

Abstract
Background:
Previous research has suggested the therapeutic potential of glutamate-modulating agents for severe mood and anxiety disorders, potentially due to enhancement of neuroplasticity. We used proton magnetic resonance spectroscopic imaging (1H MRSI) to examine the acute and chronic effects of the glutamate-release inhibitor riluzole on hippocampal N-acetylaspartate (NAA), a neuronal marker, in patients with generalized anxiety disorder (GAD), and examined the relationship between changes in NAA and clinical outcome.
Methods:
Fourteen medication-free GAD patients were administered open-label riluzole and then evaluated by 1H MRSI before drug administration, and 24 hours and 8 weeks following treatment. Patients were identified as responders (n = 9) or non-responders (n = 5). Seven untreated, medically healthy volunteers, comparable in age, sex, IQ, and body mass index to the patients, received scans at the same time intervals. Molar NAA concentrations in bilateral hippocampus and change in anxiety ratings were the primary outcome measures.
Results:
A group-by-time interaction was found, with riluzole responders showing mean increases in hippocampal NAA across the three time points, while non-responders had decreases over time. In GAD patients at Week 8, hippocampal NAA concentration and proportional increase in NAA from baseline both were positively associated with improvements in worry and clinician-rated anxiety.
Conclusions:
These preliminary data support a specific association between hippocampal NAA and symptom alleviation following riluzole treatment in GAD. Placebo-controlled investigations that examine hippocampal NAA as a viable surrogate endpoint for clinical trials of neuroprotective and plasticity-enhancing agents are warranted.
INTRODUCTION
There is growing evidence to support the efficacy of glutamate-modulating agents in the treatment of severe mood and anxiety disorders. Although the bulk of this work has focused on mood disorders (e.g., 1-5), substantial overlap between the neurobiology and therapeutics of mood and anxiety disorders suggests that glutamate modulators may also be promising anxiolytics. We previously reported that riluzole, a glutamate-release inhibitor approved for the treatment of amyotrophic lateral sclerosis (ALS), may be effective for generalized anxiety disorder (GAD) (6). In an 8-week open-label trial, 8 of 15 trial completers achieved remission, as indexed by Hamilton Anxiety Rating Scale (HAM-A) (7) scores ≤ 7 at endpoint.
Glutamate, the primary excitatory neurotransmitter in the brain (8), has been implicated as a key contributor to stress-induced impairments in neuroplasticity. When stress-related glutamatergic dysregulation results in excessive glutamate accumulation in the synapse, glutamatergic neurotoxicity or “excitotoxicity,” culminating in neuronal death, has been observed (e.g., 9, 10). Riluzole has a complex mechanism of action, including: (1) inhibition of voltage-dependent sodium channels in central nervous system (CNS) neurons (11, 12); (2) inhibition of excitotoxic injury (13); (3) increased glutamate reuptake (14); (4) stimulation of growth factor synthesis, including brain-derived neurotrophic factor (BDNF) (15, 16); (5) promotion of neuritogenesis, neurite branching, and neurite outgrowth (17); and (6) enhancement of hippocampal AMPA receptor subunit (GluR1 and GluR2) expression (18).
Proton magnetic resonance spectroscopy (1H MRS) was previously used to measure riluzole's effect in ALS on neurometabolites (19,20), including N-acetylaspartate (NAA), a mitochondrial amino acid frequently characterized as a marker of neuronal integrity (21). In these reports, concentrations of NAA were reported in relation to total creatine (Cr), an index of cellular energetics previously viewed as a stable internal standard but more recently observed to have low reproducibility in medial temporal lobe (22), with poor correlations with absolute measures of NAA (23). Three weeks of riluzole treatment resulted in increased (+6.1%) NAA/Cr ratios in the motor cortex of 11 patients with ALS, while untreated ALS patients showed NAA/Cr decreases (−4.1%) in the same region (19). In a follow-up ALS study by the same group, administration of two doses of riluzole (50 mg) was associated with increased NAA/Cr ratios (+5% in the precentral gyrus and +8% in the supplementary motor area), suggesting that increased NAA/Cr reflected metabolic, rather than structural, change (20). Thus, the therapeutic action of riluzole in ALS may hinge on its ability to promote neuronal function via reversal of glutamatergic excitotoxicity and/or rapid restoration of mitochondrial metabolic function in sublethally injured neurons.
GAD is a common condition with high lifetime mood disorder comorbidity (24), and is a risk factor for major depression (25). Despite its prevalence, GAD's underlying neurochemical substrates remain obscure (26). To clarify the relationship between GAD, glutamatergic function, and regional neuronal viability, we used 1H MRSI to test the acute and chronic impact of riluzole on neurochemical concentrations in GAD, focusing on hippocampal NAA. The hippocampus was selected as the primary region of interest (ROI) because of its putative role in stress-related glutamatergic neurotoxicity (27, 28). We measured NAA concentrations at three time points in a subgroup of GAD patients recruited for a clinical trial (6): (a) at baseline, (b) after 24 hours (2 doses) of riluzole (50 mg b.i.d.), and (c) after 8 weeks of riluzole treatment. We hypothesized that for GAD responders, both acute and chronic riluzole treatment would be associated with increased hippocampal NAA, and that NAA increases would correspond to reductions in pathological worry and anxiety. In exploratory analyses, we investigated patterns of acute and chronic change in NAA in additional brain regions subserving anxiety and worry, including dorsolateral prefrontal cortex (DLPFC) (29), anterior cingulate cortex (ACC) (30-32), and prefrontal white matter (33).
