

Abstract
The three-dimensional structure of the N-terminal domain (residues 18–112) of α2-macroglobulin receptor-associated protein (RAP) has been determined by NMR spectroscopy. The structure consists of three helices composed of residues 23–34, 39–65, and 73–88. The three helices are arranged in an up-down-up antiparallel topology. The C-terminal 20 residues were shown not to be in a well defined conformation. A structural model for the binding of RAP to the family of low-density lipoprotein receptors is proposed. It defines a role in binding for both the unordered C terminus and the structural scaffold of the core structure. Pathogenic epitopes for the rat disease Heymann nephritis, an experimental model of human membranous glomerulonephritis, have been identified in RAP and in the large endocytic receptor gp330/megalin. Here we provide the three-dimensional structure of the pathogenic epitope in RAP. The amino acid residues known to form the epitope are in a helix–loop–helix conformation, and from the structure it is possible to rationalize the published results obtained from studies of fragments of the N-terminal domain.
The α2-macroglobulin receptor-associated protein (RAP) is a 39- to 40-kDa intracellular glycoprotein (1–3) that binds to the α2-macroglobulin receptor (α2MR/LRP) and other members of the low density lipoprotein receptor family (4). The protein inhibits binding of all currently known ligands of these receptors. It has been suggested that binding to α2MR/LRP serves to prevent receptor aggregation and degradation in the endoplasmatic reticulum (5–7), and as such RAP seems to serve a function as a molecular chaperone. Recently, two additional RAP binding receptors have been identified (8, 9). Furthermore, RAP has been shown to bind calmodulin and to become phosphorylated by calmodulin-dependent kinase II, suggesting a regulatory mechanism by which control of RAP function could be exerted by calmodulin-dependent phosphorylation (10).
Previously, distinct functional fragments of RAP have been defined for binding to heparin as well as to gp330/megalin, which belongs to the low density lipoprotein receptor family (11). In addition, the primary structure of RAP shows evidence of internal triplicate sequence homology first noticed by Warshawsky et al. (12). Recently, a new set of domain boundaries was established, and human RAP was shown to contain three structural and functionally autonomous domains comprising residues 18–112 (domain 1), 113–218 (domain 2), and 219–323 (domain 3) (13). Binding studies have shown that the C-terminal 15 residues of domain 1 are required to inhibit binding of activated α2-macroglobulin, whereas the C-terminal 15 residues of both domains 1 and 3 are important for binding of RAP to the α2-macroglobulin receptor (13, 14). A common receptor binding motif shared among many of the ligands of the low density lipoprotein receptor family members recently has been proposed on the basis of biochemical analysis, sequence comparisons, and mutagenesis studies (15). This motif rationalizes the importance for receptor binding of the C-terminal segments of RAP domains 1 and 3.
Heymann nephritis (HN) is an experimentally induced autoimmune disease in rats (16) used widely as a valid model for human membranous glomerulonephritis, a common human glomerular disease known to cause proteinuria and loss of renal function (17), with which it shares evident similarities at the pathophysiological and immunohistological levels. The disease is characterized by immune deposits initially located in coated pits at the base of the glomerular epithelium, subsequently shed from the cell surface to adhere tightly to the glomerular basement membrane (18). HN exists in an active form traditionally induced by immunization with crude antigen preparations such as the proximal renal tubular brush border extract Fx1A (19), and in a passive form that can be induced with antibodies raised against renal antigens. An improved understanding at the molecular level of the pathogenesis of HN is essential for immunotherapeutic intervention with the disease, based on e.g., synthetic peptides mimicking pathogenic epitopes, as previously proposed (20, 21).
Recent efforts have been directed at defining pathogenic epitopes for HN in both of the known antigenic targets of the disease, gp330/megalin and RAP. In gp330/megalin, a fragment containing amino acid residues 1,114–1,250 of the molecule was shown to be able to induce active HN (22). Furthermore, the participation of residues 1,160–1,205 in formation of passive HN has been demonstrated (23). In RAP, a major pathogenic nonlinear epitope for the induction of passive HN has been mapped to the fragment comprising amino acid residues 31–53 (21).
