

Abstract
Lactacystin, a microbial metabolite that inhibits protease activity only in the proteasome, was used to study the role of the proteasome in the activation-induced cell death (AICD) of T cells. Lactacystin induces DNA fragmentation and apoptosis in a T cell hybridoma (DO.11.10) in a dose-dependent manner. Between 1 and 10 μM, the mildly cytotoxic lactacystin inhibited the AICD of DO.11.10 cells cultured in anti-CD3-coated wells. Degradation of IκBβ and the translocation of the NF-κB (p50/RelA) into the nucleus, which occurred at 1.5 hr after anti-CD3 activation, were inhibited by lactacystin. Lactacystin did not inhibit the expression of nuclear transcription factor Oct-1. The activation-induced expression of the immediate–early gene, Nur77, and the T cell death genes, CD95 (Fas) and CD95 ligand (FasL), were inhibited. Functional expression of FasL cytotoxicity and the increase of cell surface Fas were also inhibited. Lactacystin must be added within 2 hr of activation to efficiently block AICD. In addition, lactacystin failed to inhibit the killing of DO.11.10 by FasL-expressing allo-specific cytotoxic effector cells. These observations strongly suggest a direct link between the proteasome-dependent degradation of IκBβ and the AICD that occurs through activation of the FasL gene and up-regulation of the Fas gene.
Upon crosslinking T cell receptors, hybridoma T cells are activated and eventually die of apoptosis (1–3). This process requires activation of CD95 ligand (FasL) gene and the up-regulation of the CD95 (Fas) gene (1–3). The interaction between the two transmembrane proteins activates an apoptotic death program in Fas-expressing cells. Activation-induced cell death (AICD) is a critical mechanism for self-tolerance and lymphocyte homeostasis. We have previously shown that both the activation of FasL gene and the up-regulation of Fas gene can be inhibited by dexamethasone (Dex) (4). Dex binds to the glucocorticoid receptor and the receptor complex translocates into the nucleus, inducing IκBα gene activation (5, 6). IκBα binds to NF-κB and prevents NF-κB from translocating into the nucleus for gene activation. However, the complex of Dex and its receptor also binds AP-1 and inhibits its function, which is required for T cell activation (7).
To convincingly determine the role of NF-κB in AICD, a highly specific inhibitor of NF-κB translocation must be used. The translocation of NF-κB depends on the protease activity in the proteasome, which degrades IκBs (8–10). Although reagents capable of blocking NF-κB translocation are available, none of these inhibitors match the exquisite specificity offered by the microbial metabolite, lactacystin (LC) (11–13). LC specifically inhibits the protease activity of the proteasome and does not inhibit other well-known cellular proteases (13).
In this study, we first demonstrated that LC, at low doses that are mildly cytotoxic, is a suitable inhibitor of AICD. We then demonstrated that LC inhibits a T cell activation pathway that starts with the activation-induced degradation of IκBβ and translocation of the p50/RelA heterodimer of NF-κB. These early proteasome-dependent events are followed by the activation of the immediate–early gene, Nur77, and T cell death genes, FasL and Fas. The functional expression of FasL cytotoxicity, the up-regulation of Fas surface expression, and the final expression of the Fas-mediated death program are also inhibited. LC must be added within 2 hr of activation to efficiently block AICD, demonstrating the critical role of the early events in AICD. Our study strongly suggests a direct link between the proteasome-dependent degradation of IκBβ, translocation of NF-κB and AICD through activation of the FasL gene, and up-regulation of the Fas gene.
MATERIALS AND METHODS
Reagents and Cells.
N-acetyl-l-leucinyl-l-leucinyl-l-norleucinal (LLnL), Dex, and human Ig were obtained from Sigma. LC was produced as previously described (12). The oligonucleotide probe bearing the κB site sequence of the Igκ enhancer (GATCCAGAGGGGACTTTCCGAGAG, consensus sequence underlined) was prepared as described (14). The oligonucleotide probe for Oct-1 (AGTTGAATGCAAATAGGC) and rabbit antibodies specific to IκBα, IκBβ, p50, RelA, c-Rel, and p52 were obtained from Santa Cruz Biotechnology. Radioisotopes (Na251CrO4, [α-32P]-dCTP, [γ-32P]-dATP) were purchased from New England Nuclear. The preparation of Fas-Ig fusion proteins has been described (3). L1210 lymphoma cells transfected with Fas (LF+) were obtained from W. R. Clark (University of California, Los Angeles) (15). The T cell hybridoma DO.11.10 was obtained from P. Marrack (National Jewish Hospital, University of Colorado).
Cell Activation.
