

Abstract
How transcription affects the way specific genes are arranged within the nucleus remains to be fully understood. We examine here whether transcription occurs in discrete sites (factories) containing the required machinery and whether these sites specialize in transcribing different genes. We cotransfected plasmids encoding a common origin of replication but different transcription units into cells, where they are assembled into minichromosomes that the cellular machinery replicates and transcribes. In cells containing thousands of minichromosomes, we found (using fluorescence in situ hybridization) active templates concentrated in only a few factories that transcribe particular units depending on the promoter type and the presence of an intron. Close proximity between similar transcription units, whether on two different minichromosomes or on host chromosomes and minichromosomes, is confirmed using chromosome conformation capture. We conclude that factories specialize in producing a particular type of transcript depending on promoter type and whether or not the gene contains an intron.
Introduction
It is widely assumed that RNA polymerases diffuse to their target promoters, wherever those promoters might be in the nuclei, before initiating. However, recent evidence suggests that polymerases active on different genes cluster into “factories.” Clustering ensures high local concentrations and, thus, efficient interaction, and a promoter would then have to diffuse to a factory before it could be transcribed (Cook, 1999). Nucleolar factories containing polymerase I provide the prototypic example; only ribosomal DNA associated with a nucleolus is transcribed, and newly made ribosomal RNA is surrounded by all the machinery required for assembly into a ribosome. Active forms of polymerases II and III are also concentrated in factories (Pombo et al., 1999), and the application of new techniques show that their templates tend to cluster when transcribed. For example, the mouse Hbb-b1 (β-globin) gene lies 40–60 kbp from its locus control region (LCR) on the genetic map and ∼25 Mbp from Eraf. But in 3D nuclear space, chromosome conformation capture (3C) and FISH reveal that Hbb1 lies close to the LCR and Eraf, as well as many other parts of the genome, but only when contacting regions are transcribed (Osborne et al., 2004; Simonis et al., 2006). The LCR seems to nucleate a “hub” or factory that ties the locus in loops and facilitates expression of globin-related genes (Chakalova et al., 2005; de Laat, 2007; Ohlsson and Gondor, 2007). Other evidence points to factories specializing in the transcription of specific gene subsets (Pombo et al., 1998; Frey et al., 1999; Thompson et al., 2003; Bartlett et al., 2006; Osborne et al., 2007).
We used replicating minichromosomes as probes to examine whether transcription occurs in factories. Using FISH and 3C, we found that although a cell may contain thousands of minichromosomes, essentially all nascent minichromosomal RNA is concentrated in a few foci: the factories. These factories specialize in producing particular types of transcripts depending on the promoter type and whether or not the gene contains an intron.
Results
Strategy
Our strategy (Fig. 1 A) was to cotransfect plasmids encoding the SV40 origin of replication (ori) and different transcription units into a monkey line that expresses the SV40 T antigen (Mellon et al., 1981). Previous work has shown that the plasmid DNA is assembled into nucleosomes, and the resulting minichromosomes are replicated and transcribed by the cellular machinery (Mellon et al., 1981; Jackson and Cook, 1993; Dean, 1997). We then examined if transcribing minichromosomes are spread throughout nuclei or concentrated in the same or different factories. Plasmids are named by promoter type, coding sequence, and whether or not they contain an intron and a 3′ end (Fig. 1 B). For example, plasmid IICMV,EGFPV,pA possesses the polymerase II cytomegalovirus (CMV) promoter driving EGFP with an intron (V) and polyadenylation (pA) signal, whereas 0,EGFP,pA lacks a promoter and intron.
Figure 1.

Approach. (A) Plasmids were cotransfected into cells, where they replicate, and we asked: Are the resulting minichromosomes spread throughout nuclei, or targeted to the same or different factories? (B) Different promoters, coding regions, introns, and 3′ ends (gray regions) are inserted into a common backbone (with SV40 ori + early promoter driving Neor).
Minichromosomes and their transcripts are concentrated in foci
Quantitative “blotting” and PCR show that the minichromosomal copy number increases progressively from the level seen at 8 h (when some naked input DNA remains), and by 24 h, at least 8,000 new copies are generated (Fig. 2 A). All plasmids replicate similarly (Fig. 2 A). Transcript copy numbers also increase, but promoters and introns affect levels (Fig. 2 B). DNA FISH reveals that most minichromosomes are concentrated in ∼20 bright nuclear foci that increase in intensity (Fig. 2, C and D; Dean, 1997). Intensities are normalized relative to plastic beads (Fig. 2 C, inset), so results from different experiments can be compared. Fewer than 5% of the minichromosomes were concatenated and/or replicating (unpublished data; Jackson and Cook, 1993) and therefore bound either to one another or to replication factories. Therefore, something must confine the ∼8,000 minichromosomes seen at 24 h to a few foci. As most are not diffusely spread, we can eliminate possibility 1 in Fig. 1 A.
Figure 2.