METHODS
The domains were obtained by heterologous expression in Escherichia coli (13). A two-dimensional 1H nuclear Overhauser effect (NOE) spectroscopy spectrum recorded with a mixing time of 90 ms with 15N decoupling (24, 25) using a 2.5 mM 15N-labeled sample of RAP truncated domain 1 (RAPd1T) was used to determine the strength of the distance restraints. A total of 1,178 nontrivial upper-distance restraints obtained from NOEs in this spectrum were used for the structure calculations (26). These were 218 intraresidue, 296 sequential, 363 medium-range, and 301 long-range constraints. NOE distance constraints were divided into three classes, low, medium, and strong, all with a lower distance limit of 1.8 Å and with upper distance limits of 5.0, 3.6, and 2.7 Å, respectively. For methyl protons 0.5 Å was added (27). By using both the method of Ludvigsen et al (28). and Neri et al (29), it was possible to obtain 3JHNHα coupling constants for all residues, except for glycines and prolines, resulting in 54 φ angle constraints. For residues with a 3JHNHα below 4 Hz a φ angle constraint of −57 ± 40 degrees was applied, and for residues with coupling constants above 8 Hz a φ angle constraint of −120 ± 40 degrees was applied (30). One positive φ angle was identified for His-20, satisfying the criteria of Ludvigsen and Poulsen (31). Also 41 χ angles were constrained to one of the three staggered conformations ±60 based on NOE intensities, relative coupling constants judged from double quantum filtered correlation spectroscopy (32) spectra and JNHβ coupling constants measured in undecoupled two-dimensional 1H NOE spectroscopy spectrum of a 15N-labeled sample. Distance constraints for backbone hydrogen bonds defining the helices from the first round of structure calculations were included as NOE distance restraints in the final calculations with the distance constraints being 2.85 ± 0.15 Å for the donor N to acceptor O distance and 1.9 ± 0.1 Å for the donor HN to acceptor O distance. This did not change the structure significantly but greatly improved convergence in the structure calculations. The structures were calculated using the x-plor program (33) by a standard distance geometry, restrained simulated annealing, and restrained energy minimization protocol. The mnmr software package was used for NMR data processing, and NOE assignments were performed using the pronto 3d2 program (34). Hydrogen bonds were identified using the naomi program (35). The coordinates of the three-dimensional structures and the assignments of the NMR spectra have been deposited in Protein Data Bank with reference nos. 1NRE and 1LRE.
RESULTS AND DISCUSSION
RAP Domain 1 Structure.
In the present study the three-dimensional solution structure of domain 1 is reported for the complete domain, (RAPd1), comprising residues 18–112 of the human RAP sequence, and for the truncated domain, (RAPd1T), in which the C-terminal 15 amino acid residues of domain 1 have been removed. The structure determination of RAPd1T was based on two-dimensional 1H NOE spectroscopy and 1H, 15N correlated spectra recorded at 298 K at pH 6.4. Of 100 calculated structures 31 had no NOE violations greater than 0.5 Å and no angle violations greater than 5 degrees (Table 1). The 20 structures with the lowest energy are reported (Fig. 1). The pairwise rms deviation is 0.48 ± 0.08 Å for the backbone atoms (N, Cα, C) for residues 18–91 and 1.22 ± 0.11 Å for all heavy atoms in this ordered part (Fig. 2). The spin systems of the C-terminal residues give very sharp NMR signals, suggesting that these are indeed unstructured in solution.
Table 1.
