cells grow [i.e., kidney, lung and ovaries (41)] but had little effect on spinal lymphoma, which is the site of lethal tumor growth. This suggests that the end point in our animal model (paralysis due to spinal tumor) may actually give an underestimate of the antitumor activity of the dimers.
Figure 6.
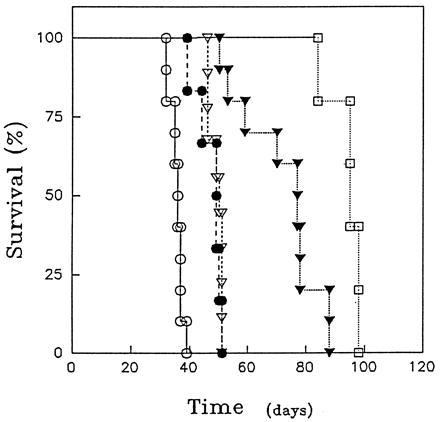
The survival of SCID/Daudi mice treated with HD37 monomers or dimers ± doxorubicin. SCID mice were inoculated i.v. with 5 × 106 Daudi cells. One day after tumor cell inoculation, the following treatments were given to groups of five to seven mice: control (injected with saline) (○); mice treated with doxorubicin (80 μg/mouse) (•); mice treated with HD37 monomer (1 mg) (▿); mice treated with HD37 dimer (1 mg) (▾); mice treated with both HD37 dimer (1 mg) and doxorubicin (80 μg) (□). Agents were injected i.v. in four equal doses on days 1–4 after tumor inoculation. This represents the averages of two experiments. The difference between the effects of the dimer vs. monomer and between the HD37 dimer and the HD37 dimer + doxorubicin are significant with P values of 0.004 and 0.0033, respectively.
Summary and Implications.
Our data suggest that at least some ineffective mAbs can be converted into effective antitumor agents by dimerization and that this increased potency can be attributed, in some cases, to increased negative signaling, presumably due to hypercrosslinking and/or a slower dissociation rate. In this regard, we have previously proposed (20) that antitumor mAbs be selected first for their negative signaling capacity and, subsequently, for their effector function. The present results support the use of this strategy and lead us to suggest that many other tumor-reactive mAbs which have little or no antigrowth activity should be reevaluated as homodimers. Furthermore, since in the case of HD37 (and perhaps other mAbs), the Fc portion does not appear necessary for cytotoxicity, some homodimers could be made even smaller by enzymatically removing the Fc portion or, alternatively, by generating recombinant Fv multimers. In this regard, recombinant oligomers (48), tail-to-tail covalent dimers (49–51) and IgM-like polymers (52) have all been generated, and these offer the promise of increased therapeutic potential. Finally, our experiments in lymphoma/SCID mice suggest that multivalent mAbs might be particularly effective when given in combination with cytotoxic agents.
Acknowledgments
We thank Drs. Amlot, Dorken, Möldenhauer, and Ledbetter for the RF84, HD37, and ZH7 hybridemas. We thank Dr. Victor Ghetie for suggestions concerning the preparation of homodimers and for critical discussions and Ms. C. Self for secretarial assistance. We thank Dr. Octavio Ramilo for his critical review of the manuscript. These studies were supported by National Institutes of Health Grant CA-28149.