METHODS AND MATERIALS
Subjects
Eighteen patients with GAD entered the clinical trial, which consisted of 8 weeks of open-label monotherapy with riluzole (100 mg/day, given 50 mg b.i.d.) (6). Fifteen of these patients (6 males, 9 females; mean age ± SD, 31.7 y ± 9.6) completed the neuroimaging protocol, which comprised three 1H MRS scans. Eight untreated, medically healthy volunteers (3 males, 5 females; mean age: 27.4 y ± 4.2) received 1H MRS scans at the same time intervals as patients. All subjects were recruited by advertising or clinician referral. The baseline scans of one GAD patient and one healthy volunteer were uninterpretable due to motion-degraded spectral quality. These two individuals were excluded from analyses, yielding a final sample of 14 GAD patients and 7 healthy volunteers. All patients met DSM-IV-TR criteria for GAD as established by the Structured Clinical Interview for DSM-IV (SCID) (34). GAD patients had a chronic course of illness (mean duration of illness: 14.3 y ± 9.9), with moderate anxiety severity (mean baseline HAM-A: 20.0 ± 3.6; mean baseline Penn State Worry Questionnaire (PSWQ; 35) score: 64.6 ± 8.3), and mild-to-moderate depressive symptoms (Hamilton Rating Scale for Depression, 24-item version (HRSD24; 36):14.9 ± 4.3). Comorbid diagnoses, determined by SCID, included panic disorder (n = 6), dysthymia (n = 5), social anxiety disorder (n = 3), past major depressive disorder (n = 1), and past depressive disorder NOS (n = 1). Exclusion criteria for GAD patients included: major depressive episode or substance abuse/dependence within 6 months of study entry; lifetime histories of psychosis, bipolar disorder, obsessive-compulsive disorder (OCD), eating disorder, or PTSD; or significant medical or neurological conditions requiring daily medication treatment. Four patients had received 1 or more adequate previous trials of psychotropic medication, and 6 patients were psychotropic-medication naïve. One patient was taking psychiatric medication at the time of initial screening. Medication was halted two weeks before the initial scan; based on the medication's elimination half-life, this was judged sufficient to prevention interaction with riluzole.
Healthy volunteers had no lifetime history of axis I psychiatric disorders, according to SCID-NP interview (37). GAD patients and healthy volunteers did not differ in mean age, sex, IQ, or body mass index (p > 0.20 for all). All participants had unremarkable screening laboratory evaluations, including urine toxicology. Written informed consent was obtained and all study procedures were approved by the New York State Psychiatric Institute Institutional Review Board.
Timing of 1H MRSI Scans and Clinical Trial Procedures
Immediately following the baseline scan, the first dose of riluzole (50 mg p.o.) was administered by a study physician. The following morning, patients were instructed to take the second dose of riluzole (50 mg) 3 hours prior to the second (24-hour) MRSI scan. Patients were re-evaluated by the study psychiatrist prior to the 24-hour scan; clinical global impression revealed no acute behavioral effects or changes in diagnostic status after 2 doses (100 mg) of riluzole treatment in any patient. After the 24-hour scan, patients continued riluzole (50 mg b.i.d.) and had weekly 30-minute visits with the study psychiatrist for medication management (6). The third MRSI scan was performed at endpoint (Week 8), prior to medication taper. Primary efficacy measures were the HAM-A, administered by a single trained rater, and the PSWQ, which was collected at the baseline and week 8 scans.
1H MRSI Data Acquisition Protocol
All neuroimaging studies were conducted on a 1.5 T GE Horizon 5.× Signa MR system. Following sagittal scout images, a 4-section T1-weighted axial/oblique localizer imaging series, angulated parallel to the Sylvian fissure (Figure 1A), was acquired, with a slice thickness of 15 mm and an inter-slice gap of 3.5 mm, matching the subsequent multislice 1H MRSI scan. Next, the 1H MRSI scan was performed using the method of Duyn et al. (38), with TE/TR 280/2300 ms, FOV 240 mm, 32×32 circularly sampled k-space phase-encoding steps with 1 excitation per phase-encoding step, and 256 time-domain points. The strong pericranial lipid resonances from the skull, scalp and calvarial marrow were suppressed using octagonally tailored outer-volume suppression pulses, and water was suppressed with a single chemical shift-selective (CHESS) pulse followed by spoiler gradients. The entire neuroimaging protocol required approximately 60 minutes to complete. The raw data were separated into individual slices and then processed by the standard fast Fourier transform algorithm, as previously described (29). The actual MRSI voxel, estimated from the integral of the point-spread function (PSF) following spatial filtering with a Hamming window and Fermi window and then Fourier transformation, was 1.13 cm3 or approximately 40% larger than the nominal voxel size that would be derived from the acquisition parameters.
Figure 1.

[A] T1-weighted MR brain image in a generalized anxiety disorder patient showing prescription of the 4 investigated brain sections on a sagittal view, with the most inferior slice parallel to the Sylvian fissure and containing the hippocampal region of interest (ROI); [B] axial/oblique view of the most inferior of the four slices showing voxels selected to cover the right (R) and left (L) hippocampus ROIs. The voxels are depicted in actual rather than “nominal” size, after taking into account a ∼40% broadening due to point-spread function (PSF); several such voxels can be seen to be fully contained within the hippocampal ROIs, which would minimize tissue heterogeneity and partial volume averaging. [C] Sample frequency-domain nonlinear least-squares Lorentzian lineshape model fitting of spectra from representative right (R) and left (L) hippocampal voxels, showing [a] the measured spectra, [b] the calculated “best-fit” spectra, [c] individual components of the “best-fit” spectra, and [d] residuals of the difference between the measured and calculated “best-fit” spectra. Spectral resonances identified are for total choline-containing compounds (tCho), creatine + phosphocreatine (tCr), and N-acetylaspartate moieties (NAA). All the spectra are plotted using the same vertical-axis scale.
1H MRSI Data Analysis and Quantitation
The raw MRSI data were processed and analyzed voxel by voxel offline on a Sun Microsystems (Mountain View, CA) workstation, using Interactive Data Language (IDL, ITT Visual Information Solutions, Boulder, CO) software package developed in-house by two of the investigators (XM, DCS). Figure 1C shows a representative spectrum and sample spectral fit for a hippocampal MRSI voxel. Voxels that best covered the primary ROIs (right and left hippocampus) in each subject were selected on the basis of their location on the matching high-resolution MR localizer images (Figure 1B). Data analysis was performed by a trained investigator blinded to diagnosis and scan number. The mean of the peak areas for each metabolite within the ROIs was computed from fitted spectral data. The a priori measure of interest was the concentration of NAA, which was derived as described below. Concentrations of creatine + phosphocreatine (tCr) and total choline-containing compounds (tCho, an index of myelin turnover) were also obtained. Peak areas derived from spectral fitting were converted to “absolute” (i.e., molar) metabolite concentrations using phantom replacement methodology (39). See Supplementary Information for absolute quantification methods.