Replicating minichromosomes are concentrated in few foci. Cells were cotransfected with IICMV,DsRed,pA (so transfection efficiencies could be monitored by FACS using DsRed fluorescence) and another encoding EGFP as indicated; after 8 h, cells were replated (to wash away input) and regrown for 4, 16, and 28 h. (A) The DNA copy number of IICMV,EGFP,pA per transfected cell—determined by reference to known amounts of pure plasmid DNA (by blotting using a “Hirt” extract and qPCR using total DNA)—increases above the maximum possible background due to input (upper limit of gray area). Numbers (24 h) obtained in analogous experiments for I45S,EGFP,pA, III7SK,EGFP,pT, and IICMV,EGFPV,pA were similar (i.e., 23,000 ± 2,000, 18,000 ± 3,000, and 21,000 ± 3,000, respectively; one-way ANOVA, P > 0.05). (B) Promoters and introns affect the RNA copy number per transfected cell (determined using qRT-PCR by reference to known amounts of pure RNA). (C) DNA FISH shows that minichromosomes are concentrated in ∼20 foci. (inset) Fluorescent bead used for normalization. Bar, 2.5 μm. (D) Mean intensities (+SD) of pixels in or outside foci in 100 images like those in C were normalized relative to the beads; most signal is in the foci, and this increases with time.
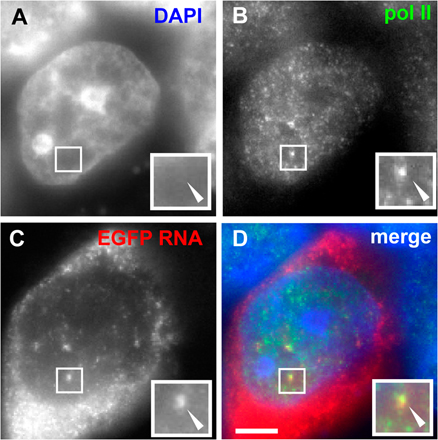
We next examined whether minichromosomes are transcribed in factories. We transfect IICMV,DsRed,pA, which encodes both a CMV promoter (driving DsRed) and an SV40 early promoter, on the common backbone (driving Neor; Fig. 1 B); the latter is down-regulated under these conditions (Myers et al., 1981; Dynan and Tjian, 1985). RNA FISH shows that both DsRed and Neor transcripts are found mainly in the cytoplasm, but some are concentrated in nuclear foci against a general nuclear background (Fig. 3 A, i–iv; foci can only be seen clearly in the insets). Many studies have shown that such nuclear foci mark the high concentration of RNA at transcription sites, whereas the general background represents completed transcripts on their way to the cytoplasm (Dirks et al., 1993; Levsky et al., 2002). Immuno-FISH confirms that such foci colocalize with high concentrations of the active form of RNA polymerase II, which is detected using an antibody directed against phospho-Ser2 in the heptad repeats in the C-terminal domain of the largest catalytic subunit (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200710053/DC1). Most RNA in such foci is nascent (i.e., still associated with engaged polymerases), as transcripts leave transcription sites so soon after the polymerase terminates (Iborra et al., 1998).
Figure 3.

RNA FISH shows nascent minichromosomal RNA in few nuclear foci. Cells were transfected and grown for 24 h, minichromosomal transcripts were detected by RNA FISH, DNA was counterstained with DAPI, and images were collected. (A) Two sets of four views of one field are shown. (bottom) Percentages (± SD) of green foci that overlap red foci, and vice versa. (i–iv) Transfection with IICMV,DsRed,pA. An untransfected cell (arrowhead) contains no DsRed or Neor transcripts. The other contains many cytoplasmic DsRed transcripts but few Neor transcripts; its nucleus contains some red and green foci (marking nascent RNA at transcription sites) against a general background (marking transcripts on their way to the cytoplasm). (insets) Two foci with both red and green fluorescence; this is expected, as the plasmid encodes both DsRed and Neor (on the backbone). (v–viii) Cotransfection with IICMV,DsRed,pA and IICMV,EGFP,pA, which differ solely in coding region. The two central (transfected) cells contain DsRed and EGFP RNA mainly in the cytoplasm, with some in the nuclear foci. (insets) Nuclear focus with both types of RNA. Insets show an enlarged view of the boxed portions. (B) Discriminating between nuclear foci and background. Intensities are expressed relative to those given by fluorescent reference beads, and the fraction of foci in 200 cells with relative intensities of 0–0.05, 0.06–0.1, etc., is indicated. Promoterless 0,EGFP,pA gives faint EGFP signal due to autofluorescence (equivalent to that seen in mock-transfected cells, not depicted; gray bar) and read-through from the SV40 early promoter into EGFP (red bars). For plasmids with promoters (e.g., IICMV,EGFP,pA), only foci with intensities greater than the maximum read-through (green dotted line) were considered. (C) Cells were transfected with IICMV,EGFP,pA and incubated with or without α-amanitin and RNase, DNA was stained with DAPI, and EGFP transcripts were detected; the treatments abolish EGFP signal. Three sets of two views of one field are shown. Bars: (A) 5 μm; (C) 10 μm.
