

Abstract
Evidence from postmortem studies suggest an involvement of oxidative stress in the degeneration of dopaminergic neurons in Parkinson disease (PD) that have recently been shown to die by apoptosis, but the relationship between oxidative stress and apoptosis has not yet been elucidated. Activation of the transcription factor NF-κB is associated with oxidative stress-induced apoptosis in several nonneuronal in vitro models. To investigate whether it may play a role in PD, we looked for the translocation of NF-κB from the cytoplasm to the nucleus, evidence of its activation, in melanized neurons in the mesencephalon of postmortem human brain from five patients with idiopathic PD and seven matched control subjects. In PD patients, the proportion of dopaminergic neurons with immunoreactive NF-κB in their nuclei was more than 70-fold that in control subjects. A possible relationship between the nuclear localization of NF-κB in mesencephalic neurons of PD patients and oxidative stress in such neurons has been shown in vitro with primary cultures of rat mesencephalon, where translocation of NF-κB is preceded by a transient production of free radicals during apoptosis induced by activation of the sphingomyelin-dependent signaling pathway with C2-ceramide. The data suggest that this oxidant-mediated apoptogenic transduction pathway may play a role in the mechanism of neuronal death in PD.
Keywords: apoptosis, transcription factor, neurodegenerative disease, postmortem, in vitro
Parkinson disease (PD), one of the most common neurological disorders in the elderly, is characterized by a progressive and massive loss of midbrain dopaminergic neurons. The mechanism by which these neurons degenerate is still unknown. However, the intensity of neuronal loss varies from one dopaminergic cell group of the mesencephalon to another. Several factors may be involved in this selective vulnerability among which oxygen free radical metabolism may play a particularly important role (1). Morphological studies on postmortem brain, by both light (2) and electron (3) microscopy, have shown that mesencephalic dopaminergic neurons ultimately die by apoptosis in patients with PD. Other studies suggest that oxidative stress might be involved in the apoptotic process (1). At a late stage of the disease, the substantia nigra (SN) exhibits (i) increased lipid peroxidation (4), (ii) increased manganese-dependent superoxide dismutase activity (5), (iii) decreased glutathione levels (6), and (iv) increased iron content, which may favor formation of hydroxyl radicals (7, 8). However, the molecular mechanism triggering apoptosis in PD and its relationship with oxidative stress has not been established.
Oxidative stress is most often defined as an excessive production of reactive oxygen species (ROS), which directly damage cells by peroxidation of lipids and alteration of proteins and nucleic acids (9). However, ROS may also take part in signal transduction pathways that mediate apoptosis, one of which is the sphingomyelin- or ceramide-dependent signaling pathway, first described in the immune system (10, 11), but which has recently been shown to induce apoptosis in neurons as well (12). This signaling system may be of interest with respect to the pathophysiology of PD, because it can be activated by tumor necrosis factor α (TNF-α) (11, 13), which has been detected in microglial cells in the SN of PD patients in the vicinity of dopaminergic neurons that express receptors for this cytokine (14). TNF-α is also known to stimulate the production of oxygen radicals from the mitochondria of transformed fibroblasts (15, 16), which are required for apoptosis to occur, and we have recently shown, in a culture model of catecholaminergic neurons, that these radicals are produced within the ceramide-dependent signaling pathway (17). We wished to determine whether this oxidant-mediated apoptogenic signaling pathway is also activated in the dopaminergic neurons that degenerate in patients with PD.
Since it is not possible to show the formation of free radicals in postmortem brain, we have investigated a downstream element in the signaling cascade, the transcription factor NF-κB, which is translocated from the cytoplasm to the nucleus when activated. Under normal conditions, NF-κB, a heterodimer composed of 50 kDa (p50) and 65 kDa (p65) subunits, is bound to an inhibitor protein, I-κB, which sequesters NF-κB in the cytosol. Activation of NF-κB involves its dissociation from I-κB followed by translocation of the p50–p65 heterodimer to the nucleus, where it directly binds to its cognate DNA sequences (for review see ref. 18). Immunohistochemical techniques were used to study the expression and cellular location of NF-κB in dopaminergic neurons in the mesencephalon of control subjects and PD patients. In addition, to determine the possible relationship between free radical formation, activation of NF-κB, and the apoptosis of dopaminergic neurons, we analyzed free radical production and NF-κB translocation in cultures of rat mesencephalic neurons induced to die by apoptosis by activation of the sphingomyelin-dependent signaling pathway with a cell-permeant analogue of ceramide, its second messenger.
MATERIALS AND METHODS
Human Brain Tissue.