Statistical Analyses
Response was defined a priori as a Week 8 HAM-A ≤ 7 or PSWQ ≤ 45. A HAM-A cut-off score of 7 is common in psychopharmacological trials of GAD (40); a PSWQ cut-off score of 45 has previously been found to maximize sensitivity and specificity in differentiating GAD patients from non-anxious controls (41). There were no significant interactions between brain side (left or right) and group or between the effect of neurometabolites on anxiety and side; thus right- and left-sided metabolite concentrations were averaged to obtain a mean bilateral concentration for each ROI. Repeated measures analyses of variance (ANOVA) was performed on regional metabolite concentrations, with time of scan (baseline, 24 hours, 8 weeks) as the within-subjects factor and group (responder vs. non-responder vs. healthy volunteer) as the between-subjects factor. Post-hoc group comparisons used the Tukey HSD test. Differences among group proportions showing increases in hippocampal NAA were assessed by the Freeman-Halton extension of the Fisher exact probability test. Post-hoc 2 × 2 Fisher exact tests compared individual groups. Tests of association were examined using Pearson's product-moment correlations. All tests were two-tailed, with significance level set at p ≤ 0.05.
RESULTS
Clinical Trial Outcome
One patient's dosage was decreased to 50 mg/day after 4 weeks due to excessive sedation. All other patients completed the protocol at the target dose of 100 mg/day. Riluzole was generally well-tolerated, with no significant adverse events (6). Nine patients (64.3%) met the a priori definition of responder at Week 8; 5 patients (35.7%) were classified as nonresponders. The mean HAM-A score of patients decreased from 20.0 (SE = .95) to 7.2 (SE = 1.44) at Week 8 (paired t[13] = 8.71, p = < .001), while mean PSWQ scores decreased from 64.6 (SE = 2.21) to 51.0 (SE = 3.37) (paired t[13] = 4.10, p < .001). There were no differences between responders and non-responders in clinical or demographic measures (all p's > .25) (see Table). At study endpoint, responders were significantly less symptomatic than nonresponders on all three clinical measures: HAM-A (responders = 4.2 [SE = 1.0], non-responders = 12.3 [SE = 1.6], t[13] = 4.49, p < .001); PSWQ (responders = 44.9 [SE = 3.5], non-responders = 62.5 [SE = 2.9], t[13] = 3.58, p < .01); and HRSD24 (responders = 4.9 [SE = 1.7], nonresponders = 16.3 [SE = 1.5], t[13] = 4.75, p < .001).
Table.
Baseline Characteristics of Responders and Non-Responders to Riluzole and Healthy Volunteers
| Responders (n = 9) | Non-Responders (n= 5) | Healthy Volunteers (n= 7) | |
|---|---|---|---|
| Age | 32 (3.9) | 31 (2.5) | 27 (1.6) |
| Female | 5/9 (56%) | 3/5 (60%) | 5/7 (71%) |
| w/ ≥ 1 comorbid AD | 6/9 (67%) | 3/5 (60%) | 0/7 (0%) |
| Psychotropic-naive | 4/9 (44%) | 2/5 (40%) | 0/7 (0%) |
| Abuse history | 1/9 (11%) | 2/5 (40%) | 2/7 (29%) |
| Body Mass Index | 23.4 (4.4) | 21.1 (1.2) | 21.9 (.61) |
| IQ | 117.0 (3.5) | 119.5 (6.2) | 123.0 (3.8) |
| Age of onset | 16.1 (3.6) | 19.7 (4.6) | N/A |
| Duration of illness | 15.9 (3.8) | 11.5 (3.1) | N/A |
| HAM-A | 19.2 (1.0) | 21.4 (2.0) | 1.7 (.64)* |
| PSWQ | 66.0 (2.6) | 62.0 (4.2) | 31.0 (3.6)* |
| HRSD24 | 14.8 (1.8) | 15.2 (0.9) | 1.3 (.57)* |
Note: Standard errors are in parentheses. AD = anxiety disorder (panic disorder, social phobia, or specific phobia); self-reported childhood abuse history as per modified version of Early Trauma Inventory (see 33); IQ = Wechsler Abbreviated Scale of Intelligence full scale score; HAM-A = Hamilton Anxiety Rating Scale; PSWQ = Penn State Worry Questionnaire; HRSD24 = 24-item Hamilton Rating Scale for Depression.
Healthy volunteers differ from responders and non-responders, p < .001.
Primary Region of Interest Analyses
Repeated measures ANOVA revealed no main effects of time or group (responders, non-responders, healthy volunteers) on hippocampal NAA concentration. However, a significant interaction effect was found (F[4, 36] = 3.26, p = .02). As seen in Figure 2, NAA concentrations increased over time in responders, and decreased over time in the non-responder group. Post-hoc analyses revealed that between-group differences in NAA concentration were not significant at any time point (Baseline scan: responders vs. non-responders, p = .134; responders vs. healthy, p = .549, non-responders vs. healthy, p = .576; Week 8 scan: responders vs. non-responders, p = .292; responders vs. healthy, p = .991, non-responders vs. healthy, p = .276). Similarly, post-hoc comparisons of hippocampal NAA in GAD patients (responders + non-responders) vs. healthy volunteers yielded no significant group differences at any time point.
Figure 2.

Changes in N-acetylaspartate concentration in bilateral hippocampus across Baseline MRSI scan, 24-hour MRSI scan (after 2 doses of riluzole, 50 mg b.i.d.), and Week 8 MRSI scan (after 8 weeks of riluzole, 50 mg b.i.d.). Bars represent standard error of the mean.
