

Abstract
Obesity is a complex disease, and multiple genes contribute to the trait. The description of five genes (ob, db, tub, Ay, and fat) responsible for distinct syndromes of spontaneous monogenic obesity in mice has advanced our knowledge of the genetics of obesity. However, many other genes involved in the expression of this disease remain to be determined. We report here the identification of an additional class of genes involved in the regulation of adipose tissue mass. These genes encode receptors mediating leukocyte adhesion. Mice deficient in intercellular adhesion molecule-1 became spontaneously obese in old age on normal mouse chow or at a young age when provided with a diet rich in fat. Mice deficient in the counterreceptor for intercellular adhesion molecule-1, the leukocyte integrin αMβ2 (Mac-1), showed a similar obesity phenotype. Since all mice consumed approximately the same amount of food as controls, the leukocyte function appears to be in regulating lipid metabolism and/or energy expenditure. Our results indicate that (i) leukocytes play a role in preventing excess body fat deposition and (ii) defects in leukocyte adhesion receptors can result in obesity.
Intercellular adhesion molecule-1 (ICAM-1) is a well-characterized receptor with five immunoglobulin (Ig)-like extracellular domains, a transmembrane domain, and a short cytoplasmic tail (1). ICAM-1 is expressed on leukocytes, endothelium, epithelium, hepatocytes, myocytes, and fibroblasts, and can be up-regulated by cytokines in all of these cell types (2–5). The primary cellular counterreceptors for ICAM-1 are the β2 leukocyte integrins, e.g., LFA-1 (αLβ2, CD11a/CD18) and Mac-1 (αMβ2, CD11b/CD18). LFA-1 is expressed on all types of leukocytes and binds to extracellular domain 1 of ICAM-1 (1). In contrast, the expression of Mac-1 is restricted to monocytes/macrophages, granulocytes, natural killer cells, and a subpopulation of T cells and it binds to domain 3 of ICAM-1 (1). It has been recognized that through binding and interacting with LFA-1 and/or Mac-1, ICAM-1 functions as a major cell–cell adhesion molecule in inflammatory and immune systems (2, 6, 7).
As expected, mice deficient in ICAM-1 (ICAM-1 −/−) have both inflammatory and immune defects (8, 9). These include a decreased emigration of neutrophils in chemically induced peritonitis, reduced contact hypersensitivity, diminished ability of spleen cells to act as stimulators in mixed lymphocyte responses, and resistance to septic shock. Similarly, Mac-1-deficient (Mac-1 −/−) mice exhibit defects in several neutrophil functions including adhesion to the endothelium, phagocytosis, and neutrophil apoptosis (10). However, while experimenting with ICAM-1 −/− mice and subsequently with Mac-1 −/− mice, we recently noticed an unexpected new phenotype of these mice, i.e., obesity. Our data identify a novel function of leukocyte adhesion receptors, namely, regulation of body weight and adipose tissue mass.
MATERIALS AND METHODS
Animals.
ICAM-1-deficient (ICAM-1 −/−) mice used in our study were obtained by targeted disruption of exon 4 of the ICAM-1 gene (9). These mice were originally on a mixed background (9) and were backcrossed four times to C57BL/6J. The Mac-1-deficient (Mac-1 −/−) mice, generated by targeted deletion of the exon encoding the signal peptide, were on a mixed C57BL/6J × 129sv background (10). Mice were maintained on 12-hr dark/12-hr light cycles. Water and food were available ad libitum. The care of the experimental mice was in accordance with the guidelines of the Center for Blood Research, Harvard Medical School.
Mouse Food.
Mouse chow (Prolab 3000; PMI Feeds, St. Louis) contained 5.0% (wt/wt) fat, 55% (wt/wt) carbohydrate, and 22% (wt/wt) protein. The Western-type diet (Harlan Teklad Adjusted Calories Western-Type Diet No. 88137, Madison, WI) contained 21% (wt/wt) fat (42% of calories), 49.2% (wt/wt) carbohydrate, and 19.8% (wt/wt) protein.
Body Weight and Food Measurements.