Activation of DO.11.10 cells was carried out as previously described with slight modification (3). In brief, DO.11.10 cells (1.5 × 106) were added to uncoated and anti-CD3-coated dishes (60 × 15 mm) and cultured for various times. Cells treated for 1.5 hr were used to assay for cytoplasmic IκB levels by Western blot analyses and for nuclear levels of NF-κB and Oct-1 by electrophoretic mobility shift assay (EMSA). Cells activated for 5 hr were assayed for Nur77, Fas, and FasL messages by reverse transcription (RT)–PCR. Cells activated for 10 hr were assayed for FasL cytotoxicity by a 5-hr 51Cr release assay (16). Cells treated for 14 hr were used for DNA fragmentation assay by agarose gel electrophoresis as described (17). Cells activated for 14 hr in the presence of 10 μg/ml of Fas-Ig were assayed for Fas by flow cytometry as described (4). The effect of various inhibitors on cell activation was carried out either by pretreating cells with inhibitors for 30 min or by adding various amounts of inhibitors at the beginning of culture.
Western Blot Analysis.
Western blot analyses for IκBα and IκBβ were carried out as previously described (18).
EMSA.
EMSA were carried out as described (14), using nuclear extracts of DO.11.10 cells treated under various conditions as described above. Oligonucleotide probes were labeled either by T4 polynucleotide kinase or by Klenow fragment (New England Biolabs). The binding reaction mixture contained labeled probe (0.2 ng), 1–3 μg of nuclear extract protein, poly(dI⋅dC) (1 μg), in a final volume of 15–20 μl of EMSA buffer (14). The reaction mixtures were incubated at room temperature for 25 min and then electrophoresed through 5% polyacrylamide gels under nondenaturing conditions. Supershift analysis was carried out by adding 1.5 μg of purified rabbit antibodies against RelA, c-Rel, p50, or p52 to the reaction mixtures. Normal rabbit Ig was used as a control. Gels were dried and developed with Kodak OXR film.
RT-PCR.
The primers for Nur77 are: 5′-CTTCTTCAAGCGCACAGTAC-3′ (forward) (positions 986 to 1005 of the cDNA sequence), and 5-′GGTTAGAAGAGTGAAGGGAG-3′ (reverse) (position 1223 to 1204). The RT-PCR assays for Fas, FasL, and β-actin were carried out as previously described and the same condition was used for Nur77 (3).
FasL Cytotoxicity.
Various numbers of hybridoma T cells, treated under the conditions described above, were used as effectors in a 5-hr cytotoxicity assay using 51Cr-labeled LF+ cells as target (2 × 104/w) and in the presence of 6 mM EGTA and 3 mM MgCl2. Target cells cultured in the absence of effectors were used as controls. The [Ca2+]ext-independent condition specifically measures FasL-mediated cytotoxicity (16). In separate experiments, perforin knockout mice were used to generate allo-specific cytotoxic T cells against H-2d as described (17). They were first activated in anti-CD3-coated wells for 5 hr and then used as effector cells against DO.11.10 target under the [Ca2+]ext-independent condition. The 5- and 10-hr activation conditions were used because they induced maximal expression of FasL cytotoxicity (not shown). Cytotoxicity, expressed as percent specific killing, was determined 5 hr later.
AICD.
AICD of the 51Cr-labeled DO.11.10 cells was carried out in 96-well plates coated with anti-CD3 mAb as previously described (3). Cells cultured in uncoated wells were used as controls. The percent cell death was determined at 12–16 hr after culture. The effect of inhibitors on AICD was determined by adding various doses of inhibitors at the beginning of culture.
The percent cell death for AICD and FasL cytotoxicity was calculated by the formula: 100 × (cpm released by experimental group − cpm released by control)/(total cpm added − cpm released by control).
Flow Cytometry.
To assess surface Fas expression, 5 × 105 cells were washed in PBS containing 5% fetal calf serum and 20 mM azide, resuspended in 30 μl buffer, and incubated for 45 min on ice with 1 μl phycoerythrin-conjugated anti-Fas, fluorescein isothiocyanate (FITC)–anti-Thy-1 antibody, or an isotype control antibody (PharMingen). Cells were washed and analyzed with a Becton Dickinson FACScan.
All experiments were repeated 2–5 times with similar results.
RESULTS
LC Inhibits AICD.
The effect of LC on AICD was studied using plate-bound anti-CD3 to induce the death of DO.11.10 cells (Fig. 1). LLnL and Dex were included for comparison because they inhibit NF-κB translocation (5, 6, 19). At noncytotoxic doses, both LC and LLnL caused a modest inhibition of AICD. In contrast, noncytotoxic doses of Dex strongly inhibited AICD. At doses that were mildly toxic, both LC and LLnL caused a significant degree (50–80%) of inhibition of AICD. However, complete inhibition of cell death was not observed, suggesting that anti-CD3 activation did not protect hybridoma T cells from the inhibitor’s toxicity. This “one-way” protection is different from the “mutual” protection observed with both moderately and highly toxic doses of Dex (Fig. 1).
Figure 1.