We distinguished a nuclear focus from the background using two criteria. A focus should have an intensity greater than a threshold defined as follows. As some DsRed transcripts might be made by read-through from the SV40 promoter on the backbone (despite down-regulation and the presence of an efficient polyadenylation signal), we determined the extent of this using 0,EGFP,pA. As EGFP lacks a promoter, any EGFP RNA seen must result from read-through from the SV40 promoter. EGFP signal is again normalized relative to the intensity given by reference beads; it is just above (Fig. 3 B, red bars) the irreducible background caused by the autofluorescence seen in mock-transfected cells (Fig. 3 B; gray bars). We require foci to have intensities above a threshold just above the maximum signal given by this read-through (i.e., above the green dotted line in Fig. 3 B). We also require a focus to occupy >4 pixels (where a pixel is 100 × 100 nm and little signal is then discarded, as such foci contain >85% nuclear signal above the threshold). A nucleus transfected with IICMV,EGFP,pA typically contains 23 ± 6 foci (mean area of 7 ± 3 pixels) defined in this way. Such foci are not seen if cells are treated after fixation with RNase or before fixation with an inhibitor of RNA polymerase II: α-amanitin (Fig. 3 C).
Inspection of the two nuclear foci in the inset in Fig. 3 A (ii–iv) reveals that each contains red and green signal. We deem signals to colocalize if ≥25% of a focus of either color overlaps a focus of the other color; then, 95% EGFP foci colocalize with DsRed foci, and vice versa (Fig. 3 A, iv, bottom). As expected, two units on one minichromosome driven by similar viral promoters are transcribed in the same place. Transcripts from the (down-regulated) SV40 promoter are not considered from now on, as Neor probes are not used and read-through is below our threshold.
Minichromosomes with identical promoters are targeted to the same factories
Two minichromosomes identical except for coding region were now compared: IICMV,EGFP,pA and IICMV,DsRed,pA (Fig. 3 A,v–viii). As before, transfected cells contained high concentrations of both types of mature mRNA in the cytoplasm as well as nuclear foci against the nuclear background. 78% of DsRed foci colocalize with EGFP foci, and vice versa; minichromosomes with identical promoters are transcribed in the same factories.
Different polymerase II units target minichromosomes to different factories
We next examined two very different polymerase II units. IICMV,DsRed,pA encodes a “standard” intronless unit, whereas IIU2,U2G,3′box encodes a structural U2 RNA (which can be differentiated from host U2 RNA because it is marked by a globin sequence) and possesses its own special enhancers, promoter, and 3′ end (Cuello et al., 1999; Smith and Lawrence, 2000; Matera et al., 2007). After cotransfection, most standard (DsRed) transcripts are cytoplasmic, whereas most U2G transcripts are nuclear. This is expected; standard messages are exported to accumulate in the cytoplasm, whereas U2G transcripts are detected using a probe complementary to sequences in the preU2 RNA that are removed rapidly and degraded (Medlin et al., 2003). Most nuclear RNA of both types is again found in foci, but now ≤6% foci colocalize (Fig. 4, A–D); U2G units are transcribed in different factories from CMV units, which is consistent with possibility 3 in Fig. 1 A. There are 23 ± 6 and 15 ± 4 DsRed and U2G nuclear foci of similar area (7 ± 3 pixels), respectively.
Figure 4.

Promoters and introns target minichromosomes to specific factories. Cells (cotransfected with IICMV,DsRed,pA and the plasmid indicated) were grown (24 h), transcripts were detected by RNA FISH, and DNA was counterstained with DAPI. Four views of one field for each pair are shown. (bottom) Percentages (± SD) of green foci that overlap red foci, and vice versa. Arrows indicate noncolocalizing foci and arrowheads indicate colocalizing foci. Insets show an enlarged view of the boxed portions. (A–D) Different polymerase II units. One transfected cell contains DsRed but not U2G transcripts in the cytoplasm; both are found in nuclear foci. (insets) Nuclear foci do not colocalize (confirmed by low percentages shown on the bottom). (E–H). Plus/minus intron. Both cells contain DsRed and EGFP transcripts in cytoplasm and nucleus. (insets) The top focus contains both types, whereas the bottom shows only EGFP RNA (infrequent colocalization is reflected by low percentages on the bottom). (I–L) I + II. The central transfected cell contains DsRed but not EGFP RNA in the cytoplasm; nuclear foci do not colocalize. (insets) Foci do not colocalize (as confirmed by the low percentages shown below). (M–P) II + III. The transfected cell (left) contains both types of RNA in cytoplasm and nucleus. (insets) Foci do not colocalize (as confirmed by the low percentages shown below). Bar, 5 μm.
This experiment was repeated using IICMV,DsRed,pA and IIU2p−,U2G,3′box, the only difference being the replacement of 12 bp in the U2 promoter with an irrelevant sequence that destroys promoter activity (Cuello et al., 1999). No U2G transcripts were then detected (unpublished data), so those seen previously cannot be produced from a cryptic promoter.