Mice were housed two to four per cage. Body weight was measured by the same individual every 2 weeks using a scale (Mettler–Toledo). To measure food consumption, a sufficient amount of food for 2 weeks was weighed and provided to the mice ad libitum. After 1 or 2 weeks, the remaining food was weighed and the difference divided by the number of mice per cage. The result was expressed as grams per mouse per 1 or 2 weeks for food consumption. Mean values per cage were used for comparison.
Quantitative Phenotype Measurements.
Mice were fasted overnight prior to transport to the laboratory and collection of blood through retroorbital venous plexus. Glucose concentrations in total plasma were determined using the enzymatic assay according to the manufacturer’s instructions (Sigma). Insulin was measured by a radioimmunoassay system (Linco Research Immunoassay, St. Charles, MO). Body mass index was calculated as body weight (g) divided by the square of the anal–nasal length (cm). The subcutaneous, inguinal, two intra-abdominal (retroperitoneal and omental) white fat pads, and a brown fat pad located between the two scapulas were dissected and weighed. The internal organs including heart, liver, spleen, and kidneys were also removed and weighed.
Statistical Analysis.
Data are presented as mean ± SE of the mean. Statistical significance was assessed by Student’s t test.
RESULTS
ICAM-1 −/− Mice Spontaneously Become Obese Without an Obvious Increase in Food Intake.
On normal chow diet, ICAM-1 −/− male mice maintained a body weight comparable to wild-type animals until 16 weeks of age. Thereafter, we observed that ICAM-1 −/− mice gained more weight than control mice (Fig. 1). The difference in body weight between the two groups became highly significant starting at 20 weeks of age (P < 0.004). Despite the fact that ICAM-1 −/− mice gained significantly more weight than controls, we found that consumption of chow food by both groups of mice was comparable at 16–24 weeks of age (20–25 g per week). A limited number of female mice were also examined and displayed a similar growth difference. At 12 weeks of age their weights were identical (23.57 ± 0.64 g in ICAM-1 −/− vs. 23.95 ± 0.44 g in wild type, P = 0.64), but with time the ICAM-1 −/− females became significantly heavier (39.87 ± 2.54 g in ICAM-1 −/− vs. 33.02 ± 1.2 g in wild type at 24 weeks of age, P < 0.05; 54.10 ± 6.20 g in ICAM-1 −/− vs. 36.70 ± 1.20 g in wild type at 45 weeks of age, P < 0.03).
Figure 1.
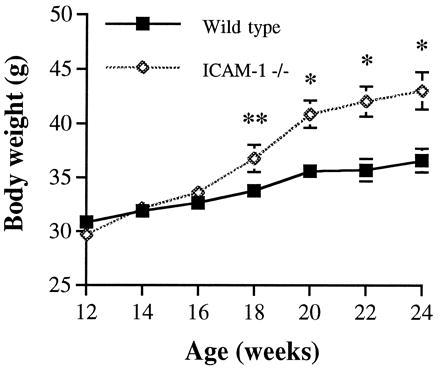
Growth curve of wild-type and ICAM-1 −/− mice on normal chow diet. Each group contained 13–14 male mice. Data were combined from two independent experiments showing similar results. ∗, P < 0.004; ∗∗, P = 0.05.
Most of the excess body weight in ICAM-1 −/− male mice was due to the increased weight of combined white fat pad (Table 1). The excess 5.2 g of white fat (8.81 ± 0.47 g in ICAM-1 −/− mice vs. 3.66 ± 0.36 g in wild-type mice, P < 0.0001) was composed predominantly of subcutaneous fat (54%). The weight of interscapular brown fat pad was also significantly increased in ICAM-1 −/− mice (Table 1). The weights of kidney, spleen, and heart in the 24-week-old ICAM-1 −/− mice were similar to those of wild-type mice. However, ICAM-1 −/− mice tended to have a slightly heavier liver (1.82 ± 0.13 g vs. 1.53 ± 0.09 g, P = 0.06). Hematoxylin-stained liver sections counterstained with oil-red-O revealed abnormal lipid deposits in the hepatocytes of ICAM-1 −/− mice, suggesting that these mice develop fatty livers (not shown).
Table 1.