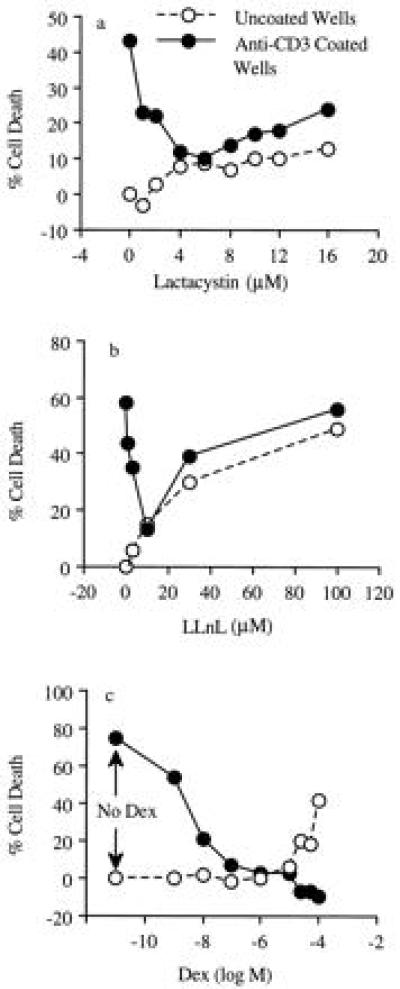
Effect of LC on AICD. AICD was carried out with the 51Cr-labeled DO.11.10 cells in anti-CD3-coated wells. Various doses of LC, LLnL, and Dex were added at the beginning of culture. AICD (•) was determined at 12 hr for LC and 14–16 hr for LLnL and Dex, respectively. Drug cytotoxicity (○) was determined in uncoated wells. The results were expressed as the percent of cell death. Similar results were obtained in two other experiments.
The effect of LC on AICD was also determined by DNA fragmentation analysis using agarose gel electrophoresis. As shown in Fig. 2, LC by itself induced DNA fragmentation in DO.11.10 cells in a dose-dependent manner. The extent of DNA fragmentation induced by 100 μM was slightly weaker than plate-bound anti-CD3 mAb. LC, at 10 μM and 0.5 μM, induced weak and undetectable DNA fragmentation, respectively. When cultured in anti-CD3-coated wells, DNA fragmentation was inhibited significantly by 10 μM LC. Both 100 μM and 0.5 μM LC did not inhibit DNA fragmentation. In the former case, the toxicity of LC dominated over AICD, whereas in the latter situation the concentration of LC was insufficient to block AICD. As shown by the 51Cr release assay, 10 μM LC was a proper dose to inhibit AICD.
Figure 2.
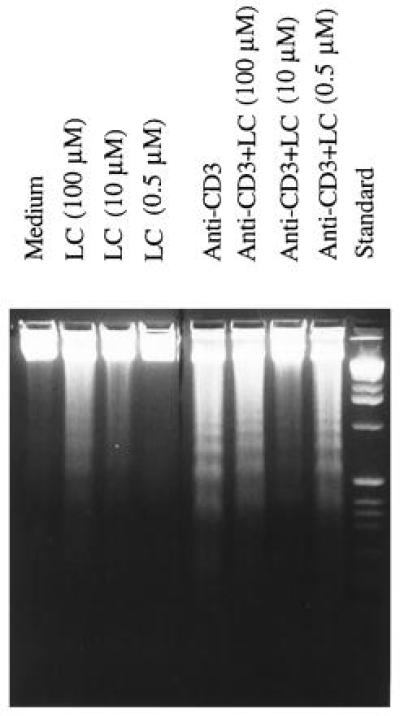
DNA fragmentation analysis of DO.11.10 cells. DNA fragmentation was carried out as described. AICD was inhibited by 10 μM of LC by comparing lanes (from left) 3, 5, and 7.
LC Inhibits IκBβ Degradation.
Next, we determined the effect of LC on IκB levels during anti-CD3-mediated activation of DO.11.10 cells. The results showed that IκBβ was degraded following CD3 crosslinking (Fig. 3a). LC did not affect IκBβ levels in unactivated cells (not shown), but blocked the anti-CD3-induced IκBβ degradation. There was little degradation of IκBα in activated DO.11.10 cells and its level remained unchanged in the presence of LC. The preferential degradation of IκBβ has been demonstrated in another hybridoma (data not shown).
Figure 3.