Following acute riluzole treatment (baseline to 24-hour scan), hippocampal NAA increased by 5.3% (SE = 5.1) in healthy volunteers and by 9.4% (SE = 7.5) in responders, while decreasing by 9.5% (SE = 5.7) in non-responders. From baseline MRSI scan to week 8 MRSI scan, hippocampal NAA increased by 6.2% (SE = 3.6) in healthy volunteers and by 17.0% (SE = 8.4) in riluzole responders; hippocampal NAA decreased by 15.6% (SE = 7.2) in non-responders. Coefficient of variation in this region for control subjects was 4.1%.
Significant group differences in direction of change in hippocampal NAA emerged after 8 weeks of riluzole treatment. Hippocampal NAA concentrations increased in 5 of 7 (71.4%) control subjects and in 7 of 9 (77.8%) responders across the 8-week treatment interval, in comparison to 0 of 5 non-responders (0.0%) (p = .018, Fisher exact test for all three groups) (Figure 3). Post-hoc two-group comparisons by Fisher exact tests found that nonresponders differed both from responders (p = .021) and from healthy volunteers (p = .028) in direction of change. Responders and healthy volunteers did not differ on this dimension (p > .99).
Figure 3.
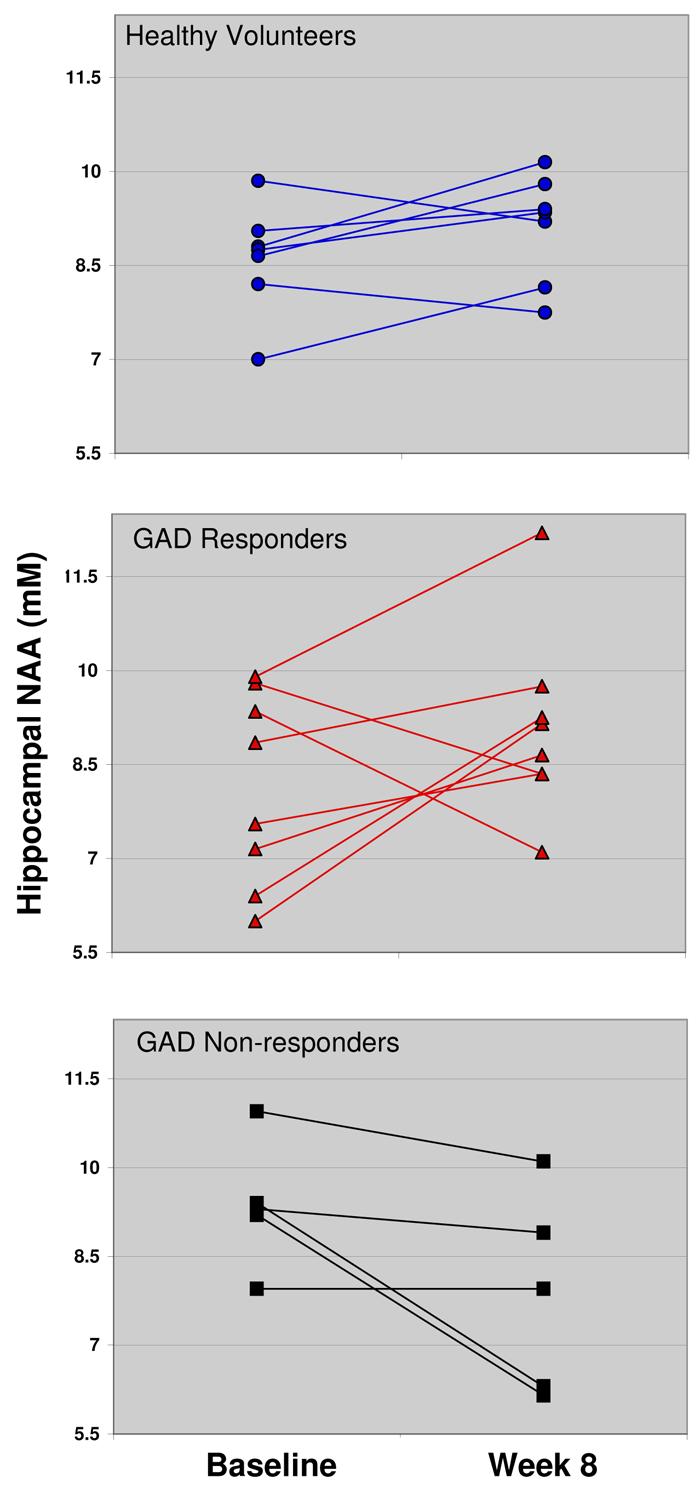
Hippocampal N-acetylaspartate of healthy volunteers (untreated) and generalized anxiety disorder patients at Baseline MRSI scan and Week 8 MRSI scan (8 weeks of riluzole, 50 mg b.i.d.).
Similar repeated measures ANOVAs were performed using tCho and tCr concentrations as dependent measures; there were no significant main or interaction effects for time or responder status.
Correlational Analyses
The relationship between changes in hippocampal NAA and changes in anxiety, as measured by percent change of HAM-A and PSWQ, was assessed in GAD patients. Percent change in hippocampal NAA from baseline scan to Week 8 scan was positively correlated with percent improvement (decrease) in HAM-A (r = .71; p < .01) and PSWQ (r = .56; p < .05) scores (Figure 4). Similarly, percent change in hippocampal NAA from the 24-hour scan to the Week 8 scan was positively correlated with improvement in HAM-A (r = .63; p < .05) and PSWQ (r = .61; p < .05) from baseline to trial completion.
Figure 4.

Scatter plots for percent change in hippocampal N-acetylaspartate vs. percent decrease in symptom measures at Week 8 (generalized anxiety disorder patients only). PSWQ = Penn State Worry Questionnaire; HAM-A = Hamilton Anxiety Rating Scale.
Baseline hippocampal NAA did not correlate with percent change from baseline to endpoint for HAM-A (r = −.410, p = .145), PSWQ (r = −.332, p = .246), or HRSD24 (r= .198, p = .497). Likewise there were no significant correlations between baseline hippocampal NAA and baseline PSWQ (r = −.38; p = .184), HAM-A (r = −.11; p = .71), or HRSD24 (r= −.12; p = .69). However, at Week 8, a significant relationship emerged for anxiety symptoms, such that hippocampal NAA was inversely related to both HAM-A (r = −.58; p < .05) and PSWQ (r = −.79; p < .001) scores. Hippocampal NAA concentrations at Week 8 were also correlated with percent improvement (i.e., decrease from baseline) in HAM-A (r = .64; p < .05) and PSWQ (r = .59; p < .05). Baseline HAM-A or PSWQ was not associated with change in NAA from Baseline to Week 8 (r = .023, p = .94; r = −.045, p = .877). No additional correlations for hippocampal NAA were observed with any other relevant clinical or demographic variable.