Characteristics of ICAM-1 −/− mice on chow diet
| Wild type | ICAM-1 −/− | % of wild type | P value | |
|---|---|---|---|---|
| Body weight (g) | 36.06 ± 1.19 | 43.74 ± 1.60 | 121 | <0.001 |
| Body length (cm) | 9.52 ± 0.03 | 9.45 ± 0.05 | 99 | 0.24 |
| Body mass index (g/cm2) | 0.40 ± 0.01 | 0.49 ± 0.01 | 123 | <0.0001 |
| Subcutaneous fat pad weight (g) | 1.39 ± 0.16 | 4.15 ± 0.27 | 299 | <0.0001 |
| Inguinal fat pad weight (g) | 0.27 ± 0.02 | 0.66 ± 0.06 | 244 | <0.0001 |
| Omental fat pad weight (g) | 1.38 ± 0.15 | 2.62 ± 0.17 | 190 | <0.0001 |
| Retroperitoneal fat pad weight (g) | 0.62 ± 0.06 | 1.37 ± 0.12 | 221 | <0.0001 |
| White fat pad weight (% body weight) | 9.89 ± 0.65 | 20.10 ± 0.69 | 203 | <0.0001 |
| Brown fat pad weight (g) | 0.27 ± 0.04 | 0.43 ± 0.05 | 159 | <0.02 |
| Plasma insulin (ng/ml) | 1.14 ± 0.18 | 1.48 ± 0.42 | 130 | 0.43 |
Each group contained 12–14 male mice at 24 weeks of age. Mice were given a standard chow diet. White fat pad included the subcutaneous, inguinal, omental, and retroperitoneal fat pad. Brown fat pad was taken from the interscapular site.
ICAM-1 −/− Mice Demonstrate Increased Susceptibility to Obesity Induced by High Fat Diet.
The regulation of body weight and composition involves input from both genes and the environment (11). This is demonstrated, for example, by the variable susceptibility to obesity of inbred strains of mice when offered a high-fat diet (11). Since genes involved in sensitivity to dietary obesity in mice have not been identified (12), we were interested in examining whether ICAM-1 −/− mice were more sensitive to diet-induced obesity than control mice. To obtain this information, 7-week-old ICAM-1 −/− and wild-type controls were fed a Western-type diet containing 21% fat. Both male and female mutant mice rapidly gained more body weight than controls (Fig. 2 A and B). These results suggest that the ICAM-1 gene could contribute to reduced susceptibility of mice to diet-induced obesity. Despite gaining significantly more body weight, both male and female ICAM-1 −/− mice consumed the same amount of Western-type food as controls for the length of the experiment (Fig. 2 E and F).
Figure 2.
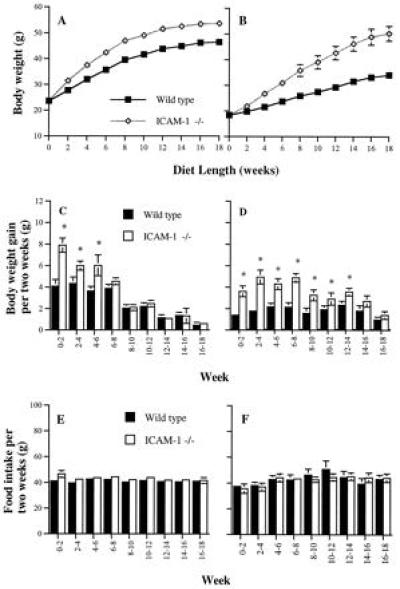
Growth curves (A and B), weight gains (C and D), and food intake per 2 weeks (E and F) of wild-type and ICAM-1 −/− mice on Western-type diet. (A, C, and E) Male mice. (B, D, and F) Female mice. Nine to 11 mice in each group were fed Western-type diet starting at 7 weeks of age. Body weight gain and food intake (grams per mouse) were measured at 2-week intervals. “0–2” refers to feeding time from the start of the diet to 2 weeks of feeding, etc. The experiment for female groups was repeated with a similar result. (A) From 2 to 18 weeks, P < 0.02; (B) from 4 to 18 weeks, P < 0.01; (C and D) ∗, P < 0.05; (E and F) no statistically significant differences between groups.