Structural statistics for the 20 structures of RAPd1T
| Parameter | Value |
|---|---|
| Distance restraints (all) | 1178 |
| Intraresidue | 218 |
| Sequential (|i − j| = 1) | 296 |
| Medium range (1 < |i − j|24) | 363 |
| Long range (|i − j| > 4) | 301 |
| Hydrogen bonds | 41 |
| Dihedral angle restraints (all) | 95 |
| φ | 54 |
| χ | 41 |
| Deviations from experimentally derived restraint | |
| Distance restraints | |
| No. of violations in the interval | |
| 0.1–0.2 Å | 16.5 |
| 0.2–0.3 Å | 1.7 |
| 0.3–0.4 Å | 0.1 |
| Hydrogen bonds > 0.2 Å | 0 |
| rmsd, Å | 0.023 ± 0.001 |
| Deviations from dihedral angle restraints, ° | |
| No. of violations larger than 3 | 0 |
| rmsd | 0.12 ± 0.06 |
| Deviations from idealized geometry | |
| Impropers, ° | |
| >5 | 0 |
| rmsd | 0.49 ± 0.01 |
| Bonds, Å | |
| >0.05 | 0 |
| rmsd | 0.0027 ± 0.001 |
| Angles, ° | |
| >5 | 0.1 |
| rmsd | 0.58 ± 0.01 |
| Energies, kcal·mol−1* | |
| NOE | 32.1 ± 3.8 |
| Dihedral angle restraint | 0.097 ± 0.081 |
| Bond | 9.94 ± 0.96 |
| Angle | 130.2 ± 5.8 |
| Improper | 25.2 ± 1.2 |
| Repel | 26.5 ± 3.8 |
| van der Waals | −145.0 ± 15.3 |
| Hydrogen bond restraint | 8.98 ± 0.67 |
| Hydrogen bond | −69.1 ± 3.5 |
| Pairwise rmsd of atomic positions, ņ | |
| Backbone (N, Cα, C) | |
| residues 18–91 | 0.48 ± 0.08 |
| All heavy atoms | |
| residues 18–91 | 1.22 ± 0.11 |
The number of experimentally derived restraints used in the structure calculations, and average values per structure of the deviations between the 20 accepted structures, and the experimental restraints or idealized geometry are shown.
Energies were calculated using the final force constants of: kNOE = 50 kcal·mol−1·Å−2, kcdih = 200 kcal·mol−1·rad−2, kbond = 1,000 kcal·mol −1·Å−2, kangle = 500 kcal·mol−1·rad−2, kimproper = 500 kcal·mol−1·Å −2, krepel = 4.0 kcal·mol−1·Å−2, and with the final van der Waals hard sphere radius set to 0.75 of the values calculated from the x-plor parallhdg_new.pro parameter file. The van der Waals energy and hydrogen bond energies were calculated using the x-plor switched Lennard-Jones van der Waals energy function and hydrogen bond function using the parmallh3x.pro parameter file, but they were not included in the target functions used in the simulated annealing procedure.
The pairwise rmsds between the 20 reported structures are also listed.
Figure 1.

The structural parameters for the 20 presented structures of RAPd1T. The deviations are taken to an average structure and the vertical bars correspond to one SD. In the panels displaying the dihedral angle data for φ, ψ, and χ1 the frames show the applied dihedral angle constraints. The average structure is calculated from residue 19 to 91. The NOE display shows the number of intra (black bar), sequential (red), medium-range (green), and long-range (yellow) NOEs assigned to protons of the residues.
Figure 2.

The solution structure of RAPd1T. The 20 structures are superpositioned based on the backbone heavy atoms of residues 19–91 (red). Their side chains are shown in blue. The part of the backbone colored black corresponds to the residues not used in the structural alignment. The figure has been prepared using molmol (36).
Secondary structure identification was carried out by use of procheck (37), using the definitions of Kabsch and Sander (38) for the helices and by use of the program naomi (35) for the turns.