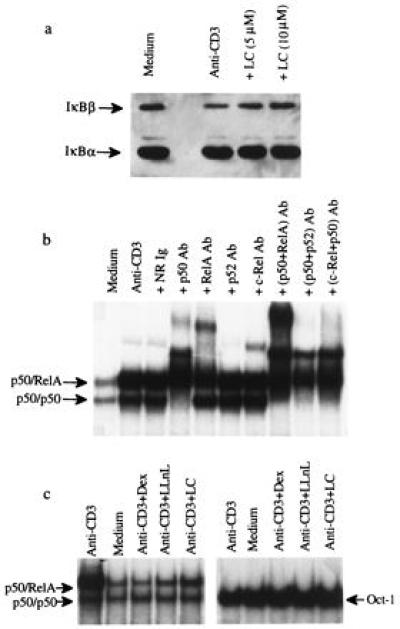
LC inhibits IκBβ degradation and NF-κB translocation induced by plate-bound anti-CD3 mAb. (a) LC inhibits IκBβ degradation. DO.11.10 cells were cultured for 90 min in uncoated wells (Medium), wells coated with anti-CD3 mAb (Anti-CD3), or coated wells with LC (Anti-CD3+LC). Cytoplasmic extracts were analyzed for IκBα and IκBβ by Western blot analysis as described. Scanning results showed that there was a 51% degradation of IκBβ by anti-CD3 activation and 10 μM LC inhibited the degradation to 8%. (b) Supershift analysis of NF-κB. The nuclear extracts were analyzed for NF-κB with antibodies specific to p50, RelA, c-Rel, and p52. A combination of anti-p50 and anti-RelA, anti-p50 and anti-p52, and anti-p50 plus anti-c-Rel were also tested. Normal rabbit Ig (NR Ig) was included as a control. Free probe band at the bottom of the gel was not presented. (c) LC inhibits NF-κB translocation. DO.11.10 cells were cultured for 90 min in uncoated wells (Medium), wells coated with anti-CD3 mAb (Anti-CD3), or anti-CD3-coated wells with 5 μM Dex (+Dex), 20 μM LLnL (+LLnL), or 10 μM LC (+LC). Nuclear extracts were assayed for NF-κB and Oct-1 by EMSA. Scanning results showed that anti-CD3 activation increases 250% of p50/p65 expression and 30% of the p50/p50 expression. The inhibition of p50/p65 expression by Dex, LLnL, and LC was 111%, 95%, and 80%, respectively. No inhibition of the p50/p50 expression was observed. There was no inhibition of Oct-1 expression in all cases.
LC Inhibits NF-κB Translocation.
The effect of LC on NF-κB translocation into the nucleus was determined by EMSA (Fig. 3b). The nuclear extract of unactivated cells expressed a basal level of NF-κB. By contrast, the nuclear extract from anti-CD3-activated DO.11.10 cells strongly expressed the p50/RelA heterodimer and moderately expressed the p50/p50 homodimer of NF-κB, as demonstrated by antibody-mediated supershift assays (Fig. 3b). There was little c-Rel- or p52-containing NF-κB in the nuclear extracts. In the presence of LC, translocation of p50/RelA was strongly inhibited. The anti-CD3-induced NF-κB translocation was also inhibited by LLnL and Dex (Fig. 3c). As controls, these extracts were tested for Oct-1, a constitutively expressed transcription factor, and no inhibition was observed (Fig. 3c).
LC Inhibits FasL and Fas Gene Expression.
The effect of LC on the the expression of Nur77, FasL, and Fas at the mRNA level was determined by RT-PCR (Fig. 4). Anti-CD3 induced the expression of Nur77, an immediate–early gene required for AICD (20, 21). Both LC and LLnL, but not Dex, inhibited Nur77 gene activation. Dex has been shown to be a weak inhibitor of Nur77 expression (22). The data suggest that the expression of Nur77 is essential, but additional events that can be blocked by Dex must have occurred to activate FasL gene. By contrast, FasL mRNA induced by plate-bound anti-CD3 was strongly inhibited by LC, LLnL, and Dex (Fig. 4). These inhibitors also blocked the anti-CD3-induced up-regulation of Fas mRNA. Interestingly, treated cells retained the basal expression of Fas mRNA.
Figure 4.
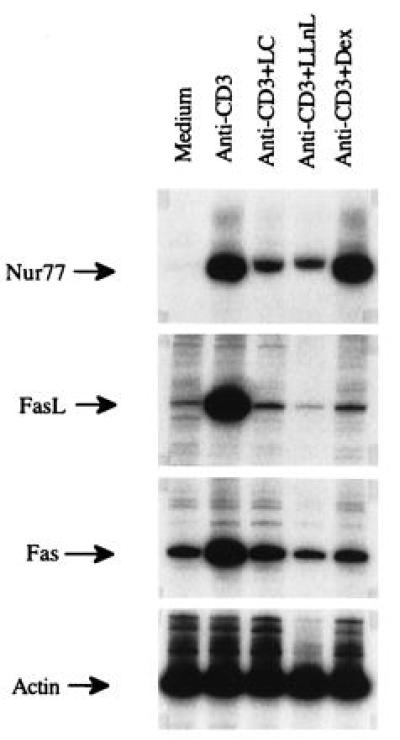
Effect of LC on the mRNA expression of Fas and FasL. DO.11.10 cells were cultured in uncoated wells, anti-CD3-coated wells, and anti-CD3-coated wells with 10 μM LC, 20 μM LLnL, or 5 μM Dex for 5 hr. Expression of Nur77, FasL, Fas, and β-actin was determined by RT-PCR as described.