This study was performed on autopsy brainstem tissue from seven control subjects and five PD patients well characterized clinically and neuropathologically. PD patients and control subjects did not differ significantly for their mean age at death (mean ± SEM: PD, 74 ± 6; controls, 86 ± 4 years) and the mean interval from death to freezing of tissue (mean ± SEM: PD, 28.6 ± 4.6; controls, 20.6 ± 6.8 hr). Brainstem tissue was dissected and fixed as described (19).
Immunohistochemistry.
Immunohistochemical labeling for NF-κB was performed on free-floating (40 μm thick) sections of the mesencephalon taken in the caudal third of the SN (19). Sections were then incubated at 4°C for 96 hr under gentle agitation, with a rabbit polyclonal antiserum (1 μg/ml) raised against a peptide corresponding to amino acids 3–19 of the amino-terminal region of the human p65 subunit of NF-κB (Santa Cruz Biotechnology). The primary antiserum was revealed using the avidin–biotin complex method (Vector Laboratories) (19) and developed using 0.04% (wt/vol) diaminobenzidine in acetate buffer with 1.28% (wt/vol) nickel ammonium sulfate to produce a blue reaction product. To test the specificity of the antiserum some sections were incubated with the primary antiserum preadsorbed for 6 hr at room temperature with a 2 × 104 excess of the corresponding peptide and Western blotting for NF-κB was performed on tissue homogenates of human SN and ventral tegmental area (VTA) as described (20).
Image and Data Analysis.
The total number of pigmented dopaminergic neurons presenting a cytoplasmic, nuclear, or no staining for NF-κB were counted on each stained section using a computer-based image analysis system (Biocom, Les Ulis, France). The analysis was performed in the VTA, the substantia nigra pars compacta (SNpc), and the nucleus of the third cranial nerve defined as previously described (21). Because the degree of neuronal loss varies among PD patients the number of pigmented neurons differed. The results were therefore expressed as the number of pigmented neurons stained in the cytoplasm or in the nucleus for NF-κB with respect to the total number of pigmented neurons. The proportions were compared between control subjects and PD patients using nonparametric statistical test because distributions differed significantly from normality (Mann–Whitney U test; SigmaStat Statistical Software, Jandel, San Rafael, CA).
Electron Microscopy.
Ultrastructural analysis of NF-κB-immunopositive nuclei in dopaminergic neurons of the SN from additional brains was performed as previously described with minor modifications (22). In brief, small blocks of mesencephalon containing SN were fixed in a mixture of 4% paraformaldehyde and 2.5% glutaraldehyde. Sections (50 μm) were cut and labeled for NF-κB as described above. Small areas of sections were then excised and were postfixed in 1% osmium tetroxide for 30 min, rinsed in distilled water, dehydrated in a graded series of ethanol solutions, and embedded in Epon. Semithin (1 μm) and ultrathin (50–60 nm) sections were cut and analyzed after counterstaining with conventional techniques.
Primary Cultures of Rat Mesencephalon.
Rat embryos were recovered at day 15.5 from gestating Wistar rats (Centre d’Elevage R. Janvier, Le Genest St. Isles, France). The ventral midbrain was dissected as described (23), mechanically dissociated, and plated on polyethyleneimine (1 mg/ml) precoated culture wells, in N5 medium, supplemented with 5% horse serum and 2.5% fetal calf serum, at a density of 0.8–1.2 × 105 cells/cm2. After 2 days in culture, fetal calf serum was reduced to 0.5% to halt astrocyte proliferation.
Experimental Procedures for Cell Cultures.
Apoptosis was induced in primary cultures of rat mesencephalon after 8 days of differentiation, with cell permeant C2-ceramide (25 μM; N-acetyl sphingosine; Biomol, Plymouth Meeting, PA) as described (12). In this study, the time point at which C2-ceramide-induced apoptosis became irreversible was determined. In subsequent experiments, C2-ceramide-containing medium was replaced, at this time point, with C2-ceramide-free medium. The number of surviving neurons was determined 36 hr after initiation of treatment, when, according to Brugg et al. (12), a loss of about 75% was expected.
Free radicals in the cultured cells were detected with 5- and 6-carboxy-2′7′-dichlorodihydrofluoresceine diacetate bis(acetomethyl)ester (DCDHF-DA, Molecular Probes), which produces a green fluorescence when oxidized (24). Cultures were incubated with 5 μM DCDHF-DA for 15 min at 37°C, rinsed three times with phosphate-buffered saline, and visualized by fluorescence microscopy (excitation 475 nm; emission 525 nm). The intensity of fluorescence was quantified by image analysis (morphostar Program, Imstar, Paris).