Exploratory Analyses in Prefrontal ROIs
There were no main effects or interaction effects for NAA concentration in any region (DLPFC, ACC, prefrontal white matter). Baseline NAA concentrations did not differ between GAD patients and healthy volunteers in any region. No significant correlations between NAA concentrations and symptom measures were observed at any time point.
DISCUSSION
In GAD patients treated with the glutamate-modulating agent riluzole for 8 weeks, a strong relationship was found between changes in anxiety symptoms and changes in hippocampal NAA from baseline to endpoint. A significant group-by-time interaction was evident, signifying that the pattern of change in hippocampal NAA across the three assessment points (Baseline, 24 hours, Week 8) differed for responders, non-responders, and non-anxious healthy volunteers. In most patients who responded to riluzole (7 of 9), hippocampal NAA increased from baseline to study endpoint (+17.0% mean increase), while in all non-responders (5 of 5), hippocampal NAA remained stable or decreased (−15.6% mean decrease). A similar proportion of healthy volunteers displayed increased hippocampal NAA as the riluzole responders.
The acute effect of riluzole on NAA concentrations was consistent with the overall trajectory of increase (responders) or decrease (non-responders) over the course of the study. However, the relative acute change in NAA did not differ significantly between responders and non-responders, and significant relationships between NAA and symptoms did not emerge until study endpoint. This distinction between riluzole's acute and chronic effects suggests that riluzole's anxiolytic properties might be dependent on longer-term processes associated with enhanced neuronal viability and neuroplasticity. However, given that acute trends mimicked chronic effects, rapid restoration of mitochondrial function in extant neurons may also be associated with symptom alleviation, as suggested previously in ALS (20).
Riluzole's effect on hippocampal NAA in responders is consistent with the view that neuroplasticity-enhancing therapies may benefit subgroups of patients with GAD and mood disorders. Modulation of the glutamatergic system for stress-related mood disorders may confer neuroprotection (42) and enhance neuroplasticity (e.g., 43), which encapsulates a range of neural processes (e.g., dendritic function, axonal sprouting, synaptic remodeling, long-term potentiation) that support the brain's ability to perceive, adapt to, and respond to internal and external stimuli (44). Riluzole's effect on neural and behavioral plasticity in hippocampus is mediated in part by its role in AMPA receptor trafficking, a process implicated in the regulation of activity-dependent synaptic strength and postsynaptic receptor responsiveness (18). Riluzole administered chronically and at therapeutically relevant concentrations in cultured hippocampal neurons enhanced surface expression of the AMPA receptor subunits GluR1 and GluR2 (18). Although we did not find baseline evidence of impaired neuroplasticity, GAD patients who responded to riluzole after 8 weeks showed a robust increase in hippocampal NAA, suggesting that this putative marker of neuroplasticity may signify a surrogate endpoint for clinical response. Long-tem outcome data are needed to test whether hippocampal NAA increases during treatment can predict sustained clinical benefit. Conversely, decreased hippocampal NAA in riluzole non-responders may reflect illness-associated impairments in neuronal viability and/or mitochondrial function. It is noteworthy that the 2 GAD non-responders at Week 8 with the greatest decreases in hippocampal NAA from baseline (>25%) had NAA concentrations below the range for healthy volunteers at any time point, representing a pathological NAA deficit. No baseline clinical, demographic, or MRSI variables distinguished riluzole responders from non-responders, and baseline NAA did not predict change in anxiety, nor did baseline anxiety measures predict change in NAA. Additional studies are thus necessary to determine the value of hippocampal NAA as a biomarker of clinical anxiety.
In interpreting the neurobiological significance of riluzole's impact on hippocampal NAA, several salient issues regarding NAA merit discussion. First, it is now accepted that NAA is present in immature oligodendrocytes and is not neuron-specific (45). As riluzole has been demonstrated to modulate extracellular glutamate levels through glial reuptake mechanisms (14), increased hippocampal NAA may reflect increased non-neuronal activity. Second, genetic variation in the regulation of synaptic glutamate concentrations has been found to impact NAA concentrations (46) while polymorphisms in neurotrophic factors contribute to individual differences in hippocampal volume (47). Neuroimaging investigations that assess genetic moderators of hippocampal plasticity such as the brain-derived neurotrophic factor Val66Met polymorphism (47) would enable further scrutiny of the relationship between riluzole response, hippocampal NAA, and neuroplasticity. Third, while the NAA resonance peak in MRSI consists predominantly of NAA (20), there are contributions of up to 25% from other N-acetyl compounds, including the dipeptide N-acetylaspartylglutamate (NAAG) (48). Thus, it is possible, although unlikely, that the increased hippocampal NAA in GAD responders with chronic riluzole administration may reflect increased NAAG with normal NAA, a pattern observed in normal-appearing white matter in multiple sclerosis (49). Finally, studies designed to measure the glutamate resonance (which includes glutamate and glutamine), using either high-field 1H MRS or appropriate spectral editing techniques, could also advance our understanding of the effects of riluzole on this metabolic pathway. Notably, glutamate-glutamine contamination of the NAA peak has been observed at short echo times (TE = 35 ms) (50) but not at long TEs, as were used in this study.
The lack of baseline group differences in hippocampal NAA concentrations is consistent with the only previous published MRSI study in GAD (29). Increased NAA/Cr ratios were found in right DLPFC of 15 GAD patients compared to healthy controls, although the use of ratio analyses hinders direct comparability between studies. In major depression, most studies have failed to detect cortical NAA abnormalities (51), although in PTSD, hippocampal NAA reductions have been reported even in the absence of volumetric reductions (52-54). Our correlational results at study endpoint add to an emerging literature relating NAA concentrations to anxiety variables, though the directionality and regional localization of the findings have been inconsistent across studies. In a non-clinical sample, NAA concentrations in the orbital frontal cortices were positively related to a composite measure of state and trait anxiety (55), while in social phobics, NAA/Cr in the ACC was found to be elevated and positively related to symptom severity (32). These divergent findings underscore the need for additional research, using consistent metabolite measures and ROIs, in discrete patient populations.