Introducing an intron modifies targeting
The introduction of an intron also affects localization. Thus, transcripts copied from IICMV,DsRed,pA and IICMV,EGFPV,pA are both mainly cytoplasmic (Fig. 4, E–H), and quantitative RT-PCR (qRT-PCR) shows that those from the intron-containing unit are present in higher quantities (Fig. 2 B; Brinster et al., 1988). Protein levels are also higher, as the ratio of EGFP–DsRed (protein) fluorescence (detected using FACS) in cells transfected with IICMV,EGFPV,pA + IICMV,DsRed,pA is greater than that given by IICMV,EGFP,pA + IICMV,DsRed,pA (unpublished data). Both intron-containing and intronless nascent RNA is also found in ∼20 nuclear foci of similar size and intensity (there are 21 ± 4 and 23 ± 6 intron-containing and intronless nuclear foci of 6 ± 3 and 7 ± 3 pixels, respectively.) Although flanked by the same promoter and 3′ signals, they are usually found in different factories (Fig. 4, E–H). Immuno-FISH shows that intron-containing foci colocalize more often with splicing factor SC-35 than their counterparts without introns; however, neither type of transcript generally colocalizes with the very brightest “splicing speckles,” which are known to be transcriptionally inactive (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200710053/DC1; see Lamond and Spector, 2003; Xie et al., 2006).
Polymerase I, II, and III units are transcribed in different factories
We next analyzed promoters transcribed by the three different nuclear polymerases. Few transcripts copied using the polymerase I promoter in I45S,EGFP,pA are found in prominent nucleoli (Fig. 4 K) or the cytoplasm; instead, most are found in 38 ± 13 large nuclear foci (area of 30 ± 13 pixels). These nuclear polymerase I transcripts often colocalize with upstream binding factor (UBF) and are still seen when transcription by polymerase II is inhibited with α-amanitin (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200710053/DC1). This is consistent with what has been seen previously. When UBF-binding sites are inserted ectopically into host chromosomes, they organize many nonnucleolar foci—or “pseudonucleoli”—which, in this case, are transcriptionally inactive, as they lack polymerase I promoters (Mais et al., 2005). However, few of our (nascent) polymerase I transcripts colocalize with (nascent) polymerase II transcripts (Fig. 4, I–L). Similarly, few transcripts copied from a polymerase III unit are cytoplasmic (Fig. 4 O), and nuclei typically contain 16 ± 5 small foci (area of 6 ± 3 pixels). Few of these colocalize with polymerase II foci, and vice versa (Fig. 4, M–P). There are more polymerase II transcripts than polymerase I or III transcripts (Fig. 2 B), so it was possible that we might have missed some polymerase I or III foci. However, differential detection is unlikely to underlie the low colocalizations seen here, as the same values are obtained when we selected green foci and then determined how many also contain red foci, and vice versa (Fig. 4, values given at the bottom). These results are again consistent with possibility 3 in Fig. 1 A, and with the finding that some factories contain nascent RNA made by polymerase II (but not III), whereas others contain transcripts made by polymerase III (but not II; Pombo et al., 1999).
Chromatin immunoprecipitation (ChIP)-3C confirms that minichromosomes with similar promoters lie together
We now used 3C to compare the relative proximity of two plasmids when they encode similar or different promoters. Preliminary experiments (unpublished data) revealed that DsRed and EGFP lie closer (P < 0.05, Mann-Whitney test) in a II + II combination (i.e., IICMV,DsRed,pA + IICMV,EGFP,pA) than in a II + III combination (IICMV,DsRed,pA + III7SK,EGFP,pT). As inactive DNA (i.e., naked input and/or minichromosomes in the diffuse nuclear background) probably masks larger differences, we repeated the experiment after selecting the transcriptionally active fraction by ChIP using an antibody directed against trimethyl K4 in histone H3 (Barski et al., 2007). Then, DsRed–EGFP 3C products were seen in both the supernatant and pellet (Fig. 5 A, rows 1 and 3), which is consistent with the presence of inactive DNA in the supernatant. More such products were seen in the pellet (the active fraction) when promoters were similar (Fig. 5 A, row 3). In contrast, ALDOA is an active housekeeping gene on the host chromosome and no ALDOA–ALDOA 3C products are seen in the supernatant (Fig. 5 A, row 2); moreover, pellets from both combinations yield similar levels (Fig. 5 A, row 5). The mean of four independent experiments confirms that the II + II combination gives sixfold more DsRed–EGFP 3C product than the II + III combination (Fig. 5 A, row 7). We conclude that minichromosomes bearing similar promoters are more likely to be together, which confirms the FISH results.
Figure 5.