The domain is a three-helix protein where the three α helices are arranged in an up-down-up pattern (Fig. 3a). Amino acid residues 23–34 form the first helix, H1, an amphipathic helix with a highly charged surface. Helix two, H2, consists of the residues 39–65, and H3 contains residues 73–88. H1 is connected to H2 by a sharp bend centered on His-36 and Leu-37. H2 is highly hydrophilic with only short stretches of hydrophobic surface. The amino acid residues that connect H2 to H3 form two turns, 66–69 and 68–71 (a type IV and a type I turn, respectively), the last turn being less well defined than the rest of the core structure. The structured part of RAPd1T ends with a type IV turn formed by residues 89–92. The N terminus is arranged along the first helix in a well ordered fashion determined by many interactions. Asn-26 HND2 and OD1 hydrogen bond to the O and NH of Phe-20, respectively, and the Phe-20 side chain forms interactions with residues both on H1 and H2 (Fig. 4), in addition the side chain of Glu-19 is packed between the side chains of His-46 and Trp-29. Furthermore, the methyl group of Met-22 interacts with H2. These interactions give the five N-terminal amino acids of domain 1 a stable conformation. Residues 18–24 have been reported as important for the binding of domain 1 to the receptors (14). Analysis of the presented structure suggests that these residues are important for the structural integrity of the domain.
Figure 3.

(a) A stereo view of the backbone of RAPd1T structure. Helix H1 is red, H2 is yellow, and H3 is green. The unordered parts (see legend to Fig. 2) of the molecule are shown as thin gray lines. (b) The water-accessible surface of the RAPd1T structure. The color code is as in a. The arrow points to the position of the groove that may be the structural component involved in receptor binding. The figure has been prepared using insight ii (Biosym Technologies, San Diego).
Figure 4.

The interactions of Phe-20 (shown by spacefilling of the side chain) with residues from H1 and H2 are shown. The red (oxygen) and white (hydrogen) spacefillings mark the hydrogen bonds between the side chain of Asn-26 and the backbone of Phe-20. The figure has been prepared using Insight II (Biosym Technologies).
A search for structural homologues to RAPd1T domain, performed using the program dali (39), produced a list of topologically homologous proteins, most of which were four-helix bundle proteins. In particular, the search results demonstrated that RAPd1T has the very common topology of cytochrome b562, a four-helical bundle with a left-handed twist (40, 41). The three helices in RAPd1T align in a sequential manner with helices 2, 3, and 4 of the four helical bundles with H1 aligning with the C-terminal half of helix 2.
The HN Pathogenic Epitope in RAP.
The structure-sensitive epitope previously mapped by Kerjaschki et al. (21) to residues 31–53 in the homologous rat protein corresponds to an integral structural part of RAPd1T: It consists of the C terminus of H1, the N-terminal half of H2, and the turn connecting these helices. It is apparent from the structure that a peptide composed of these residues might form a native-like structure stabilized by a hydrophobic core composed of Ala-32 and the leucine residues 35, 37, 42, 45, and 49 (Fig. 5). The structure also provides a basis for the rational design of therapeutic compounds that could bind to and mask the epitope, or alternatively the antibody combining site. One strategy shown to interfere with different autoimmune diseases in rats and mice involves the use of synthetic peptides corresponding to antigenic epitopes for immunization (43, 44). A variant of this strategy is the use of designed peptide analogues containing one or more amino acid substitutions as compared with the native sequences. This approach has been demonstrated to be able to generate nonpathogenic peptides capable of blocking the immuneresponse to the autoantigen (45–47). As deduced from the structure of human RAPd1T the HN epitope contains several charged surface residues displayed within a surface area 650 Å2 compatible with that of an antibody combining site (48). These surface residues would be primary targets for amino acid substitution in the design of synthetic peptide analogues, aimed at interfering with the pathogenesis of HN.
Figure 5.
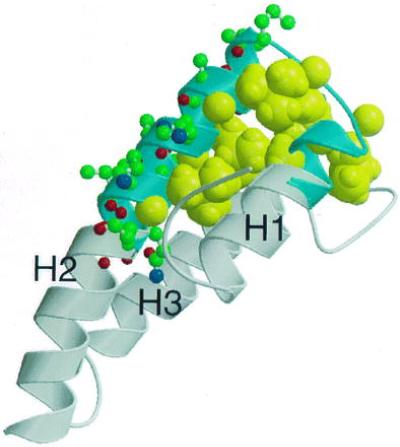
Uncovering of the structural basis for conformational sensitivity of the HN pathogenic epitope in RAP. The structure elements marked in blue corresponding to residues 31–53 are sufficient to induce immune deposits in passive HN. Aliphatic residues Ala-32, Leu-35, Leu-37, Leu-42, Leu-45, and Leu-49 that might stabilize the helical structure in the isolated peptide are shown as by spacefilling (yellow) of their atoms. Residues that are part of the proposed antibody binding site and not involved in the hydrophobic core are shown as ball-and-stick models. The figure has been prepared using molscript (42).