This study is the first to examine the neurochemical effects of riluzole in a clinically anxious patient group. We studied a well-characterized GAD sample with no substance abuse comorbidity or concurrent major depressive episodes. Collecting 1H-MRSI data at three time points allowed us to differentiate between the acute and chronic effects of riluzole on metabolite measures, and the use of a reliable multi-slice multi-voxel spectroscopic acquisition (38) with absolute quantitation mitigated limitations of ratio analyses of MRSI data. We employed an absolute quantitation scheme for NAA rather than report NAA/Cr ratios due NAA/Cr's poor association with absolute levels of NAA (23), and its relatively low test-retest reliability reported in medial temporal lobe (22). In our report, we found low variability of hippocampal NAA for healthy volunteers (CV = 4.1%), which compares favorably to the CV (> 10%) using the same acquisition H-MRSI acquisition procedure reporting hippocampal NAA/Cr ratios (56).
Nevertheless, several methodological limitations are noted. First, we cannot determine whether the changes in hippocampal NAA reflected a specific mechanism of riluzole or epiphenomenal symptom improvement, due to lack of placebo control. This concern is particularly relevant for GAD clinical trials, which are associated with high placebo responsivity (40). Secondly, the relatively small sample size of healthy comparison subjects and riluzole non-responders likely limited the power to detect significant differences in hippocampal (and prefrontal cortical) neurochemistry at any time point. Finally, the confounding effects of partial volume averaging cannot be ruled out, since tissue segmentation was not performed due to lack of volumetric MRI data on the subjects at each of the three time points. However, even after taking into account a 40% broadening due to PSF, our voxels were still sufficiently small to be contained within the hippocampal ROIs, thereby minimizing tissue heterogeneity. Furthermore, since this study compared within-subject NAA changes over time, with each subject effectively serving as his or her own control, the possibility that the variability in NAA over these 3 time points would be due to significant partial volume effects appears remote.
In conclusion, we have identified hippocampal metabolic correlates of anxiolytic response to the glutamate-modulating agent riluzole in GAD. We suggest that riluzole might be efficacious for GAD (and subtypes of mood disorders) in part due to reduced glutamate excitotoxicity and enhancement of hippocampal neuroplasticity. Further investigation of neuroimaging biomarkers of response remains an important goal for development of novel treatments for these conditions.
Acknowledgments
Support/Acknowledgments: Supported by Young Investigators Award, the National Alliance for Research in Schizophrenia and Depression, Sackler Institute of Columbia University, and National Institute of Mental Health Career Development Award K23-MH-069656. We thank Jonathan Amiel, M.D., Steven Dashnaw, Heidi Fitterling, M.P.H., Jack Gorman, M.D., Joy Hirsch, Ph.D., Kathryn Keegan, and Harold Sackeim, Ph.D., for their valuable contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Previous Presentation: An earlier version of this study was presented at the annual meetings of the International Society for Magnetic Resonance in Medicine, Miami, May 10-14, 2005, and the Society of Biological Psychiatry, Atlanta, May 19-21, 2005.
Financial Disclosures:
Dr. Mathew reports having received lecture or consulting fees over the last two years from AstraZeneca, Cephalon Inc., Pfizer Pharmaceuticals, and Takeda Industries. He has also accepted research grant support from Alexza Pharmaceuticals and Predix Pharmaceuticals. He has filed for a patent for the use of ketamine for the treatment of depression.
Dr. Coplan reports having received lecture or consulting fees over the last two years from AstraZeneca, Bristol-Meyers Squibb, Glaxo-SmithKline, and Pfizer Pharmaceuticals, and has accepted research grant support from Alexza Pharmaceuticals, Glaxo-SmithKline, and Pfizer Pharmaceuticals.
Dr. Charney reports consulting fees over the last two years from AstraZeneca, Bristol-Myers Squibb Company, Cyberonics, Forest Laboratories, Inc., GeneLogic, Inc., Institute of Medicine, Neuroscience Education Institute, Novartis Pharmaceuticals Corporation, Organon International Inc., and Quintiles, Inc. He has filed for a patent for the use of ketamine for the treatment of depression.
Ms. Price, Ms. Mao, and Drs. Smith and Shungu report no biomedical financial interests or potential conflicts of interest.