Immunocytochemistry in primary cultures of rat mesencephalon was performed as described (12). NF-κB was detected with the same polyclonal antiserum used on postmortem brain tissue, diluted 1:250. Neurons were identified with a mAb directed against microtubule-associated protein 2 (MAP-2) (25), diluted 1:50, the dopaminergic neurons with a mAb against tyrosine hydroxylase (Boehringer Mannheim), diluted 1:100. Primary antisera were detected with biotinylated antisera (1:250, Amersham). Immunolabeling was revealed with streptavidin sulforhodamine (Boehringer Mannheim), diluted 1:1,000. The morphology of normal and apoptotic (condensed, fragmented) nuclei was evidenced by the cell permeant fluorescent marker Hoechst 33258 (1 μM, Boehringer Mannheim), which intercalates into DNA. Cultures were analyzed by phase contrast and standard epi-illumination fluorescence microscopy and by computer-assisted image analysis (Imstar).
RESULTS
Specificity of Antibodies Directed Against Human NF-κB.
Western immunoblotting of proteins extracted from human SN and VTA showed a major band of 65 kDa (Fig. 1A) corresponding to the expected molecular weight of NF-κB-p65. On tissue sections, staining intensity decreased with lower antibody dilutions, and no staining was observed when the primary antiserum were omitted (data not shown) and when preadsorption tests were performed with a large excess of the homologous peptide (Fig. 1 B and C).
Figure 1.

Specificity of the NF-κB-p65 antiserum. (A) Western blot of NF-κB-p65 from SNpc (lane 1) and VTA (lane 2) of a normal human mesencephalon after SDS/PAGE, blotting on nitrocellulose membranes, and immunodetection with a rabbit polyclonal antiserum (1:250 dilution) against the p65 subunit of NF-κB. After revelation by enhanced chemiluminescence, a single band at about 65 kDa was observed. NF-κB-p65 immunoreactivity is shown in neurons of normal human SNpc, with (C) and without (B) adsorption of the antiserum for 6 hr at room temperature with a 2 × 104 excess of the corresponding peptide. Open arrows, neuromelanin-containing dopaminergic neurons; solid arrows, NF-κB immunostaining in neurons and processes. (Bar = 30 μm.)
Immunohistochemical Detection of NF-κB in Human Mesencephalon.
NF-κB immunostaining was observed in all subregions of the mesencephalon, with regional variations in staining intensity. Among dopaminergic regions, NF-κB immunoreactivity was most intense in the SNpc and VTA, moderate in the perirubral region (catecholaminergic cell group A8), and low in the central gray substance. At the cellular level, both pigmented and nonpigmented neurons were stained (Fig. 2). Low-to-high NF-κB immunoreactivity was detected in the perikarya and processes of neurons in the SNpc (Fig. 2 A and B), the VTA, the A8 region, and the central gray substance. Staining intensity was slightly lower, particularly in the SNpc and VTA, in parkinsonian patients (Fig. 2C) than in control subjects (Fig. 2A). NF-κB immunoreactivity was also detectable in the nucleus of a small proportion of neurons that were frequently characterized by an absence of stained processes (Fig. 2 C and D). These neurons were more numerous in parkinsonian patients than in controls. NF-κB labeling was also observed in glial cells in almost all sections from controls and PD patients. Immunoreactive glial cells, with the morphology and size of astrocytes and microglial cells, were observed in all subregions of the mesencephalon with strongest staining in the red nucleus and the cerebral peduncle (data not shown).
Figure 2.

Immunohistochemical detection of NF-κB-p65 in transverse sections of control SNpc (A and B) and parkinsonian SNpc (C and D). Low- (A and C) and higher-power (B and D) photomicrographs of melanized dopaminergic neurons (large arrows) and nonmelanized neurons (arrowheads) and their processes (small arrows), with dark-blue NF-κB immunolabeling. Note in C and D neurons with strong immunoreactivity in the nucleus (open arrows), which can clearly be distinguished from the labeled perikarya (solid arrows). (Bar: A and C, 60 μm; B and D, 30 μm.)
Quantitative Analysis of NF-κB-Positive Pigmented Neurons in the SNpc and the VTA of Control and Parkinsonian Patients.
The proportion of melanized neurons with cytoplasmic NF-κB staining was similar in PD patients and in control subjects in all the catecholaminergic regions studied (Table 1). In contrast, the proportion of melanized neurons with nuclear NF-κB staining was significantly increased in the group of parkinsonian patients compared with control subjects, in the SNpc (71-fold increase, P < 0.003), as well as in the VTA (51-fold increase, P < 0.005) (Table 1). In the nucleus of the third cranial nerve, a structure in which no neuronal loss is observed in parkinsonian patients, the proportion (%) of neurons with nuclear NF-κB staining was similar in controls (0.27 ± 0.22) and in parkinsonian patients (0.35 ± 0.35; P = 0.93).