Overall, the three-dimensional structure of the N-terminal domain of RAP provides a structural platform delineating the residues important to preserve structural integrity of the epitope, which in turn simplifies the mapping of residues actually involved in the formation of pathogenic immune deposits. The sequence location of the HN epitope in RAP domain 1 appears to correspond to that of a nonlinear epitope detected in RAP domain 2 (49).
Receptor Binding.
Binding studies show that the C-terminal 15 amino acid residues of RAP domain 1 are important for inhibition of α2-macroglobulin binding to α2-macroglobulin receptor (13, 14). According to 1H, 13C, and 15N NMR spectroscopic analyses of the intact domain 1 these residues are not organized in a regular structure neither are they part of the tertiary structure in the separate protein domain 1. Other binding studies have shown that mutation or deletion of residues near the hydrophobic core around Phe-20 eliminate the ability of domain 1 to bind to the receptor (12). Inspection of the three-dimensional structure suggests that these mutations could exert their effect on binding by destabilizing the tertiary structure. Taken together, these two sets of results suggest that binding of RAP domain 1 to the receptor is mediated by at least two components, the C-terminal receptor binding motif and a second component defined by the tertiary structure of RAPd1T. Further studies are required to determine whether the C-terminal binding motif engages in an intramolecular interaction with the structure-dependent component or the receptor interacts in a bidentate interaction with both components. We speculate that a groove on one side of the surface could be a binding site for the receptor or for the C-terminal residues of domain 1 harboring the proposed binding motif (Fig. 3b). This groove is a prominent feature in the tertiary structure formed by the three helices, which are arranged almost parallel to each other at angles of 163, 12, and −163 degrees for H1/H2, H1/H3, and H2/H3, respectively (50).
The three-dimensional structure determination of RAP domain 1 contributes two important pieces of information. It provides a structural outline of the pathogenic epitope of rat RAP in the homologous human protein, and it provides a working model for understanding the binding site of RAP for the family of low density lipoprotein receptors at the structural level.
Acknowledgments
We thank Pia Skovgaard and Ove Lillelund for skilled technical assistance. The work presented has been supported by the Danish Biotechnology Programme.
ABBREVIATIONS
- RAP
α2-macroglobulin receptor-associated protein
- RAPd1T
RAP truncated domain 1
- HN
Heymann nephritis
- NOE
nuclear Overhauser effect
Footnotes
Data deposition: The coordinates of the three-dimensional structures and the assignments of the NMR spectra reported in this paper have been deposited in the Protein Data Bank, Brookhaven National Laboratory, Upton, NY 11973 (reference nos. 1NRE, 1LRE).