References
- 1.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 2.Zarate CA, Jr, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161:171–174. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- 3.Zarate CA, Jr, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57:430–432. doi: 10.1016/j.biopsych.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Sanacora G, Kendell SF, Fenton L, Coric V, Krystal JH. Riluzole augmentation for treatment-resistant depression. Am J Psychiatry. 2004;161:2132. doi: 10.1176/appi.ajp.161.11.2132. [DOI] [PubMed] [Google Scholar]
- 5.Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, Krystal JH. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry. 2007;61:822–825. doi: 10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew SJ, Amiel JM, Coplan JD, Fitterling HA, Sackeim HA, Gorman JM. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. 2005;162:2379–2381. doi: 10.1176/appi.ajp.162.12.2379. [DOI] [PubMed] [Google Scholar]
- 7.Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32:50–55. doi: 10.1111/j.2044-8341.1959.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 8.Coyle J, Leski M, Morrison J. The diverse roles of L-glutamic acid in brain signal transduction. In: Davis K, Charney D, Coyle J, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. Lippincott, Williams, & Wilkins; Philadelphia: 2002. pp. 71–90. [Google Scholar]
- 9.Moghaddam B, Bolinao ML, Stein-Behrens B, Sapolsky R. Glucocorticoids mediate the stress-induced extracellular accumulation of glutamate. Brain Res. 1994;655:251–254. doi: 10.1016/0006-8993(94)91622-5. [DOI] [PubMed] [Google Scholar]
- 10.Sapolsky RM. The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry. 2000;48:755–765. doi: 10.1016/s0006-3223(00)00971-9. [DOI] [PubMed] [Google Scholar]
- 11.Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12:3567–3574. doi: 10.1046/j.1460-9568.2000.00242.x. [DOI] [PubMed] [Google Scholar]
- 12.Benoit E, Escande D. Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflugers Arch. 1991;419:603–609. doi: 10.1007/BF00370302. [DOI] [PubMed] [Google Scholar]
- 13.Risterucci C, Coccurello R, Banasr M, Stutzmann JM, Amalric M, Nieoullon A. The metabotropic glutamate receptor subtype 5 antagonist MPEP and the Na+ channel blocker riluzole show different neuroprotective profiles in reversing behavioral deficits induced by excitotoxic prefrontal cortex lesions. Neuroscience. 2006;137:211–220. doi: 10.1016/j.neuroscience.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 14.Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Kuno S. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. 2001;310:117–120. doi: 10.1016/s0304-3940(01)02098-5. [DOI] [PubMed] [Google Scholar]
- 16.Katoh-Semba R, Asano T, Ueda H, Morishita R, Takeuchi IK, Inaguma Y, Kato K. Riluzole enhances expression of brain-derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. FASEB J. 2002;16:1328–1330. doi: 10.1096/fj.02-0143fje. [DOI] [PubMed] [Google Scholar]
- 17.Shortland PJ, Leinster VH, White W, Robson LG. Riluzole promotes cell survival and neurite outgrowth in rat sensory neurones in vitro. Eur J Neurosci. 2006;24:3343–3353. doi: 10.1111/j.1460-9568.2006.05218.x. [DOI] [PubMed] [Google Scholar]
- 18.Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z, et al. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32:793–802. doi: 10.1038/sj.npp.1301178. [DOI] [PubMed] [Google Scholar]
- 19.Kalra S, Cashman NR, Genge A, Arnold DL. Recovery of N-acetylaspartate in corticomotor neurons of patients with ALS after riluzole therapy. NeuroReport. 1998;9:1757–1761. doi: 10.1097/00001756-199806010-00016. [DOI] [PubMed] [Google Scholar]
- 20.Kalra S, Tai P, Genge A, Arnold DL. Rapid improvement in cortical neuronal integrity in amyotrophic lateral sclerosis detected by proton magnetic resonance spectroscopic imaging. J Neurol. 2006;253:1060–1063. doi: 10.1007/s00415-006-0162-7. [DOI] [PubMed] [Google Scholar]
- 21.Barker PB. N-acetyl aspartate—a neuronal marker? Ann Neurol. 2001;49:423–424. [PubMed] [Google Scholar]
- 22.Traber F, Block W, Freymann N, Gur O, Kucinski T, Hammen T, et al. A multicenter reproducibility study of single-voxel H-MRS of the medial temporal lobe. Eur Radiol. 2006;16:1096–1103. doi: 10.1007/s00330-005-0108-y. [DOI] [PubMed] [Google Scholar]
- 23.Jessen F, Traeber F, Freymann N, Maier W, Schild H-H, Heun R, Block W. A comparative study of the different N-acetylaspartate measures of the medial temporal lobe in Alzheimer's disease. Dement Geriatr Cogn Disord. 2005;20:178–183. doi: 10.1159/000087095. [DOI] [PubMed] [Google Scholar]
- 24.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hettema JM, Kuhn JW, Prescott CA, Kendler KS. The impact of generalized anxiety disorder and stressful life events on risk for major depressive episodes. Psychol Med. 2006;36:789–795. doi: 10.1017/S0033291706007367. [DOI] [PubMed] [Google Scholar]
- 26.Mathew SJ, Steinbugler M, Smith ELP. Neurochemistry of generalized anxiety disorder. In: Kinrys G, Renshaw PF, editors. Understanding Anxiety: Its Neurobiological Basis and Treatment. Taylor & Francis Group, LLC; New York: In press. [Google Scholar]
- 27.Sapolsky RM, Uno H, Rebert CS, Finch CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci. 1990;10:2897–2902. doi: 10.1523/JNEUROSCI.10-09-02897.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sapolsky RM. Why stress is bad for your brain. Science. 1996;273:749–750. doi: 10.1126/science.273.5276.749. [DOI] [PubMed] [Google Scholar]
- 29.Mathew SJ, Mao X, Coplan JD, Smith ELP, Sackeim HA, Gorman JM, Shungu DC. Dorsolateral prefrontal cortical pathology in generalized anxiety disorder: a proton magnetic resonance spectroscopic imaging study. Am J Psychiatry. 2004;161:1119–1121. doi: 10.1176/appi.ajp.161.6.1119. [DOI] [PubMed] [Google Scholar]
- 30.McClure EB, Monk CS, Nelson EE, Parrish JM, Adler A, Blair RJ, et al. Abnormal attention modulation of fear circuit function in pediatric generalized anxiety disorder. Arch Gen Psychiatry. 2007;64:97–106. doi: 10.1001/archpsyc.64.1.97. [DOI] [PubMed] [Google Scholar]