Table 1.
Proportions (%) of pigmented neurons with cytoplasmic or nuclear NF-κB staining in the SNpc and the VTA of control subjects and parkinsonian patients
| Cytoplasmic staining
|
Nuclear staining
|
|||
|---|---|---|---|---|
| SNpc | VTA | SNpc | VTA | |
| Control | 37.8 ± 7.3 | 30.6 ± 6.7 | 0.07 ± 0.03 | 0.14 ± 0.14 |
| Parkinsonian | 28.9 ± 5.2 | 31.3 ± 2.3 | 4.96 ± 1.91* | 7.17 ± 3.40* |
Values represent the mean ± SEM. ∗, Significantly higher than controls (Mann–Whitney U test, P < 0.005).
NF-κB Activation During C2-Ceramide-Induced Apoptosis in Cultured Mesencephalic Neurons.
The neurons in primary cultures of mesencephalon began to die about 12 hr after initiation of treatment with 25 μM C2-ceramide. Nuclear translocation of NF-κB occurred during the 12-hr lag period, associated with ROS production (Fig. 3). In untreated cultures, neurons, identified with an antibody against MAP-2, did not emit ROS-activated DCDHF fluorescence (Fig. 3A) and immunofluorescent NF-κB was located in the cytoplasm (Fig. 3B), both of the MAP-2-positive neurons and of tyrosine hydroxylase-positive dopaminergic neurons (Fig. 3B Inset). In C2-ceramide-treated cultures, the neurons, which were still morphologically normal except for retraction of neurites, emitted DCDHF fluorescence (Fig. 3C) and NF-κB immunoreactivity was located principally in their nuclei (Fig. 3D). Astrocytes in C2-ceramide-treated cultures did not produce DCDHF-detectable ROS (data not shown).
Figure 3.
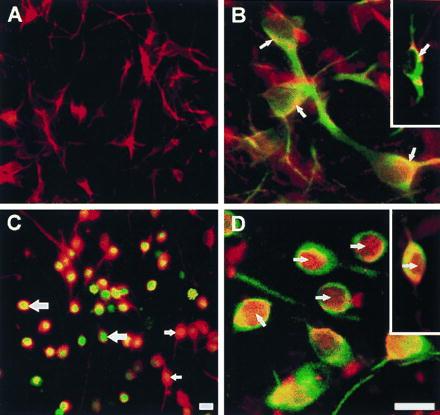
ROS-induced DCDHF fluorescence and translocation of NF-κB in ceramide-treated primary cultures of mesencephalon. (A) Neurons in untreated cultures, identified by rhodamine (red) immunofluorescence with an antibody against MAP-2. (B) NF-κB in untreated cultures, identified by rhodamine (red) immunofluorescence in neurons (arrows) is preferentially localized in the cytoplasm, and appears yellow because of superimposition with fluorescein (green) MAP-2 or tyrosine hydroxylase (Inset) immunoreactivity. (C) ROS-activated DCDHF green fluorescence that appears orange/yellow/green due to superimposition on red MAP-2 staining in neurons in cultures treated with 25 μM C2-ceramide for 5 hr. (D) NF-κB (red) labeling is primarily in the nuclei (arrows) of green MAP-2 and tyrosine hydroxylase-immunolabeled neurons in ceramide-treated cultures (Inset). [Bar = 20 μm (30 μm for Insets)].
ROS production in neurons slightly preceded the translocation of NF-κB; both were transient events, with their respective peaks at 5 and 8 hr after the beginning of C2-ceramide treatment (Fig. 4A). Neuronal cell death occurs approximately 6 hr later, but in parallel with NF-κB translocation. Once NF-κB was activated, neuronal death was irreversible: after 8 hr of treatment, elimination of C2-ceramide from the culture medium could no longer reverse the apoptotic process (Fig. 4B).
Figure 4.