LC Inhibits FasL Function and Fas Surface Expression.
The effect of inhibitors on the anti-CD3-induced FasL cytotoxicity was determined as described in Materials and Methods. The results showed that the induction of FasL cytotoxicity by anti-CD3 activation was strongly inhibited by LC, LLnL, and Dex (Fig. 5a). To determine the effect of LC on Fas expression, we carried out anti-CD3-mediated activation in the presence of Fas-Ig. Cells activated in the presence of Fas-Ig did not die of AICD and after washing, stained strongly with FITC–anti-Fas over the control cells cultured in uncoated wells in an otherwise identical condition (Fig. 5b). In the presence of LC, the up-regulation of Fas was strongly inhibited (Fig. 5c). LC did not inhibit the basal level of Fas expression, in agreement with the Fas mRNA study. There was no difference in Thy-1 staining in untreated, anti-CD3, or LC treated cells (not shown).
Figure 5.
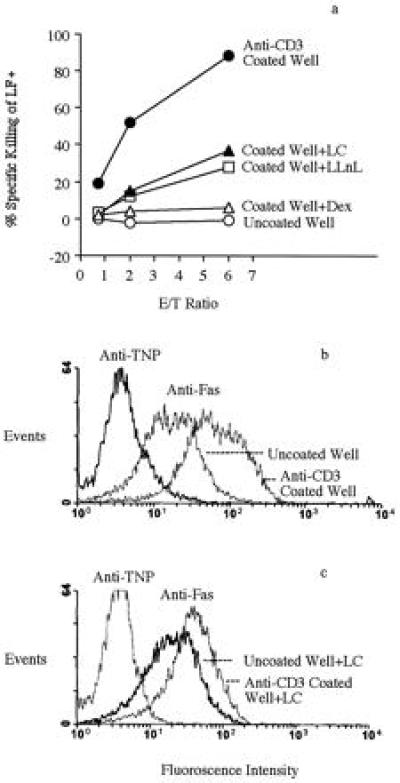
LC inhibits the anti-CD3-induced FasL cytotoxicity and Fas surface expression. (a) LC inhibits the anti-CD3-induced FasL cytotoxicity. DO.11.10 cells were cultured in uncoated wells, anti-CD3-coated wells, or anti-CD3-coated wells with inhibitors for 10 hr. The inhibitors tested were LC (10 μM), LLnL (20 μM), and Dex (5 μM). Treated cells were collected, washed three times, and tested as effectors against 51Cr-labeled LF+ targets (2 × 104) at various effector-to-target cell (E/T) ratios under the [Ca2+]ext-independent condition. There were 30% dead cells in the anti-CD3-treated group. The strong cytotoxicity of this group indicates that this level of AICD has little effect on FasL cytotoxicity. Cytotoxicity was determined after 5 hr of culture. In the absence of effector cells, nonspecific 51Cr release was less than 15%. The data presented are representative of three experiments. (b and c) LC inhibits the anti-CD3-induced cell surface expression of Fas. The condition was the same as in a, except that 10 μg/ml Fas-Ig were added so that hybridoma T cells expressing high Fas remained viable for analysis. Cells were extensively washed and stained with FITC–anti-Fas mAb and isotype control FITC–anti-2,4,6,-trinitrophenyl (TNP) mAb.
LC Must Be Added Early to Inhibit AICD.
That the early activation events inhibited by LC are critical for AICD was demonstrated by the rapid loss of the ability of LC to inhibit AICD when added at 2 hr after addition of DO.11.10 cells to anti-CD3-coated wells (Fig. 6a). Moreover, LC did not inhibit the DO.11.10 cell death mediated by the FasL-expressing allo-specific effectors (see Materials and Methods). Death of DO.11.10 cells was completely inhibited by Fas-Ig but not human Ig (Fig. 6b). These observations show that LC acts upon the early events of T cell activation and becomes ineffective once the crosslinking of Fas by FasL has occurred.
Figure 6.
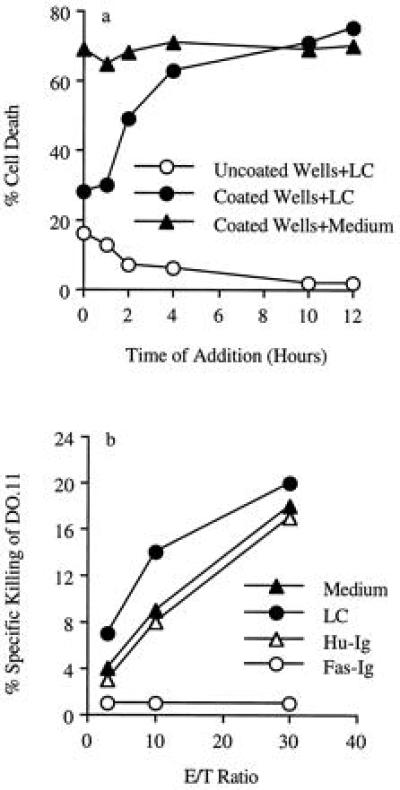
LC must be added early to inhibit AICD. (a) LC (10 μM) was added at various times after addition of 51Cr-labeled DO.11.10 cells to uncoated wells or anti-CD3-coated wells. Control was done by the addition of medium. AICD was determined 12 hr after the addition of DO.11.10 cells. (b) Various numbers of allo-specific effectors, prepared and activated as described in Materials and Methods, were cultured with labeled DO.11.10 cells (2 × 104 per well) at various effector-to-target cell (E/T) ratios. LC (10 μM), Fas-Ig (10 μg/ml), or human Ig (Hu-Ig, 10 μg/ml) was added at the beginning of culture. Cytotoxicity assays were carried out under the [Ca2+]ext-independent condition.