An interesting difference between male and female ICAM-1 −/− mice in their capacity to regulate body weight gain induced by the Western-type diet was observed. After 6 weeks on a Western-type diet, the rate at which ICAM-1 −/− male mice gained weight no longer exceeded that of the controls (Fig. 2C). Therefore, the major difference in body weight between male ICAM-1 −/− and wild-type mice was established after 6 weeks of Western-type diet intake. In contrast, female ICAM-1 −/− mice gained more weight than controls for 14 weeks on the Western-type diet (Fig. 2D). At 14 weeks, female ICAM-1 −/− mice were 14.6 g heavier than controls, whereas males weighed only 7.6 g more than controls. Our results suggest that the ICAM-1-dependent mechanism of body weight regulation plays a more important role in female than in male mice. At the end of the experiment shown in Fig. 2, the excess white fat in males was composed mainly of subcutaneous fat (56%). In females, the major component (64%) was intra-abdominal fat (omental + retroperitoneal) resulting in prominent abdominal obesity (Fig. 3). Intra-abdominal fat has a higher correlation with fatty liver, hyperglycemia, and a higher associated risk of atherosclerosis than other fat depots (12, 13). Such correlation may explain the increased liver weight and plasma glucose observed in females (Table 2). Mice of both sexes and genotypes on the Western-type diet had a significantly higher level of plasma insulin (Table 2) than those on chow food (Table 1). The result is consistent with the fact that hyperinsulinemia develops in mice with high fat diet-induced obesity (12). However, both wild-type and ICAM-1 −/− mice on either diet tended to have similar levels of plasma insulin. This indicates that the higher level of plasma glucose observed in ICAM-1 −/− female mice on Western-type diet, as compared with their wild-type controls (Table 2), is likely due to a difference in insulin sensitivity between the two groups.
Figure 3.
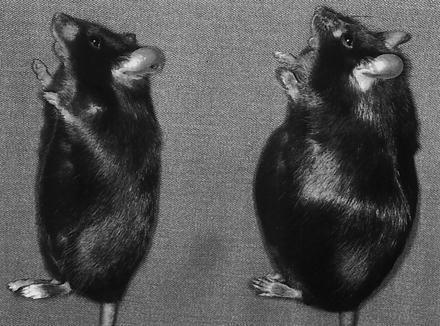
Photograph of ICAM-1 −/− (Right) and wild-type (Left) female mice after being fed the Western-type diet for 20 weeks starting at 7 weeks of age.
Table 2.
Characteristics of ICAM-1 −/− mice on Western-type diet
| Body weight, g | Body mass index, g/cm2 | White fat pad weight, % body weight | Brown fat pad weight, g | Liver weight, g | Plasma glucose, mg/dl | Plasma insulin, ng/ml | |
|---|---|---|---|---|---|---|---|
| Male | |||||||
| Wild type | 46.24 ± 1.24 | 0.49 ± 0.01 | 17.72 ± 0.59 | 0.59 ± 0.04 | 4.75 ± 0.40 | 167 ± 14 | 2.66 ± 0.47 |
| ICAM-1 −/− | 54.96 ± 1.64 | 0.56 ± 0.01 | 25.16 ± 1.03 | 0.77 ± 0.05 | 5.64 ± 0.38 | 193 ± 11 | 4.66 ± 0.96 |
| % of wild type | 119% | 114% | 142% | 131% | 119% | 116% | 175% |
| P = 0.0004 | P = 0.0003 | P < 0.0001 | P < 0.006 | P = 0.125 | P = 0.178 | P = 0.072 | |
| Female | |||||||
| Wild type | 38.18 ± 2.11 | 0.44 ± 0.02 | 18.76 ± 1.22 | 0.26 ± 0.03 | 2.63 ± 0.23 | 269 ± 13 | 2.76 ± 0.41 |
| ICAM-1 −/− | 56.57 ± 2.89 | 0.62 ± 0.02 | 35.48 ± 0.92 | 0.51 ± 0.10 | 3.91 ± 0.49 | 331 ± 21 | 3.40 ± 0.66 |
| % of wild type | 148% | 141% | 189% | 196% | 149% | 123% | 123% |
| P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.02 | P < 0.03 | P = 0.02 | P = 0.41 |
Nine to 11 mice in each group were fed a Western-type diet for 20 weeks (in male groups) or 24 weeks (in female groups) starting at 7 weeks of age. White fat pad included the subcutaneous, inguinal, omental, and retroperitoneal fat pad. Brown fat pad was taken from the interscapular site.