Time course of ROS-induced DCDHF fluorescence, nuclear translocation of NF-κB, and neuronal death in ceramide-treated primary cultures of mesencephalon. (A) Time course of ROS-induced DCDHF fluorescence (○), translocation of NF-κB (•), and neuronal cell death (▴) during treatment with 25 μM C2-ceramide. At each time point after the initiation of treatment, the number of cells with levels of DCDHF fluorescence (mean gray level per cell) 2 SD or more above the control level, the number of MAP-2-positive neurons with NF-κB immunoreactivity in the nucleus, and the total number of MAP-2-positive neurons were counted. (B) The numbers of MAP-2-positive neurons surviving at 36 hr were counted in cultures treated with C2-ceramide for the durations indicated: an 8-hr treatment, corresponding to the peak translocation of NF-κB in A, was both necessary and sufficient to provoke maximal neuronal death at 36 hr. Data represent the mean ± SEM of three independent experiments. ∗, Significantly different compared with untreated cultures (P < 0.0001, two-tailed t test).
The antioxidant N-acetyl-cysteine prevented C2-ceramide-induced ROS production, the translocation of NF-κB in neurons, and neuronal death (Fig. 5), indicating that ROS were necessary for both the activation of NF-κB and the death of the cells. Even if N-acetyl-cysteine was eliminated once the translocation of NF-κB was prevented, its protective effect was maintained, indicating that neuronal cell death was not directly a result of the ROS but perhaps of ROS-stimulated NF-κB activation.
Figure 5.

The thiol antioxidant N-acetyl-cysteine prevents ROS-induced DCDHF fluorescence, NF-κB translocation, and neuronal death induced by C2-ceramide. N-acetyl-cysteine (20 mM) was added 1 hr before and during an 8-hr exposure to C2-ceramide (25 μM), which was then withdrawn and the cells maintained in fresh N-acetyl-cysteine supplemented medium. The number of DCDHF fluorescent cells (see Fig. 4 legend) and the number of MAP-2-positive neurons with nuclear NF-κB staining were counted at their respective peaks, 5 and 8 hr after the initiation of C2-ceramide treatment (see Fig. 4A) and neuronal survival at 36 hr. Data represent the mean number of neurons per well (×103) ± SEM of three independent experiments. Significantly different from cultures treated with C2-ceramide alone (∗, P < 0.05; ∗∗, P < 0.01, two-tailed t test).
Morphological Analysis of NF-κB-Positive Nuclei in Patients with Parkinson Disease and in Ceramide-Treated Mesencephalic Cultures.
NF-κB-positive nuclei in neuromelanin containing dopaminergic neurons in the SN of patients with PD were examined at the ultrastructural level for the classical nuclear alterations associated with apoptotic cell death; irregular nuclear envelope, chromatin condensation and/or fragmentation. On semithin sections of parkinsonian SN, dopaminergic neurons with (Fig. 6A) or without (Fig. 6B) nuclear NF-κB labeling, identified by their neuromelanin granules, were selected and ultrathin sections were prepared and analyzed. There was no correlation between the presence of NF-κB in the nucleus and the nuclear alterations characteristic of apoptosis: the NF-κB positive nucleus (Fig. 6C) did not show chromatin condensation or fragmentation, but the nuclear envelope was highly convoluted and several autophagic vesicles were observed, suggesting that the neuron could be in an early stage of degeneration. An NF-κB negative nucleus in an apoptotic neuron on the same ultrathin section (Fig. 6D) indicates that NF-κB may not be present in nuclei at this late stage of the apoptotic process; for comparison, an NF-κB negative nucleus without apoptotic features is illustrated in Fig. 6E.
Figure 6.

Morphological characteristics of NF-κB-positive nuclei in patients with PD and in ceramide-treated mesencephalic cultures. Photomicrographs of pigmented neurons from a parkinsonian patient with (A) and without (B) NF-κB labeling in the nucleus: semithin section counterstained with toluidine blue, on which the brown diaminobenzidine immunolabeling (arrow) could be distinguished from the black neuromelanin (arrowhead). (C) Ultrastructural analysis of nucleus in A; NF-κB immunolabeling (large arrow), dispersed chromatin (small arrow), convoluted nuclear envelope (arrowheads), autophagic vesicles (stars), neuromelanin (nm). (Inset) A higher magnification of an autophagic vesicle. (D) Apoptotic pigmented neuron (same ultrathin section as C); condensed chromatin (arrow). (E) Normal pigmented neuron with chromatin dispersed throughout the nucleus (arrows). (F) Mesencephalic cultures treated for 24 hr with 25 μM C2-ceramide, doubly labeled with an antibody against NF-κB (red fluorescence) and the DNA intercalating agent Hoechst 33258 (blue fluorescence): normal appearing nucleus containing red NF-κB immunofluorescence superimposed on the blue Hoechst fluorescence (large arrows); fragmented and condensed apoptotic nucleus without NF-κB staining (small arrows). [Bar = 30 μm (A and B), 4 μm (C and D), 800 nm (Inset in C), 5.5 μm (E), 20 μm (F).]