DISCUSSION
LC, which forms a covalent bond with the highly conserved threonine residue of the mammalian proteasome subunit X (also called MB1), is the most selective proteasome inhibitor presently known. Even at high concentrations and long incubation time, LC does not inhibit other proteases, including calpain, cathepsin B, trypsin, chymotrypsin, and papain (13). This study took advantage of the exquisite specificity of LC to investigate the relationship between proteasome function and AICD. Also, our study has demonstrated that proteasomes regulate AICD. The AICD pathway that we are proposing starts, upon T cell receptor-dependent activation, with the degradation of IκBβ and translocation of p50/RelA, followed by the activation of the immediate–early gene Nur77, and the T cell death genes Fas and FasL. Coexpression of Fas and FasL results in AICD (1–3). This sequence of events is consistent with the time course kinetics of these events (3, 4, 20, 21). LC inhibits each of these events of AICD, thus linking proteasome-dependent NF-κB translocation to AICD through FasL gene activation and up-regulation of the Fas gene. The role of IκBβ and NF-κB in the regulation of AICD can be established when IκB dominant–negative hybridomas become available.
A recent study has implicated lck in the activation of the NF-κB translocation pathway through tyrosine phosphorylation of IκBα (23). Jurkat cells treated with pervanadate, an inhibitor of protein tyrosine phosphatases, was found to translocate p50/RelA without the degradation of IκBα. The effect of the treatment on IκBβ was not determined. Interestingly, phosphorylation of IκBα was dependent on lck because pervanadate did not induce tyrosine phosphorylation of IκBα in lck mutant cells. We have observed an extremely low level expression of the larger and possibly the tyrosine phosphorylated IκBα (Fig. 3a). It is possible that this pathway is efficiently blocked by a phosphatase (such as CD45), which is present during T cell activation (23). If this abortive pathway happened, then the addition of phosphatase inhibitors during anti-CD3 activation should increase the level of phosphorylated IκBα and p50/RelA translocation. Moreover, both should be resistant to LC.
The limited sequence information on the 5′ flanking region of mouse FasL gene (−350 from ATG translation initiation site) contains three potential sites for SP-1, κB, and IRF-1, respectively (24). We and others have not been able to demonstrate NF-κB binding to this putative site (25). Our study uses a probe derived from Ig-κ gene sequence to analyze NF-κB translocation during AICD. Its major significance is that it delineates the early cytoplasmic pathway of AICD at the molecular level by linking proteasome-dependent NF-κB translocation to FasL gene activation and AICD. It remains to be seen whether translocated NF-κB affects FasL gene expression by binding directly to the FasL promoter region or indirectly through immediate early genes, whose expression depends on NF-κB binding. Definitive evidence can be obtained once the entire promoter region is cloned, sequenced, and characterized by standard transfection experiments. Such a study of a 5-kb fragment flanking the 5′ region of the FasL gene is in progress and several potential κB sites have been found (A. Marshak-Rothstein, personal communication). When the same analysis was carried out with the published Fas promoter region, we were unable to find a potential κB site (25). The observation that LC inhibits the up-regulation of Fas mRNA but not the basal level of Fas, therefore, strongly suggests that NF-κB indirectly regulates Fas up-regulation.
Even if a functional κB site was identified in the FasL promoter, NF-κB could still indirectly regulate FasL gene expression. This pathway could be mediated through the immediate–early genes, c-myc and Nur77, whose expression was inhibited by LC (Fig. 4 and data not shown). It has been shown that the expression of c-myc is directly regulated by NF-κB (18), and that modulation of c-myc expression is required for AICD (26). The finding that FasL is constitutively expressed in thymocytes of Nur77 transgenic mice (27) and our observation that LC regulates Nur77, probably through modulation of NF-κB, provides further evidence that NF-κB indirectly regulates FasL gene expression.