References
- 1.Jensen P H, Moestrup S K, Gliemann J. FEBS Lett. 1989;358:73–78. doi: 10.1016/0014-5793(89)81105-6. [DOI] [PubMed] [Google Scholar]
- 2.Ashcom J D, Tiller S E, Dickerson K, Cravens J L, Argraves W S, Strickland D K. J Cell Biol. 1990;110:1041–1048. doi: 10.1083/jcb.110.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strickland D K, Ashcom J D, Williams S, Battey F, Behre E, McTigue K, Battey J F, Argraves W S. J Biol Chem. 1991;266:13364–13369. [PubMed] [Google Scholar]
- 4.Strickland D K, Kounnas M Z, Argraves W S. FASEB J. 1995;9:890–898. doi: 10.1096/fasebj.9.10.7615159. [DOI] [PubMed] [Google Scholar]
- 5.Willnow T E, Armstrong S A, Hammer R E, Herz J. Proc Natl Acad Sci USA. 1995;92:4537–4541. doi: 10.1073/pnas.92.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bu G, Geuze H J, Strous G J, Schwartz A L. EMBO J. 1995;14:2269–2280. doi: 10.1002/j.1460-2075.1995.tb07221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willnow T E, Rohlman A, Horton J, Otani H, Braun J R, Hammer R E, Herz J. EMBO J. 1996;15:2632–2639. [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobsen L, Madsen P, Moestrup S K, Lund A H, Tommerup N, Nykjær A, Sottrup-Jensen L, Gliemann J, Petersen C M. J Biol Chem. 1996;271:31379–31383. doi: 10.1074/jbc.271.49.31379. [DOI] [PubMed] [Google Scholar]
- 9.Petersen C M, Nielsen M S, Nykjær A, Jacobsen L, Tommerup N, Rasmussen H H, Røigaard H, Gliemann J, Madsen P, Moestrup S K. J Biol Chem. 1997;272:3599–3605. doi: 10.1074/jbc.272.6.3599. [DOI] [PubMed] [Google Scholar]
- 10.Petersen C M, Ellgaard L, Nykjær A, Vilhardt F, Vorum H, Thøgersen H C, Nielsen M S, Jacobsen C, Moestrup S K, Gliemann J. EMBO J. 1996;15:4165–4173. [PMC free article] [PubMed] [Google Scholar]
- 11.Orlando R A, Farquhar M G. Proc Natl Acad Sci USA. 1994;91:3161–3165. doi: 10.1073/pnas.91.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warshawsky I, Bu G, Schwartz A L. Biochemistry. 1995;34:3404–3415. doi: 10.1021/bi00010a032. [DOI] [PubMed] [Google Scholar]
- 13.Ellgaard L, Holtet T L, Nielsen P R, Etzerodt M, Gliemann J, Thøgersen H C. Eur J Biochem. 1997;244:544–551. doi: 10.1111/j.1432-1033.1997.00544.x. [DOI] [PubMed] [Google Scholar]
- 14.Warshawsky I, Bu G, Schwartz A L. J Biol Chem. 1993;268:22046–22054. [PubMed] [Google Scholar]
- 15.Nielsen K L, Holtet T L, Etzerodt M, Moestrup S K, Gliemann J, Thøgersen H C. J Biol Chem. 1996;271:12909–12912. doi: 10.1074/jbc.271.22.12909. [DOI] [PubMed] [Google Scholar]
- 16.Heymann W, Hackel D B, Harwood S, Wilson S G, Hunter J L. Proc Soc Exp Biol Med. 1959;100:660–664. doi: 10.3181/00379727-100-24736. [DOI] [PubMed] [Google Scholar]
- 17.Davison A M, Cameron J S, Kerr D N, Ogg C S, Wilkinson R W. Clin Nephrol. 1984;22:61–67. [PubMed] [Google Scholar]
- 18.Kerjaschki D, Miettinen A, Farquhar M G. J Exp Med. 1987;166:109–128. doi: 10.1084/jem.166.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edginton T, Glassock R, Dixon F. J Exp Med. 1968;127:555–572. doi: 10.1084/jem.127.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerjaschki D, Ullrich R, Diem K, Pietromonaco S, Orlando R A, Farquhar M G. Proc Natl Acad Sci USA. 1992;87:1337–1341. doi: 10.1073/pnas.89.23.11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerjaschki D, Ullrich R, Exner M, Orlando R A, Farquhar M G. J Exp Med. 1996;183:2007–2015. doi: 10.1084/jem.183.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raychowdhury R, Zheng G, Brown D, McCluskey R. Am J Pathol. 1996;148:1613–1623. [PMC free article] [PubMed] [Google Scholar]