The presence of NF-κB in the nucleus of a neuron that appears to be at the very beginning of degeneration and its absence in the final stage where chromatin condensation is observed is consistent with the transient translocation of NF-κB observed during ceramide-induced apoptosis in the mesencephalic cultures shown in Fig. 4A: it was necessary for neuronal death to ensue, but it occurred during the lag period before cell death could be detected. Double-labeling of these cultures with an antibody against NF-κB and with the fluorescent DNA intercalating agent Hoechst 33258 to evidence nuclear morphology was used to determine whether NF-κB remains in the nucleus of neurons dying by ceramide-induced apoptosis. The cultures were observed 24 hr after the beginning of ceramide treatment, when some morphologically normal neurons with nuclear NF-κB still coexist with neurons that are in the process of dying, or have already died. Neurons at all stages of the apoptotic process were observed (Fig. 6F): at the two extremes, (i) normal neurons that had cytoplasmic NF-κB and large regular nuclei and (ii) fully apoptotic neurons with fragmented nuclei, where nuclear NF-κB could no longer be detected; at an intermediary stage, (iii) neurons with NF-κB labeling superimposed on the Hoechst stain in nuclei that were apparently normal at the light microscopic level, but may correspond to the deformed NF-κB-positive nucleus from the parkinsonian SN shown ultrastructurally in Fig. 6C. Most of the neurons in the culture were of this type. These morphological observations are consistent with the biochemical data showing that the late stage of apoptotic cell death, where chromatin condensation and fragmentation occur, no longer depends on the presence of NF-κB in the nucleus, but on the effects of the genes that it activates.
DISCUSSION
This study has shown the presence of NF-κB-p65 immunoreactivity both in the cytoplasm and nucleus of a subset of melanin-pigmented midbrain neurons in normal and parkinsonian patients. The staining was specific: (i) a single band at 65 kDa, the expected molecular weight of the protein (26), was observed on Western blots of proteins extracted from human SN and VTA; (ii) no staining was observed when the primary antiserum was preadsorbed with an excess of homologous peptide; (iii) omission of the primary antibodies resulted in an absence of labeling and dilution of the antibodies from 1 μg/ml to 1 × 10−3 μg/ml caused a progressive decrease in staining.
Expression of NF-κB (p65) in brain has already been reported in neurons of the SN and other brain regions, including the hippocampus, striatum, and cerebral cortex of the rat (27), and in the hippocampus and the entorhinal cortex of control subjects and patients with Alzheimer disease (28). In these studies, as in ours, NF-κB was localized, for the most part, in the cytoplasm, indicating that it is not constitutively activated, even in pathological human brain. The increased proportion of pigmented dopaminergic neurons with nuclear NF-κB, in parkinsonian patients, suggests that translocation of the transcription factor, essential to its activation, may be related to the pathophysiology of PD: (i) the proportion of melanized neurons presenting a cytoplasmic NF-κB staining was similar in control and parkinsonian patients, ruling out the possibility of nonspecific changes in NF-κB immunoreactivity between the two groups of subjects; (ii) nuclear NF-κB in parkinsonian patients was increased in brain structures where melanized catecholaminergic neurons degenerate in PD (SNpc, VTA), but not in the cholinergic neurons of the nucleus of the third cranial nerve, on the same tissue sections, which is spared. Interestingly, the proportion of melanized neurons displaying nuclear NF-κB staining in this study was identical to that of neurons showing the characteristic features of apoptosis evidenced by ultrastructural analysis (3) or end labeling of DNA fragments (2), supporting the hypothesis that the two observations may be related.