ABBREVIATIONS
- MEM
minimal essential medium
- FACS
fluorescence-activating cell sorter
- SMCC
succinimidyl 4-(maleimidomethyl)cyclohexane-1-carboxylate
- SATA
N-succinimidyl S-acethylthio-acetate
- FCS
fetal calf serum
- SCID
severe combined immunodeficient
References
- 1.Herlyn D, Koprowski H. Proc Natl Acad Sci USA. 1982;79:4761–4765. doi: 10.1073/pnas.79.15.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riethmüller G, Schnider-Gadicke E, Schlimok G, Schmiegel W, Raab R. Lancet. 1994;343:1174–1177. [Google Scholar]
- 3.Dyer M J S, Hale G, Hayhoe F G J, Waldmann H. Blood. 1989;73:1431–1439. [PubMed] [Google Scholar]
- 4.Hale G, Clarke M R, Marcus R, Winter G, Dyer M J S, Phillips J M, Riechmann L, Waldman H. Lancet. 1988;ii:1394–1399. doi: 10.1016/s0140-6736(88)90588-0. [DOI] [PubMed] [Google Scholar]
- 5.Hooijberg E, van den Berk P C M, Sein J J, Wijdenes J, Hart A A M, de Boer R W, Melief C J M, Hekman A. Cancer Res. 1995;55:840–846. [PubMed] [Google Scholar]
- 6.Hooijberg E, Sein J J, van den Berk P C M, Hart A A M, van der Valk M A, Kast W M, Melief C J M, Hekman A. Cancer Res. 1995;55:2627–2634. [PubMed] [Google Scholar]
- 7.Morrison S L, Oi V T. Adv Immunol. 1989;44:65–91. doi: 10.1016/s0065-2776(08)60640-9. [DOI] [PubMed] [Google Scholar]
- 8.Adair J R. Immunol Rev. 1992;130:5–40. doi: 10.1111/j.1600-065x.1992.tb01519.x. [DOI] [PubMed] [Google Scholar]
- 9.Hale G, Clark M, Waldmann H. J Immunol. 1985;134:3056–3061. [PubMed] [Google Scholar]
- 10.Kaminski M S, Kitamura K, Maloney D G, Campbell M J, Levy R. J Immunol. 1986;136:1123–1130. [PubMed] [Google Scholar]
- 11.Denkers E Y, Badger C C, Ledbetter J A, Bernstein I D. J Immunol. 1995;135:2183–2186. [PubMed] [Google Scholar]
- 12.Dillman R O. J Clin Oncol. 1994;12:1497–1515. doi: 10.1200/JCO.1994.12.7.1497. [DOI] [PubMed] [Google Scholar]
- 13.Ghetie M, Ghetie V, Vitetta E S. Exp Opin Invest Drugs. 1996;5:309–321. [Google Scholar]
- 14.Hellström I, Hellström K E, Siegall C B, Trail P A. Adv Pharmacol. 1995;33:349–388. doi: 10.1016/s1054-3589(08)60674-2. [DOI] [PubMed] [Google Scholar]
- 15.Qi Y, Moyana T, Bresalier R, Xiang J. Gastroenterology. 1995;109:1950–1957. doi: 10.1016/0016-5085(95)90763-7. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Ma J, Wang J, Che X, Narula J, Bigby M, Wu M, Sy M S. Cancer Res. 1994;54:1561–1565. [PubMed] [Google Scholar]
- 17.Edward M. Curr Opin Oncol. 1995;7:185–191. doi: 10.1097/00001622-199503000-00015. [DOI] [PubMed] [Google Scholar]
- 18.Ghetie M A, Tucker K, Richardson J, Uhr J W, Vitetta E S. Blood. 1992;80:2315–2320. [PubMed] [Google Scholar]
- 19.Ghetie M A, Picker L J, Richardson J A, Tucker K, Uhr J W, Vitetta E S. Blood. 1994;83:1329–1336. [PubMed] [Google Scholar]
- 20.Vitetta E S, Uhr J W. Cancer Res. 1994;54:5301–5309. [PubMed] [Google Scholar]
- 21.Trauth B C, Klas C, Peters A M J, Matzku S, Moller P, Falk W, Debatin K, Krammer P H. Science. 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 22.Page D M, Defranco A. J Immunol. 1988;140:3717–3726. [PubMed] [Google Scholar]
- 23.Bridges S H, Kruisbeek A M, Longo D L. J Immunol. 1987;139:4242–4249. [PubMed] [Google Scholar]
- 24.Funakoshi S, Longo D L, Beckwith M, Conley D K, Tsarfaty G, Tsarfaty I, Armitage R J, Fanslow W C, Spriggs M K, Murphy W J. Blood. 1994;83:2787–2794. [PubMed] [Google Scholar]
- 25.Beckwith M, Urba W J, Ferris D K, Freter C E, Kuhns D B, Moratz C M, Longo D L. J Immunol. 1991;147:2411–2418. [PubMed] [Google Scholar]
- 26.Schreiber G J, Hellström K E, Hellström I. Cancer Res. 1992;52:3262–3266. [PubMed] [Google Scholar]