At higher doses, LC induces apoptosis in hybridoma T cells. Considering the exquisite specificity of LC, it is likely that blocking proteasome function induces apoptosis. Cell death could be induced by inhibiting the basal level of NF-κB translocation required for cell survival (28). Furthermore, down-regulation of NF-κB sensitizes targets to tumor necrosis factor cytotoxicity, whereas up-regulation protects these targets (29–31). A recent study has suggested that death of leukemic HL60 cells induced by proteasome inhibitors is dependent on cell cycle, and that the activation of CPP32, a critical cysteine protease required for apoptosis, is suppressed by a proteasome-dependent pathway during cell cycle (32). This hypothesis is supported by two earlier studies that showed that LC inhibited the death of thymocytes that had been treated with ionizing radiation or with Dex, or the death of sympathetic neurons that were deprived of nerve growth factor (33, 34). The inhibition was accompanied by the blockade of the CPP32-dependent cleavage of poly(ADP-ribose) polymerase (33, 34). The mechanism by which proteasomes regulate CPP32 has not been established. Our study showed that LC, at doses that were weakly cytotoxic, inhibited AICD, but the inhibition is at the level of FasL/Fas expression. The data underscore the role of CPP32 and NF-κB in AICD because both could have regulated target cell susceptibility to Fas-mediated apoptosis. Our observations suggest that anti-CD3-induced NF-κB translocation is not sufficient to protect hybridoma T cells from the AICD and that there are multiple ways in which proteasomes regulate cell death programs.
Finally, abnormality in the regulation of Fas and FasL genes has been linked to autoimmune diseases in mice and human (35–38). FasL, in particular, has also been implicated in a number of important immune phenomena. During T/B interaction, helper T cells of the Th1 type can down-regulate both T and B cell responses by Fas/FasL interaction (39). Moreover, B cell susceptibility to the FasL cytotoxicity of CD4+ Th1 cells is critically regulated by lymphokines and signals from CD40 and antigen receptors (40, 41). Constitutive expression of FasL in tissues or grafts may be responsible for the maintenance of immune privileged sites and transplantation tolerance, respectively (42–44). Some anti-neoplastic drugs induce tumor cells to express FasL, which may induce tumor cell apoptosis (45) or may evade immune surveillence by killing the infiltrating lymphocytes (46). This study, therefore, raises the possibility that LC or related products, by their ability to efficiently inhibit FasL/Fas expression, can be developed for the regulation of immune response and for the treatment of various immune diseases.
Acknowledgments
We thank Dr. T. L. Rothstein for providing the NF-κB probe. Dr. A. Marshak-Rothstein for sharing her unpublished work, Dr. B. Stanger for Fas-Ig transfected 3T3 cells, and Drs. A. Marshak-Rothstein and D. H. Sherr for critical review of the manuscript. This work is supported by National Institutes of Health Grants RO1–36938 (S.-T.J.) and CA-36355 (G.E.S.).
ABBREVIATIONS
- AICD
activation-induced T cell death
- Dex
dexamethasone
- EMSA
electrophoretic mobility shift assay
- Fas
CD95
- FasL
CD95 ligand
- LC
lactacystin
- LLnL
N-acetyl-l-leucinyl-l-leucinyl-l-norleucinal
- RT
reverse transcription
References
- 1.Dhein J, Walczak H, Baumier C, Debatin K-M, Krammer P H. Nature (London) 1995;373:438–440. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 2.Brunner T, Mogil R J, LaFace D, Yoo N J, Mahboubi A, Echeverri F, Martin S J, Force W R, Lynch D H, Ware C F, Green D R. Nature (London) 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 3.Ju S-T, Panka D J, Cui H, Ettinger R, El-Khatib M, Sherr D H, Stanger B Z, Marshak-Rothstein A. Nature (London) 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 4.Cui H, Sherr D H, El-Khatib M, Matsui K, Panka D J, Marshak-Rothstein A, Ju S-T. Cell Immunol. 1996;167:276–284. doi: 10.1006/cimm.1996.0036. [DOI] [PubMed] [Google Scholar]
- 5.Auphan N, DiDonato J A, Rosette C, Helmberg A, Karin M. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 6.Scheinman R I, Cogswell P C, Lofquist A K, Baldwin A S., Jr Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 7.Kerppola T K, Luk D, Curran T. Mol Cell Biol. 1993;13:3782–3791. doi: 10.1128/mcb.13.6.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baeuerle P A, Henkel T. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 9.Finco T S, Baldwin A S. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 10.Baeuerle P A, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 11.Rock K L, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg A L. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 12.Omura S, Fujimoto T, Otoguro K, Matsuzaki K, Moriguchi R, Tanaka H, Sasaki Y. J Antibiot. 1991;44:113–116. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]