Is the increased nuclear localization of NF-κB in melanized midbrain neurons in parkinsonian patients related to neurodegeneration and to oxidative stress? This question cannot be answered on the basis of the postmortem study. However, in primary cultures of mesencephalic neurons undergoing ceramide-induced apoptosis, a transient production of free radicals preceded translocation of NF-κB. The radicals were required for translocation of the transcription factor, since their elimination with the antioxidant N-acetyl-cysteine prevented both translocation and neuronal death. Activation of NF-κB seems to be necessary for the lethal outcome, however, since commitment of the neurons to the apoptotic pathway became irreversible after translocation of the transcription factor had taken place. The involvement of NF-κB translocation in the degeneration of nigral dopaminergic neurons in PD is apparently challenged by the detection of nuclear NF-κB staining and apoptotic features in different populations of neurons. Nevertheless, this result may be explained by the transient expression of NF-κB observed in mesencephalic cell cultures in which nuclear NF-κB was followed by apoptosis. Furthermore, the presence of several autophagic vesicles, an index of cell suffering, in the melanized neurons displaying nuclear NF-κB staining in PD suggests that neurons in which the NF-κB transduction pathway is activated may be in an early stage of degeneration. During the delay between translocation of NF-κB and loss of cell viability, accompanied by the classic morphological signs of apoptosis, genes participating in the physical degeneration of the cells were activated; as previously shown, ceramide-dependent apoptosis in mesencephalic neurons can be inhibited by cycloheximide, which blocks new protein synthesis (12). The identity of the genes regulated by NF-κB that are required for ceramide-induced apoptosis is as yet unknown. Some of the genes activated by this transcription factor in nonneuronal systems (29), such as manganous superoxide dismutase, are intriguing but ambiguous: this enzyme may be protective by scavenging superoxide ions, but in doing so it transforms the relatively innocuous superoxide radical into the more deleterious hydrogen peroxide, the oxygen species that is primarily detected by the ROS marker DCDHF used in these studies, and the principal activator of NF-κB (29). Other genes activated by NF-κB, in particular those implicated in immune system mediated cytotoxicity (29), are also interesting candidates, as is c-myc, a mitogenic oncogene, reported to induce apoptosis in postmitotic neurons (30).
NF-κB is most probably not the only transcription factor that plays a role in the ceramide-dependent apoptotic signaling pathway in mesencephalic neurons in culture, but it is clearly associated with the cell death process. This is consistent with previous reports of ROS-related activation of the transcription factor in the cytotoxicity of TNF-α (15, 16), a known activator of the ceramide-dependent signaling pathway, serum withdrawal (31), and glutamate excitotoxicity (32). Several recent reports, however, have attributed an anti-apoptotic function to NF-κB (33–37). The reasons for this divergence are not clear. The nature of the cells in question may prove, however, to be important. In all these studies, proliferative cells were involved (embryonic hepatocytes or fibroblasts from homozygous relA/p65 knock-out mice, normal or transformed fibroblasts or lymphocytes), whereas the neurons in primary cultures are postmitotic. Stimuli that favor proliferation in the former have been reported to provoke apoptosis in the latter (30). Furthermore, the cell lines used were selected because they were resistant to TNF-α cytotoxicity and required RNA or protein synthesis inhibitors to overcome this resistance, the opposite of the regulation of apoptosis observed in primary neuronal cultures. Elucidation of the mechanism underlying the eventual role of NF-κB in this resistance, as underlined by the authors of refs. 33–36, is crucial to the control of carcinogenesis. In the same way, elucidation of the role of NF-κB in neuronal apoptosis may be essential to our understanding of cell death in Parkinson disease and perhaps also in other neurodegenerative disorders.
In conclusion, the combined postmortem and in vitro data presented here, showing a vastly increased proportion of dopaminergic neurons in the SN with nuclear NF-κB and a cellular mechanism relating NF-κB activation to oxidant formation and neuronal death, suggests that oxidative stress might be related to the apoptotic death of these neurons in PD, through NF-κB-related signal transduction. Convergence between postmortem data and the in vitro model suggests that this mechanism may be activation of the sphingomyelin-dependent signal transduction pathway by TNF-α, but other mechanisms must still be envisaged.
Acknowledgments
We thank Dr. S. Vyas for helpful discussions and Drs. A. M. Bonnet, J. P. Bouchon, C. Duyckaerts, J.-J. Hauw, M. Laurent, R. Moulias, F. Piette, A. Sachet, and M. Verny for their cooperation in providing postmortem human brain samples. This work was supported, in part, by the French Ministry of Research and Education (S.H.), the Association Claude Bernard pour le Développement des Recherches Biologiques et Médicales dans les Hôpitaux de l’Assistance Publique à Paris (B.A.F.), and the National Parkinson Foundation (Miami) (Y.A.).