ChIP-3C shows that similar genes on mini- and host chromosomes lie together. Cells were transfected, grown for 24 (rows 1–7) or 8 h (rows 8–13), and treated with or without cross-linker. Active and inactive chromatin was separated by ChIP (using an antibody against trimethyl K4 in H3) and the proximity between selected genes was assessed by 3C. Images illustrate gels containing amplimers, whereas rows 7 and 13 give the relative cross-linking frequency (the amounts of 3C amplimers in bands were determined by reference to equivalent weights of DNA and normalized relative to ALDOA–ALDOA levels and the PCR control for amplification efficiency). ALDOA is an active housekeeping gene on the host chromosome. (A) Minichromosome–minichromosome. Cotransfection with IICMV,DsRed,pA + IICMV,EGFP,pA (II + II) or IICMV,DsRed,pA + III7SK,EGFP,pT (II + III). DsRed–EGFP 3C products were obtained from both supernatant and pellet (rows 1 and 3), which is consistent with naked input and/or active minichromosomes in the supernatant, and they were seen only after cross-linking (X-link; rows 1 and 4). More are seen in the pellet when promoters are identical (row 3, compare left and right lanes; row 7, mean of four experiments); therefore, templates lie closer together in the II + II combination than in the II + III combination. ALDOA–ALDOA 3C products are seen only in the pellet (rows 2 and 5) and in similar amounts in both combinations (row 5). Amplimers are obtained in roughly equal amounts using the same primers and synthetic 3C templates made by ligating equimolar amounts of pure DNA from relevant genes (row 6). (B) Minichromosome–host chromosome. Transfection with IIU2,U2G,3′ box (IIU2) or IICMV,EGFP,pA (IICMV). More plasmid–host 3C products (detected using primers targeting the plasmid backbone and sequences flanking host U2 genes) are seen with IIU2 than with IICMV (row 1, compare left lane with the right; row 6, mean of four experiments); therefore, IIU2 lies closer to host U2 units than IICMV. In contrast, amplimers from ALDOA–ALDOA, synthetic 3C templates, and the Neor gene on the backbone (which reflect plasmid copy number) are all obtained in roughly equal amounts (rows 3–5).
Similar units on mini- and host chromosomes lie close together
Do active minichromosomes organize their own factories or share host factories? Plasmids encoding either the U2G or CMV unit, which are transcribed in different factories (Fig. 4, A–D), were transfected, and proximity between the common plasmid backbone and host U2 units was determined. Ninefold more plasmid–host 3C product was obtained with the U2G unit (Fig. 5 B). This is consistent with minichromosomal and host genes being transcribed in the same factory.
Discussion
Using replicating minichromosomes as probes, we examined whether transcription occurs in factories and whether factories specialize in producing particular types of transcripts. Plasmids encoding the SV40 origin of replication are transfected into cos7 cells, where they are assembled into minichromosomes to be replicated and transcribed by the cellular machinery. By 24 h, there are at least 8,000 minichromosomes per cell (Fig. 2 A), and DNA FISH shows these to be concentrated in only ∼20 foci (Fig. 2 C); this is in accord with results obtained with viruses (Pombo et al., 1994; Dean, 1997; Mearini et al., 2004). When two plasmids encoding different transcription units are cotransfected, RNA FISH reveals the two kinds of nascent RNA to be concentrated again in ∼20 foci (Figs. 4 and S1); this is consistent with transcription of many templates in one factory. Moreover, the two different types of transcripts are generally found in distinct foci. For example, nascent transcripts produced from CMV and U2 promoters are seen in nonoverlapping foci (Fig. 4, A–D), as are most of those with and without an intron (even though they are produced from the same CMV promoter; Fig. 4 E–H). Similarly, nascent transcripts copied from polymerase I, II, and III units are all found in their own distinct foci (Fig. 4, I–P). 3C confirms that templates bearing similar promoters are more likely to be together than those with different promoters (Fig. 5 A). All these results are consistent with different templates being transcribed in different factories that specialize in transcribing particular types of gene.
We can envisage that these factories might form in two extreme ways from host components: either minichromosomes diffuse to preexisting host factories that then transcribe both host chromosomes and minichromosomes; or they recruit host factors, perhaps even complete assemblies (Mitchell and Fraser, 2008), to generate factories that transcribe only minichromosomes. Our results are most simply explained if both processes occur simultaneously. On the one hand, 3C results reveal that minichromosomal U2G genes lie near host U2 genes (Fig. 5 B); this suggests that a U2 promoter on a minichromosome must first diffuse to a host U2 factory before it can be transcribed (Fig. 6 A). On the other hand, nascent transcripts from polymerase I units are found in nucleoplasmic foci (Fig. 4, I–L; and Fig. S3) known as pseudonucleoli (Mais et al., 2005). As host (polymerase I) units are only active in the nucleolus, we must assume that nucleoplasmic minichromosomes recruit host factors to nucleate these pseudonucleoli, which then recruit additional minichromosomes (as in Fig. 6 A).
Figure 6.