- 23.Saito A, Yamazaki H, Rader K, Nakatani A, Ullrich R, Kerjaschki D, Orlando R, Farquhar M G. Proc Natl Acad Sci USA. 1996;93:8601–8605. doi: 10.1073/pnas.93.16.8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeener J, Meier B H, Bachmann P, Ernst R R. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 25.Anil-Kumar, Wagner G, Ernst R R, Wüthrich K. J Am Chem Soc. 1981;103:3654–3658. [Google Scholar]
- 26.Güntert P, Braun W, Wüthrich K. J Mol Biol. 1991;217:517–530. doi: 10.1016/0022-2836(91)90754-t. [DOI] [PubMed] [Google Scholar]
- 27.Clore G M, Brünger A T, Karplus M, Gronenborn A M. J Mol Biol. 1986;186:435–455. doi: 10.1016/0022-2836(85)90116-0. [DOI] [PubMed] [Google Scholar]
- 28.Ludvigsen S, Andersen K V, Poulsen F M. J Mol Biol. 1991;217:731–736. doi: 10.1016/0022-2836(91)90529-f. [DOI] [PubMed] [Google Scholar]
- 29.Neri D, Otting G, Wüthrich K. J Am Chem Soc. 1990;112:3663–3665. [Google Scholar]
- 30.Karplus M. J Phys Chem. 1959;30:11–15. [Google Scholar]
- 31.Ludvigsen S, Poulsen F M. J Biomol NMR. 1992;2:227–233. doi: 10.1007/BF01875318. [DOI] [PubMed] [Google Scholar]
- 32.Piantini U, Sørensen O W, Ernst R R. J Am Chem Soc. 1982;104:6800–6801. [Google Scholar]
- 33.Brünger A T. x-plor Version 3.1, A System for X-ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 34.Kjær M, Andersen K V, Poulsen F M. Methods Enzymol. 1994;239:288–307. doi: 10.1016/s0076-6879(94)39010-x. [DOI] [PubMed] [Google Scholar]
- 35.Brocklehurst S M, Perham R N. Protein Sci. 1993;2:626–639. doi: 10.1002/pro.5560020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- 38.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 39.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 40.Argos P, Rossmann M G, Johnson J E. Biochem Biophys Res Commun. 1977;75:83–86. doi: 10.1016/0006-291x(77)91292-x. [DOI] [PubMed] [Google Scholar]
- 41.Orengo C A, Flores T P, Taylor W R, Thornton J M. Protein Eng. 1993;6:485–500. doi: 10.1093/protein/6.5.485. [DOI] [PubMed] [Google Scholar]
- 42.Kraulis P J. J Appl Crystallog. 1991;24:946–950. [Google Scholar]
- 43.Neilson E G, Sun M J, Kelly C J, Hines W H, Haverty T P, Clayman M D, Cooke N E. Proc Natl Acad Sci USA. 1991;88:2006–2010. doi: 10.1073/pnas.88.5.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaur A, Wiers B, Liu A, Rothbard J, Fathman C G. Science. 1992;258:1491–1494. doi: 10.1126/science.1279812. [DOI] [PubMed] [Google Scholar]
- 45.Kumar V, Urban J L, Horvath S J, Hood L. Proc Natl Acad Sci USA. 1990;87:1337–1341. doi: 10.1073/pnas.87.4.1337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Wauben M H, Boog C J, van der Zee R, Joosten I, Schlif A, van Eden W. J Exp Med. 1992;176:667–677. doi: 10.1084/jem.176.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh D P, Kikuchi T, Singh V K, Shinohara T. J Immunol. 1994;152:4699–4705. [PubMed] [Google Scholar]
- 48.Janin J, Chothia C. J Biol Chem. 1990;265:16027–16030. [PubMed] [Google Scholar]
- 49.Ellgaard L, Holtet T L, Moestrup S K, Etzerodt M, Thøgersen H C. J Immunol Methods. 1994;180:53–61. doi: 10.1016/0022-1759(94)00298-b. [DOI] [PubMed] [Google Scholar]
- 50.Hutchinson E G, Thornton J M. Protein Sci. 1996;5:212–220. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]