- 13.Fenteany G, Standaert R F, Lane W S, Choi S, Corey E J, Schreiber S L. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 14.Francis D A, Karras J G, Ke X-Y, Sen R, Rothstein T L. Int Immunol. 1994;7:151–161. doi: 10.1093/intimm/7.2.151. [DOI] [PubMed] [Google Scholar]
- 15.Walsh C M, Glass A A, Chiu V, Clark W R. J Immunol. 1994;153:2506–2514. [PubMed] [Google Scholar]
- 16.El-Khatib M, Stanger B Z, Dogan H, Cui H, Ju S-T. Cell Immunol. 1995;163:237–244. doi: 10.1006/cimm.1995.1122. [DOI] [PubMed] [Google Scholar]
- 17.Ju S-T. J Immunol. 1991;146:812–818. [PubMed] [Google Scholar]
- 18.La Rosa F A, Pierce J W, Sonenshein G E. Mol Cell Biol. 1994;14:1039–1044. doi: 10.1128/mcb.14.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 20.Woronicz J D, Calnan B, Ngo V, Winoto A. Nature (London) 1994;367:277–281. doi: 10.1038/367277a0. [DOI] [PubMed] [Google Scholar]
- 21.Liu A-G, Smith S W, McLaughlin K A, Schwartz L M, Osborne B A. Nature (London) 1994;367:281–284. doi: 10.1038/367281a0. [DOI] [PubMed] [Google Scholar]
- 22.Woronicz J D, Lina A, Calnan B J, Szychowski S, Cheng L, Winoto A. Mol Cell Biol. 1995;15:6364–6376. doi: 10.1128/mcb.15.11.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imbert V, Rupec R A, Livolsi A, Pahl H L, Traenckner E B-M, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle P A, Peyron J-F. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi T, Tanaka M, Inazawa J, Abe T, Suda T, Nagata S. Int Immunol. 1994;6:1567–1574. doi: 10.1093/intimm/6.10.1567. [DOI] [PubMed] [Google Scholar]
- 25.Anel A, Simon A K, Auphan N, Buferne M, Boyer C, Golstein P, Schmitt-Verhyulst A M. Eur J Immunol. 1995;25:3381–3387. doi: 10.1002/eji.1830251227. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y, Glynn J M, Guilbert L J, Cotter T G, Bissonnette R P, Green D R. Science. 1992;257:212–214. doi: 10.1126/science.1378649. [DOI] [PubMed] [Google Scholar]
- 27.Weih F, Ryseck R-P, Chen L, Bravo R. Proc Natl Acad Sci USA. 1996;93:5533–5538. doi: 10.1073/pnas.93.11.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu M, Lee H, Bellas R E, Schauer S L, Marcello A, Katz D, Fitzgerald M J, Rothstein T L, Sherr D H, Sonenshein G E. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 29.Beg A A, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 30.Wang C-Y, Mayo M W, Baldwin A S., Jr Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 31.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 32.Drexler H C A. Proc Natl Acad Sci USA. 1997;94:855–860. doi: 10.1073/pnas.94.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grimm L M, Goldbery A L, Poirier G G, Schwartz L W, Osborne B A. EMBO J. 1996;15:3835–3844. [PMC free article] [PubMed] [Google Scholar]
- 34.Sadoul R, Fernandez P-A, Quiquerez A-L, Martinou I, Maki M, Schroter M, Becherer J D, Irmler M, Tschopp J, Martinou J-C. EMBO J. 1996;15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe-Fukunaga R, Brannan C I, Copeland N G, Jenkins N A, Nagata S. Nature (London) 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi T, Tanaka M, Brannan C I, Jenkins N A, Copeland N G, Suda T, Nagata S. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 37.Fisher G H, Rosenberg F J, Straus S E, Dale J K, Middelton L A, Lin A Y, Strober W, Lenardo M J, Puck J M. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 38.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts I A G, Debatin K M, Fischer A, DeVillartay J P. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 39.Ozdemirli M, El-Khatib M, Foote L C, Wang J K M, Marshak-Rothstein A, Rothstein T L, Ju S-T. Eur J Immunol. 1996;26:415–419. doi: 10.1002/eji.1830260222. [DOI] [PubMed] [Google Scholar]
- 40.Foote L C, Schneider T J, Fischer G M, Wang J K M, Rasmussen B, Campbell K A, Lynch D H, Ju S-T, Marshak-Rothstein A, Rothstein T L. J Immunol. 1996;157:1878–1886. [PubMed] [Google Scholar]
- 41.Rothstein T L, Wang J K M, Panka D, Foote L C, Wang Z, Stanger B, Cui H, Ju S-T, Marshak-Rothstein A. Nature (London) 1995;374:163–165. doi: 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- 42.Lau H T, Yu M, Fontana A, Stoeckert C J., Jr Science. 1996;273:109–112. doi: 10.1126/science.273.5271.109. [DOI] [PubMed] [Google Scholar]
- 43.Bellgrau D, Gold D, Selawry H, Moore J, Franzosoff A, Duke R C. Nature (London) 1995;337:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- 44.Griffin T S, Brunner T, Fletcher S M, Green D R, Ferguson T A. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 45.Friesen C, Herr I, Krammer P H, Debatin K-M. Nat Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- 46.Strand S, Hofmann W J, Hug H, Muller M, Otto G, Strand D, Mariani S M, Stremmel W, Krammer P H, Galle P R. Nat Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]