Models. (A) Targeting to factories. The promoter encoded by the minichromosome in the center can initiate only in a factory containing the appropriate machinery (i.e., one of similar color). As a result, it forms a cluster with two similar minichromosomes by association with the factory on the right. (B) Intron targeting. A newly replicated intron-containing minichromosome is “naïve” (left), and may either bind and initiate (incorrectly) in a nonsplicing (polymerase II) factory (bottom, blue) or a “splicing” factory (top, magenta), where it acquires a mark during splicing (e.g., a histone modification; right) that now targets it to the appropriate (splicing) factory. (C) Chromosome pairing. Just as similar (“homologous”) minichromosomes cluster (“pair”) in A, a similar process may underlie the pairing of homologous chromosomes in both somatic and meiotic cells. For example, during meiosis, homologues seek out and align with their partners before the close synapsis that occurs during recombination. It is now accepted that distinct mechanisms underlie alignment and synapsis, as homologues still align in mutants unable to carry out the later steps (McKee, 2004; Gerton, and Hawley, 2005). During alignment, homologues are transcriptionally active (Cook, 1997) so that each chromosome in the haploid set will possess a unique array of active transcription units running from telomere to telomere. Only the homologue will possess a similar array. Here, only one of the many loops associated with a factory is shown. The yellow promoter on maternal chromosome 2 (2m) is unlikely to bind to the green factory on maternal chromosome 1 (1m). But just as a factory of a particular type nucleates pairing between minichromosomes bearing similar transcription units, correct alignment begins when the yellow (2m) promoter binds to the yellow factory on its homologue (2p). Once transcription of the yellow promoter on 2m begins, 2m and 2p become temporarily tethered together, and this will increase the chances that adjacent promoters bind to homologous factories (i.e, the gray unit on 2m with the gray factory on 2p, etc.). As a result, 2m and 2p eventually become zipped together (arrowhead) and thus aligned.
These results beg many other questions. For example, how many different types of factory are there? In this preliminary screen, we found five different kinds (Fig. 4), and we expect to find more. How might targeting be achieved? There is some evidence that certain factories contain high concentrations of particular factors (Bartlett et al., 2006). Then, as a promoter diffuses through the nucleus, it might only bind stably to a factory containing the appropriate factors. This might then increase the frequency of initiation. But how could a gene with an intron recruit (or be recruited to) a different factory from an intronless gene (Fig. 4, E–H), especially when binding to a factory inevitably precedes synthesis of a transcript with an intron? One possibility is that the β-globin intron used, which is known to contain binding sites for transcription factors (Jackson et al., 1995; Wen et al., 2005), stabilizes binding to a factory with the appropriate factors, as described above. Another possibility, which is consistent with both the imprecise targeting of a minority of plasmids to certain factories (Fig. 4, E–H) and of the majority to another set of factories that contain the splicing component SC35 (Fig. S2), is as follows. A newly replicated intron-containing gene might be “naïve,” and bind to and initiate (incorrectly) in a “nonsplicing” (polymerase II) factory. However, on binding to a “splicing” factory, it might acquire some mark (e.g., a histone modification) that now targets it to the appropriate factory (Fig. 6 B). We also note that just as transcriptionally active minichromosomes “pair” by binding to the appropriate factories, a similar process might underlie the pairing of homologous chromosomes in both somatic and meiotic cells (Fig. 6 C; Cook, 1997).
Materials and methods
Oligonucleotides
Oligonucleotides (MWG Biotech AG; or Sigma-Aldrich) are listed in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200710053/DC1).
General procedures
General procedures have been described previously (Harlow and Lane, 1999; Sambrook and Russell, 2001).
Plasmids
Plasmids with a common backbone but encoding transcription units with different promoters, introns, and 3′ ends (Fig. 1 B) were constructed as follows: plasmid 0,EGFP,pA from pEGFP-N1 (Clontech Laboratories, Inc.) by deleting bp 8–591 (using oligo pair 3); I45S,EGFP,pA by inserting the 250-bp truncated human polymerase I promoter (prepared using oligo pair 1; Pleschka et al., 1996) between the AseI and NheI sites of 0,EGFP,pA; III7SK,EGFP,pA by inserting the 234-bp human 7SK promoter (Boyd et al., 1995) between the PstI and KpnI sites of 0,EGFP,pA; IIU2,U2G,3′ box and IIU2p−,U2G,3′box from pEGPF-C1 (Clontech Laboratories, Inc.) by deleting bp 8–1,367 and inserting the 623-bp U2G fragment (Medlin et al., 2003) or the promoterless U2G fragment (both prepared using oligo pair 4; Cuello et al., 1999) between the AseI and PstI sites; and IICMV,EGFPV,pA by inserting 925 bp of the HBB intron 2 (chromosome 11 positions 5,203,482–5,204,406 in the National Center for Biotechnology Information 36 assembly; prepared by PCR amplification of human DNA using oligo pair 2) between the HindIII and PstI sites of pEGPF-C1. Analogous constructs encoding DsRed were made from pDsRed-N1 (Clontech Laboratories, Inc.). Appropriate construction was confirmed by sequencing (using oligo pair 13). S. Murphy and N.J. Proudfoot (University of Oxford, Oxford, UK) provided constructs containing the U2G, 7SK, and HBB fragments.