- 27.Scott D W, Tuttle J, Livnat D, Haynes W, Cogswell J, Keng P. Cell Immunol. 1985;93:124–131. doi: 10.1016/0008-8749(85)90393-4. [DOI] [PubMed] [Google Scholar]
- 28.Levy R, Miller R A. J Natl Cancer Inst Monogr. 1990;10:61–68. [PubMed] [Google Scholar]
- 29.Hamblin T J, Abdul-Ahab A K, Gordon J, Stevenson F K, Stevenson G T. Br J Cancer. 1980;42:495–502. doi: 10.1038/bjc.1980.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott G K, Dodson J M, Montgomery P A, Johnson R M, Sarup J C, Wong W L, Ullrich A, Shepard M, Benz C C. J Biol Chem. 1991;266:14300–14305. [PubMed] [Google Scholar]
- 31.Marches R, Racila E, Tucker T F, Picker L, Mongini P, Hsueh R, Vitetta E S, Scheuermann R H, Uhr J W. Ther Immunol. 1996;2:125–136. [PubMed] [Google Scholar]
- 32.Ghetie V, Thorpe P E, Ghetie M, Knowles P, Uhr J W, Vitetta E S. J Immunol Methods. 1991;142:223–230. doi: 10.1016/0022-1759(91)90110-2. [DOI] [PubMed] [Google Scholar]
- 33.Ghetie M, May R D, Till M, Uhr J W, Ghetie V, Knowles P P, Relf M, Brown A, Wallace P M, Janossy G, Amlot P, Vitetta E S, Thorpe P E. Cancer Res. 1988;48:2610–2617. [PubMed] [Google Scholar]
- 34.Fraker P J, Speck J C., Jr Biochem Biophys Res Commun. 1978;80:849–857. doi: 10.1016/0006-291x(78)91322-0. [DOI] [PubMed] [Google Scholar]
- 35.Coligan J E, Kruisbeek A M, Margulies D H, Shevach E M, Strober W, editors. Current Protocols in Immunology. New York: Wiley–Interscience; 1991. [Google Scholar]
- 36.Press O W, Farr A G, Borroz K I, Anderson S K, Martin P J. Cancer Res. 1989;49:4906–4912. [PubMed] [Google Scholar]
- 37.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz M A, Lassota P, Traganos F. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 38.Kim J-K, Tsen M, Ghetie V, Ward E S. Eur J Immunol. 1994;24:542–548. doi: 10.1002/eji.1830240308. [DOI] [PubMed] [Google Scholar]
- 39.Perrier D, Mayerson M. J Pharmacol Sci. 1982;71:372–373. doi: 10.1002/jps.2600710332. [DOI] [PubMed] [Google Scholar]
- 40.Schumaker R C. Drug Metab Rev. 1986;17:331–348. doi: 10.3109/03602538608998295. [DOI] [PubMed] [Google Scholar]
- 41.Ghetie M A, Richardson J, Tucker T, Jones D, Uhr J W, Vitetta E S. Int J Cancer. 1990;45:481–485. doi: 10.1002/ijc.2910450318. [DOI] [PubMed] [Google Scholar]
- 42.Kalbfleisch J D, Prentice R L. The Statistical Analysis of Failure Time Data. New York: Wiley; 1980. [Google Scholar]
- 43.Shah S A, Halloran P M, Ferris C A, Levine B A, Bourret L A, Goldmacher V S, Blattler W A. Cancer Res. 1993;53:1360–1367. [PubMed] [Google Scholar]
- 44.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 45.Sato T, Van Dixhoorn M G A, Schroeijers W E M, Van Es L A, Daha M R. Kidney Int. 1997;51:173–181. doi: 10.1038/ki.1997.21. [DOI] [PubMed] [Google Scholar]
- 46.Wolff E A, Schreiber G J, Cosand W L, Raff H V. Cancer Res. 1993;53:2560–2565. [PubMed] [Google Scholar]
- 47.Vervoordeldonk S F, Merle P A, van Leeuwen E F, van der Schoot C E, Kr.von dem Borne A E G, Slaper-Cortenbach I C M. Blood. 1994;83:1632–1639. [PubMed] [Google Scholar]
- 48.Shuford W, Raff H V, Finley J W, Esselstyn J, Harris L J. Science. 1991;252:724–727. doi: 10.1126/science.1902593. [DOI] [PubMed] [Google Scholar]
- 49.Caron P C, Laird W, Co M S, Avdalovic N M, Queen C, Scheinberg D A. J Exp Med. 1992;176:1191–1195. doi: 10.1084/jem.176.4.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenwood J, Gorman S D, Routledge E G, Lloyd I S, Waldmann H. Ther Immunol. 1994;1:247–255. [PubMed] [Google Scholar]
- 51.Shopes B. J Immunol. 1992;148:2918–2922. [PubMed] [Google Scholar]
- 52.Smith R I F, Morrison S L. Bio/Technology. 1994;12:683–688. doi: 10.1038/nbt0794-683. [DOI] [PubMed] [Google Scholar]