Similar to spontaneously obese ICAM-1 −/− mice, both male and female ICAM-1 −/− mice on the Western-type diet had larger brown fat depots than the controls (Table 2). In addition, male ICAM-1 −/− mice acquired heavier hearts than the controls (0.312 ± 0.02 g vs. 0.249 ± 0.02 g, P < 0.05).
Mac-1 −/− Mice Are Susceptible to Diet-Induced Obesity.
Mice deficient in Mac-1, a counterreceptor for ICAM-1, were recently generated (10), and we examined their susceptibility to diet-induced obesity. Indeed, Mac-1 −/− mice displayed a striking similarity in weight gain (Fig. 4) with sex-matched ICAM-1 −/− mice (Fig. 2A). Like ICAM-1 −/− mice, Mac-1 −/− mice had a significant increase in white fat (56% subcutaneous fat) and brown fat (Table 3), while their food consumption did not differ from the controls (40–45 g every 2 weeks). The mixed background C57BL/6 × 129sv on which the Mac-1 −/− mice exist shows variability in body weight gain as the mice age on normal chow diet; thus, we could not determine if Mac-1 −/− mice become spontaneously obese.
Figure 4.
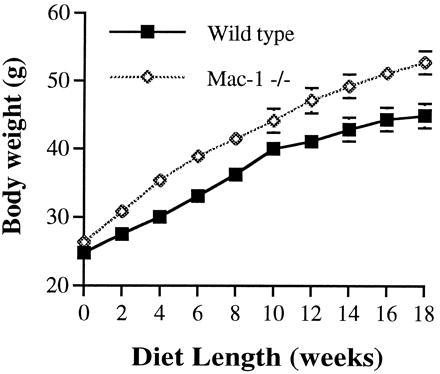
Growth curve of Mac-1 −/− mice on Western-type diet. Eight to 9 male mice in each group were fed the Western-type diet starting at 9 weeks of age; from 2 to 18 weeks, P < 0.05. A second, similar experiment was performed. Each group contained 9–10 male mice. After 6 weeks of feeding, Mac-1 −/− mice gained 13.1 ± 1.13 g and wild-type mice gained 8.68 ± 1.09 g (P < 0.02). After 10 weeks of feeding, Mac-1 −/− mice gained 20.50 ± 1.20 g and wild-type mice gained 15.06 ± 1.21 g (P < 0.01).
Table 3.
Characteristics of Mac-1 −/− mice on Western-type diet
| Body weight, g | Body length, cm | Body mass index, g/cm2 | White fat pad weight, % body weight | Brown fat pad weight, g | |
|---|---|---|---|---|---|
| Wild type | 46.56 ± 1.57 | 10.19 ± 0.09 | 0.45 ± 0.01 | 14.85 ± 0.58 | 0.43 ± 0.06 |
| Mac-1 −/− | 54.16 ± 1.66 | 10.34 ± 0.05 | 0.51 ± 0.01 | 20.23 ± 1.51 | 0.76 ± 0.08 |
| % of wild type | 116% | 101% | 113% | 136% | 177% |
| P < 0.006 | P = 0.17 | P < 0.009 | P < 0.003 | P < 0.004 |
Seven to 9 male mice in each group were fed a Western-type diet for 20 weeks starting at 9 weeks of age. White fat pad included the subcutaneous, inguinal, omental, and retroperitoneal fat pad. Brown fat pad was taken from the interscapular site.
DISCUSSION
Currently, there are five known mouse mutations that cause obesity: ob, db, Ay, fat, and tub (14). Although the resulting obesity varies in severity and time of onset, the mutations cause a common phenotype—i.e., a significant increase in food consumption (15–18). For example, ob mice, which demonstrate early-onset obesity, eat 62% more food than normal mice (15), and agouti yellow mice with maturity-onset obesity consume 36% more food than controls (16). In this study, we have shown that mice deficient in ICAM-1 develop a maturity-onset obesity without an obvious increase in food intake by these animals. Considering this difference between ICAM-1 −/− and the five known obese mutations, we expect that ICAM-1 −/− mice will become an animal model for studying a new molecular mechanism regulating adiposity without significant modification of appetite.