Cell culture and transfection
Cos7 cells (Mellon et al., 1981) were grown in DME + 10% fetal calf serum (Invitrogen). Cells in 90-mm Petri dishes were transfected using FuGene6 (Roche) with 1.2 μg of plasmid DNA plus sheared salmon sperm DNA (Sigma-Aldrich) added to 6 μg. After 8 h, cells were trypsinized, washed, and replated to reduce background of input DNA. For Fig. 5 B (where we wished to detect a few interactions between minichromosomes and host chromosomes against a high minichromosome–minichromosome background), transfected cells were grown for 6 h and washed before cross-linking. Transfection efficiencies were determined by measuring EGFP levels by microscopy and FACS. Polymerase II was inhibited by incubation (16 h at 37°C) in 50 μg/ml α-amanitin (Sigma-Aldrich).
FACS
2 × 105 cells were washed in PBS, resuspended in 2 ml PBS, and analyzed on a flow cytometer (FACScan; Becton Dickinson). Cell debris/clusters were differentiated from single cells using forward and side scattering and eliminated from the analysis. Backgrounds were determined using mock-transfected cells and those transfected with IICMV,EGFP,pA or IICMV,DsRed,pA (Fig. 3 B). Data on 10,000 cells were analyzed using CellQuest software (Becton Dickinson), and the fractions of cells expressing red fluorescence (from the reference plasmid) and green fluorescence (from the test plasmid) were calculated.
DNA copy number determined by blotting and qPCR
Plasmid DNA from a “Hirt” extract (Hirt, 1967) of ∼107 cells was digested with the appropriate restriction enzyme, separated on an agarose gel, and blotted onto nylon. Probes were labeled with DIG (digoxigenin) using a DIG labeling kit (Roche) by random priming and hybridized at 65°C for 16 h in “Church” buffer (0.5 M sodium phosphate buffer, 1 mM EDTA, 1% bovine serum albumin, and 7% SDS, pH 7.2), then bound targets were detected using a DIG detection kit (Roche).
Total DNA from ∼107 cells was purified using Trizol (Invitrogen), and the EGFP copy number was determined by qPCR using a thermal cycler (Roter-Gene 3000; Corbett Life Science); signals were normalized relative to those given by GAPDH. In brief, each 25-μl reaction contained Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen) and 5 pmol of each primer (oligo pairs 11 and 12). Cycling conditions were: 50°C for 30 min and 95°C for 10 min followed by 40 cycles at 94°C for 15 s and 57.5°C for 30 s. Samples were run in triplicate and each experiment was repeated three times.
Absolute copy numbers were obtained by reference to the known numbers of pure plasmids. Concentration and purity was determined from the absorbance using a spectrometer (Nanodrop ND-100; Thermo Fisher Scientific).
mRNA copy number determined by qRT-PCR
Total RNA was purified using Trizol, and cDNA was prepared using the SuperScript III Platinum one-step system for RT-PCR (Invitrogen) and normalized to GAPDH mRNA levels in the same sample. qRT-PCR was conducted similarly to qPCR.
Absolute copy numbers were obtained by reference to known numbers of EGFP transcripts prepared by in vitro transcription using T7 RNA polymerase (Roche). The template (pcDNA-EGFP-N1) was constructed by inserting EGFP (from pEGFP-N1) between the HindIII and BamHI sites into pcDNA3 (Invitrogen). The integrity and size of RNA were verified by gel electrophoresis, and concentration and purity were determined from the absorbance (see DNA copy number...).
DNA FISH
Plasmid DNA was detected using DNA FISH (Brown, 2002). Cells were rinsed in PBS, fixed (17 min at 20°C) in 4% paraformaldehyde and 0.05% acetic acid in 0.15 M NaCl, washed three times (5 min at 20°C) in PBS, incubated (30 min at 37°C) with 50 μg/ml RNase (which removes all detectable EGFP RNA; Fig. 3 C), permeabilized (5 min at 37°C) in 0.01% pepsin, pH 2.0, rinsed in water treated with diethylpyrocarbonate, postfixed (5 min at 20°C) in 4% paraformaldehyde in PBS, washed (10 min at 20°C) in PBS, denatured (2 min at 70°C) in 70% deionized formamide and 2× SSC (0.03 M sodium citrate and 0.3 M NaCl, pH 7.0), and immediately dehydrated (for 3 min) by passage through 70, 90, and then 100% ice-cold ethanol before hybridization (16 h at 37°C) in a moist chamber. Hybridization mix contained 50 ng of DIG- and/or biotin-tagged probe, 25% deionized formamide, 2× SSC, 200 ng/μl of sheared salmon sperm DNA, 5× “Denhardt's” solution (0.1% Ficoll 400, 0.1% polyvinylpyrolidone, and 0.1% bovine serum albumin), 50 mM phosphate buffer (20 mM KH2PO4, and 30 mM KHPO4 × 2 H2O, pH 7.0), and 1 mM EDTA. Probes were prepared by DIG random priming (Roche) of pure plasmid DNA, purified using Sephadex G-50 spin columns (GE Healthcare), and denatured (10 min at 94°C) before hybridization. Cells were then washed three times in 2× SSC (15 min at 37°C) and twice (5 min at 20°C) in TST (0.15 M NaCl, 0.1 M Tris-HCl, and 0.05% Tween 20, pH 7.5), and incubated in 1.4% blocking reagent (Roche) in TS (0.15 M NaCl and 0.1 M Tris-HCl, pH 7.5). Immunodetection was performed either using sheep anti-DIG (60 min at 20°C; 1:250; Roche) followed by donkey anti–sheep Cy3 (30 min at 20°C; 1:250; Jackson ImmunoResearch Laboratories) or using mouse anti-biotin (30 min at 20°C; 1:250; Jackson ImmunoResearch Laboratories) followed by donkey anti–mouse Alexa 488 (30 min at 20°C; 1:200; Invitrogen) or donkey anti–mouse Cy5 (30 min at 20°C; 1:150; Jackson ImmunoResearch Laboratories). Cells were washed three times (5 min at 20°C) in PBS after incubation with each antibody and mounted in Vectashield (Vector Laboratories) containing 1 μg/ml DAPI (Sigma-Aldrich).