- 31.Hasler G, Fromm S, Alvarez RP, Luckenbaugh DA, Drevets WC, Grillon C. Cerebral blood flow in immediate and sustained anxiety. J Neurosci. 2007;27:6313–9. doi: 10.1523/JNEUROSCI.5369-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phan KL, Fitzgerald DA, Cortese BM, Seraji-Bozorgzad N, Tancer ME, Moore GJ. Anterior cingulate neurochemistry in social anxiety disorder: 1H-MRS at 4 Tesla. NeuroReport. 2005;16:183–186. doi: 10.1097/00001756-200502080-00024. [DOI] [PubMed] [Google Scholar]
- 33.Coplan JD, Mathew SJ, Mao X, Smith ELP, Hof PR, Coplan PM, et al. Decreased choline and creatine concentrations in centrum semiovale in patients with generalized anxiety disorder: relationship to IQ and early trauma. Psychiatry Res. 2006;147:27–39. doi: 10.1016/j.pscychresns.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 34.First MB, Spitzer RL, Williams JBW, Gibbon M. Structured Clinical Interview for DSM-IV Axis I Disorders – Patient Edition. New York Psychiatric Institute; New York: 1995. [Google Scholar]
- 35.Meyer TJ, Miller ML, Metzger RL, Borkovec TD. Development and validation of the Penn State Worry Questionnaire. Behav Res Ther. 1990;28:487–495. doi: 10.1016/0005-7967(90)90135-6. [DOI] [PubMed] [Google Scholar]
- 36.Hamilton M. A rating scale for depression. J Neurol Neurosur. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spitzer RL, Williams JB, Gibbon M. Structured Clinical Interview for DSM-IV – Non-patient Version. New York State Psychiatric Institute; New York: 1996. [Google Scholar]
- 38.Duyn JH, Gillen J, Sobering G, van Zijl PC, Moonen CT. Multisection proton MR spectroscopic imaging of the brain. Radiology. 1993;188:277–282. doi: 10.1148/radiology.188.1.8511313. [DOI] [PubMed] [Google Scholar]
- 39.Soher BJ, van Zijl PC, Duyn JH, Barker PB. Quantitative proton MR spectroscopic imaging of the human brain. Magn Reson Med. 1996;35:356–363. doi: 10.1002/mrm.1910350313. [DOI] [PubMed] [Google Scholar]
- 40.Mathew SJ, Hoffman EJ. Pharmacotherapy of generalized anxiety disorder. In: Antony MM, Stein MB, editors. Handbook of Anxiety and the Anxiety Disorders. Oxford University Press; New York: In press. [Google Scholar]
- 41.Behar E, Alcaine O, Zuellig AR, Borkovec TD. Screening for generalized anxiety disorder using the Penn State Worry Questionnaire: a receiver operating characteristic analysis. J Behav Ther Exp Psychiatry. 2003;34:25–43. doi: 10.1016/s0005-7916(03)00004-1. [DOI] [PubMed] [Google Scholar]
- 42.Sattler R, Rothstein JD. Targeting an old mechanism in a new disease—protection of glutamatergic dysfunction in depression. Biol Psychiatry. 2007;61:137–138. doi: 10.1016/j.biopsych.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 43.Zarate CA, Jr, Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry. 2006;59:1006–1020. doi: 10.1016/j.biopsych.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 44.Mesulam MM. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 45.Baslow MH. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem Res. 2003;28:941–953. doi: 10.1023/a:1023250721185. [DOI] [PubMed] [Google Scholar]
- 46.Marenco S, Steele SU, Egan MF, Goldberg TE, Straub RE, Sharrief AZ, Weinberger DR. Effect of metabotropic glutamate receptor 3 genotype on N-acetylaspartate measures in the dorsolateral prefrontal cortex. Am J Psychiatry. 2006;163:740–742. doi: 10.1176/appi.ajp.163.4.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frodl T, Schule C, Schmitt G, Born C, Baghai T, Zill P, et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch Gen Psychiatry. 2007;64:410–416. doi: 10.1001/archpsyc.64.4.410. [DOI] [PubMed] [Google Scholar]
- 48.Pouwels PJW, Frahm J. Differential distribution of NAA and NAAG in human brain as determined by quantitative localized proton MRS. Magn Reson Med. 1998;10:73–78. doi: 10.1002/(sici)1099-1492(199704)10:2<73::aid-nbm448>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Vrenken H, Barkhof F, Uitdehaag BM, Castelijns JA, Polman CH, Pouwels PJ. MR spectroscopic evidence for glial increase but not for neuro-axonal damage in MS normal-appearing white matter. Magn Reson Med. 2005;53:256–266. doi: 10.1002/mrm.20366. [DOI] [PubMed] [Google Scholar]
- 50.Clementi V, Tonon C, Lodi R, Malucelli E, Barbiroli B, Iotti S. Assessment of glutamate and glutamine contribution to in vivo N-acetylaspartate quantification in human brain by 1H-magnetic resonance spectroscopy. Magn Reson Med. 2005;54:1333–1339. doi: 10.1002/mrm.20703. [DOI] [PubMed] [Google Scholar]
- 51.Coupland NJ, Ogilvie CJ, Hegadoren KM, Seres P, Hanstock CC, Allen PS. Decreased prefrontal myo-inositol in major depressive disorder. Biol Psychiatry. 2005;57:1526–1534. doi: 10.1016/j.biopsych.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 52.Ham BJ, Chey J, Yoon SJ, Sung Y, Jeong DU, Ju Kim S, et al. Decreased N-acetylaspartate levels in anterior cingulate and hippocampus in subjects with post-traumatic stress disorder: a proton magnetic resonance spectroscopy study. Eur J Neurosci. 2007;25:324–329. doi: 10.1111/j.1460-9568.2006.05253.x. [DOI] [PubMed] [Google Scholar]
- 53.Schuff N, Neylan TC, Lenoci MA, Du AT, Weiss DS, Marmar CR, Weiner MW. Decreased hippocampal N-acetylaspartate in the absence of atrophy in posttraumatic stress disorder. Biol Psychiatry. 2001;50:952–959. doi: 10.1016/s0006-3223(01)01245-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freeman TW, Cardwell D, Karson CN, Komoroski RA. In vivo proton magnetic resonance spectroscopy of the medial temporal lobes of subjects with combat-related posttraumatic stress disorder. Magn Reson Med. 1998;40:66–71. doi: 10.1002/mrm.1910400110. [DOI] [PubMed] [Google Scholar]
- 55.Grachev ID, Apkarian AV. Anxiety in healthy humans is associated with orbital frontal chemistry. Mol Psychiatry. 2000;5:482–488. doi: 10.1038/sj.mp.4000778. [DOI] [PubMed] [Google Scholar]
- 56.Bertolino A, Callicott JH, Nawroz S, Mattay VS, Duyn JH, Tedeschi G, et al. Reproducibility of proton magnetic resonance spectroscopic imaging in patients with schizophrenia. Neuropsychopharmacology. 1998;18:1–9. doi: 10.1016/S0893-133X(97)00090-0. [DOI] [PubMed] [Google Scholar]