ICAM-1 −/− mice are commonly used to investigate the role of this receptor in various inflammatory models (19, 20). The obesity phenotype of these mice may not have been previously noticed for two possible reasons. First, spontaneous weight gain begins only at about 4 months of age, and younger animals are preferred for studying inflammation (19, 20). Second, most investigators worked with mice mutated in exon 5 (8), whereas we analyzed mice with a targeted disruption of exon 4 (9). Exon 4 encodes Ig domain 3 of ICAM-1, which is critical for binding to Mac-1 (1). It has been shown that exon 5 mutant mice still express some isoforms of ICAM-1 due to splicing of RNA with a predominant, functional exon 4–6 spliced species (21). Production of this exon 4–6 spliced isoform would not be possible in exon 4 mutant mice. Considering this difference, it is possible that the obese phenotype observed in exon 4 targeted mice could be absent in mice mutated in exon 5.
At present, we still do not know whether the two adhesion receptors, ICAM-1 and Mac-1, act in parallel in preventing obesity, i.e, both receptors mediate leukocyte adhesion, or whether they function in their capacity as a receptor:counterreceptor pair. However, considering that both Mac-1 −/− and ICAM-1 −/− mice develop a phenotypically similar diet-induced obesity, we suspect that there is a link between leukocyte adhesion and fat storage. Since Mac-1 is leukocyte-specific it is clear that some of the leukocyte subsets play a role in preventing obesity. How might the Mac-1/ICAM-1 pathway(s) of leukocyte adhesion play a role in regulating body weight and adiposity? Our results indicate that it is not by modifying appetite. Therefore, leukocytes and their receptors have to be involved in the regulation of lipid metabolism and/or energy expenditure. Leukocytes could be directly engaged in removal of excess fat from tissues and assisting in its metabolism or indirectly by signaling to adipose tissue thus maintaining the correct adipocyte number and function. Activated leukocytes are also a major source of tumor necrosis factor α. It is known that tumor necrosis factor α levels affect adiposity (22); therefore, differences in tumor necrosis factor α metabolism might contribute to the effects we observe. However, Xu and colleagues (9) have reported similar levels of tumor necrosis factor α in the blood of both wild-type and ICAM-1-deficient mice treated with endotoxin (9). Another hypothesis comes from a recent finding that a new mechanism regulating fatty acid oxidation in the liver involves leukocytes (23, 24). Through transcellular metabolism, activated leukocytes or Kupffer cells were reported to act together with hepatocytes to produce the eicosanoid leukotriene B4. Eicosanoid leukotriene B4 was shown to activate the transcription factor peroxisome proliferator-activated receptor α in the hepatocyte, increasing transcription of enzymes involved in fatty acid oxidation (24). Leukocyte adhesion has been shown to promote efficient transcellular eicosanoid biosynthesis in other systems (25) and might therefore also play a role in the liver.
Our study identifies a previously unknown role for the leukocyte adhesion receptors ICAM-1 and Mac-1 in maintenance of normal body weight and adiposity. This function may not be restricted to these two molecules and may involve additional receptors mediating leukocyte adhesion. It will be important to examine whether, as in mice, a low expression of these leukocyte adhesion receptors is associated with obesity in humans.
Acknowledgments
We are grateful to members of Dr. Bruce M. Spiegelman’s laboratory for teaching us how to isolate fat depots, to Susan Chapman for excellent technical assistance, and to Lesley Cowan for help with preparation of the manuscript. The work was supported by National Institutes of Health Grant R01HL41002 to D.D.W.