ABBREVIATIONS
- PD
Parkinson disease
- SN
substantia nigra
- SNpc
SN pars compacta
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor α
- VTA
ventral tegmental area
- MAP-2
microtubule-associated protein 2
References
- 1.Hirsch, E. C. (1993) Eur. Neurol. 33, Suppl. 1, 52–59. [DOI] [PubMed]
- 2.Mochizuki H, Goto K, Mori H, Mizuno Y. J Neurol Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 3.Anglade P, Vyas S, Javoy-Agid F, Herrero M T, Michel P P, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch E C, Agid Y. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 4.Dexter D T, Carter C J, Wells F R, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden C D. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 5.Saggu H, Cooksey J, Dexter D, Wells F R, Lees A, Jenner P, Marsden C D. J Neurochem. 1989;53:692–697. doi: 10.1111/j.1471-4159.1989.tb11759.x. [DOI] [PubMed] [Google Scholar]
- 6.Sian J, Dexter D T, Lees A J, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden C D. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 7.Riederer, P., Dirr, A., Goetz, M., Sofic, E., Jellinger, K. & Youdim, M. B. H. (1992) Ann. Neurol. 32, Suppl., 5101–5104. [DOI] [PubMed]
- 8.Hirsch E C, Brandel J P, Galle P, Javoy-Agid F, Agid Y. J Neurochem. 1991;56:446–451. doi: 10.1111/j.1471-4159.1991.tb08170.x. [DOI] [PubMed] [Google Scholar]
- 9.Halliwell B. In: Free Radicals in the Brain. Packer L L, Prilipko L, Christen Y, editors. Berlin: Springer; 1992. pp. 21–40. [Google Scholar]
- 10.Kolesnick R, Golde D W. Cell. 1994;77:325–328. doi: 10.1016/0092-8674(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 11.Hannun Y A, Obeid L M. Trends Biochem Sci. 1995;20:73–77. doi: 10.1016/s0968-0004(00)88961-6. [DOI] [PubMed] [Google Scholar]
- 12.Brugg B, Michel P P, Agid Y, Ruberg M. J Neurochem. 1996;66:733–739. doi: 10.1046/j.1471-4159.1996.66020733.x. [DOI] [PubMed] [Google Scholar]
- 13.Kolesnick R N, Haimowitz-Friedman A, Fuks Z. Biochem Cell Biol. 1994;72:471–474. doi: 10.1139/o94-063. [DOI] [PubMed] [Google Scholar]
- 14.Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch E C. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- 15.Schulze-Osthoff K, Bakker A C, Vanhaesebroeck B, Beyaert R, Jacobs W A, Fiers W. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- 16.Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.France-Lanord, V., Brugg, B., Michel, P. P., Agid, Y. & Ruberg, M. (1997) J. Neurochem., in press. [DOI] [PubMed]
- 18.Thanos D, Maniatis T. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 19.Hunot S, Boissière F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch E C. Neuroscience. 1996;72:355–363. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
- 20.Zhang P, Anglade P, Hirsch E C, Javoy-Agid F, Agid Y. Neuroscience. 1994;61:317–330. doi: 10.1016/0306-4522(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 21.Hirsch E C, Graybiel A M, Agid Y. Nature (London) 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 22.Anglade P, Mouatt-Prigent A, Agid Y, Hirsch E C. Neurodegeneration. 1996;5:121–128. doi: 10.1006/neur.1996.0018. [DOI] [PubMed] [Google Scholar]
- 23.Michel P P, Agid Y. J Neurochem. 1996;67:1633–1642. doi: 10.1046/j.1471-4159.1996.67041633.x. [DOI] [PubMed] [Google Scholar]
- 24.Schartz M A, Lazo J S, Yalowich J C, Reynolds I, Kagan V E, Pitt B R. J Biol Chem. 1994;269:15238–15243. [PubMed] [Google Scholar]
- 25.Binder L I, Frankfurter A, Kim H, Caceres A, Payne M R, Rebhun L I. Proc Natl Acad Sci USA. 1984;81:5613–5617. doi: 10.1073/pnas.81.17.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauerle P A, Baltimore D. Genes Dev. 1989;3:1689–1698. doi: 10.1101/gad.3.11.1689. [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Otaño I, McMillian M K, Chen J, Bing G, Hong J S, Pennypacker K R. Glia. 1996;16:306–315. doi: 10.1002/(SICI)1098-1136(199604)16:4<306::AID-GLIA3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 28.Terai K, Matsuo A, McGeer P L. Brain Res. 1996;735:159–168. doi: 10.1016/0006-8993(96)00310-1. [DOI] [PubMed] [Google Scholar]
- 29.Bauerle P A, Henkel T. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 30.Bissonnette R P, Shi Y, Mahboubi A, Glynn J M, Green D R. In: Apoptosis II: The Molecular Basis of Apoptosis in Disease. Tomei L D, Cope F O, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 327–356. [Google Scholar]
- 31.Grimm S, Bauer M K A, Bauerle P A, Schulze-Osthoff K. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grilli M, Pizzi M, Memo M, Spano P F. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 33.Beg A A, Sha W C, Bronson R T, Ghosh S, Baltimore D. Nature (London) 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 34.Beg A A, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 35.Wang C-Y, Mayo M W, Baldwin A S., Jr Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 36.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z-G, Hsu H, Goeddel D V, Karin M. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]