ABBREVIATION
- ICAM-1
intercellular adhesion molecule-1
References
- 1.Diamond M S, Staunton D E, Marlin S D, Springer T A. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 2.Carlos T M, Harlan J M. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 3.Hellerbrand C, Wang S C, Tsukamoto H, Brenner D A, Rippe R A. Hepatology. 1996;24:670–676. doi: 10.1002/hep.510240333. [DOI] [PubMed] [Google Scholar]
- 4.van Oosten M, van de Bilt E, de Vries H E, van Berkel T J C, Kuiper J. Hepatology. 1995;22:1538–1546. doi: 10.1002/hep.1840220529. [DOI] [PubMed] [Google Scholar]
- 5.Ban K, Ikeda U, Takahashi M, Kanbe T, Kasahara T, Shimada K. Cardiovasc Res. 1994;28:1258–1262. doi: 10.1093/cvr/28.8.1258. [DOI] [PubMed] [Google Scholar]
- 6.Frenette P S, Wagner D D. N Engl J Med. 1996;335:43–45. doi: 10.1056/NEJM199607043350108. [DOI] [PubMed] [Google Scholar]
- 7.Springer T A. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 8.Sligh J E, Ballantyne C M, Rich S S, Hawkins H K, Smith C W, Bradley A, Beaudet A L. Proc Natl Acad Sci USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Gonzalo J A, St. Pierre Y, Williams I R, Kupper T S, Cotran R S, Springer T A, Gutierrez-Ramos J-C. J Exp Med. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coxon A, Rieu P, Barkalow F J, Askari S, Sharpe A H, von Andrian U H, Arnaout M A, Mayadas T N. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 11.Bouchard C. Mol Med Today. 1995;1:45–50. doi: 10.1016/1357-4310(95)80020-4. [DOI] [PubMed] [Google Scholar]
- 12.Warden C H, Fisler J S, Shoemaker S M, Wen P-Z, Svenson K L, Pace M J, Lusis A J. J Clin Invest. 1995;95:1545–1552. doi: 10.1172/JCI117827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vohl M-C, Lamarche B, Moorjani S, Prud’homme D, Nadeau A, Bouchard C, Lupien P-J, Després J-P. Arterioscler Thromb Vasc Biol. 1995;15:714–720. doi: 10.1161/01.atv.15.5.714. [DOI] [PubMed] [Google Scholar]
- 14.Spiegelman B M, Flier J S. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 15.Erickson J C, Hollopeter G, Palmiter R D. Science. 1996;274:1704–1707. doi: 10.1126/science.274.5293.1704. [DOI] [PubMed] [Google Scholar]
- 16.Huszar D, Lynch C A, Fairchild-Huntress V, Dunmore J H, Fang Q, Berkemeier L R, Gu W, Kesterson R A, Boston B A, Cone R D, Smith F J, Campfield L A, Burn P, Lee F. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 17.Coleman D L, Eicher E M. J Hered. 1990;81:424–427. doi: 10.1093/oxfordjournals.jhered.a111019. [DOI] [PubMed] [Google Scholar]
- 18.Noben-Trauth K, Naggert J K, North M A, Nishina P M. Nature (London) 1996;380:534–538. doi: 10.1038/380534a0. [DOI] [PubMed] [Google Scholar]
- 19.Bullard D C, Qin L, Lorenzo I, Quinlin W M, Doyle N A, Bosse R, Westweber D, Doerschuk C M, Beaudet A L. J Clin Invest. 1995;95:1782–1788. doi: 10.1172/JCI117856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doerschuk C M, Quinlin W M, Doyle N A, Bullard D C, Vestweber D, Jones M L, Takei F, Ward P A, Beaudet A L. J Immunol. 1996;157:4609–4614. [PubMed] [Google Scholar]
- 21.King P D, Sandberg E T, Selvakumar A, Fang P, Beaudet A L, Dupont B. J Immunol. 1995;154:6080–6093. [PubMed] [Google Scholar]
- 22.Spiegelman B M, Hotamisligil G S. Cell. 1993;73:625–627. doi: 10.1016/0092-8674(93)90243-j. [DOI] [PubMed] [Google Scholar]
- 23.Serhan C N. Nature (London) 1996;384:23–24. doi: 10.1038/384023a0. [DOI] [PubMed] [Google Scholar]
- 24.Devchand P R, Keller H, Peters J M, Vazquez M, Gonzalez F J, Wahli W. Nature (London) 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 25.Brady H R, Papayianni A, Serhan C N. Kidney Int. 1994;45:S90–S97. [PubMed] [Google Scholar]