RNA FISH
Transcripts were detected using RNA FISH (Osborne et al., 2004). Cells were rinsed in PBS, fixed (17 min at 20°C) in 4% paraformaldehyde and 0.05% acetic acid in 0.15 M NaCl, rinsed three times (5 min at 20°C) in PBS, permeabilized (5 min at 37°C) in 0.01% pepsin, pH 2.0, rinsed in diethylpyrocarbonate-treated H2O, postfixed (5 min at 20°C) in 4% paraformaldehyde in PBS, and washed (10 min at 20°C) in PBS before hybridization (16 h at 37°C) in a moist chamber. Probes for EGFP, Neor, and DsRed were prepared by PCR followed by nick-translation (Roche) or asymmetrical PCR using oligo pairs 5–9. In some cases, three probes for each target were mixed. Biotinylated oligonucleotide probes (oligo 10) complementary to the preU2 sequence were used for U2G (Smith and Lawrence, 2000). Hybridization, subsequent washes, and immunodetection were performed as described for DNA FISH.
In analogous experiments using nonreplicating plasmids and different fixation conditions, two patterns of nuclear signal were seen (Binnie et al., 2006). Some transfected nuclei contained plasmid transcripts concentrated in foci (as we found), but others contained diffusely spread transcripts; as plasmid input increased, the fraction with the diffuse pattern increased. We also observed two patterns in preliminary experiments and were concerned that a diffuse pool might obscure some foci and/or artifactually aggregate to create foci. But as we used progressively harsher fixation, progressively more nuclear signal was detected (up to 1.5-fold), and the cell fraction with foci increased. Therefore, we used harsh conditions where detection is optimized, >85% nuclear signal was in the foci (see Results), and it is unlikely that much diffuse pool is extracted or aggregates (as nuclear signal does not fall or remain constant).
Immuno-RNA FISH
Cells were fixed (17 min at 20°C) in 4% paraformaldehyde and 250 mM Hepes, pH 8.0, permeabilized (5 min at 37°C) in 0.5% Triton X-100 and 0.5% saponin, postfixed and hybridized as with RNA FISH, and UBF or SC-35 were detected using mouse monoclonal anti-UBF (2 h at 20°C; 1:150; Santa Cruz Biotechnology, Inc.) or anti-SC-35 (2 h at 20°C; 1:200; Sigma-Aldrich) followed by donkey anti−mouse Alexa 647 (2 h at 20°C; 1:200; Invitrogen). Alternatively, RNA polymerase II was detected using a rabbit antibody directed against phospho-Ser2 in the heptads of the C-terminal domain of the largest catalytic subunit (2 h at 20°C; 1:250; Abcam) followed by donkey anti–rabbit Alexa 488 (2h at 20°C; 1:250; Invitrogen).
Microscopy
2.5 μm of orange or green intensity calibration beads (0.1% intensity; Invitrogen) were added to the mounting medium at 6 × 104/ml. Images were collected on a microscope (Axioplan 2; Carl Zeiss, Inc.) with a charge-coupled device camera (MicroMax 1024B; Princeton Instruments) using an exposure that gave a signal intensity of the beads of 200–255 on the grayscale. Signal intensities were measured using ImageJ (W.S. Rasband, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/) and normalized relative to the intensity of the reference beads.
3C and ChIP-3C
Relative proximity between templates was assessed using 3C (Dekker et al., 2002; de Laat, 2007; Ohlsson and Gondor, 2007). 4 × 107 cells were fixed (10 min at 20°C) in 50 ml DME + 10% FCS supplemented with 2% formaldehyde before glycine was added to 0.125 M. Cells were pelleted; lysed (90 min at 4°C) with vigorous stirring in 50 ml of 10 mM Tris-HCl, pH 8, 10 mM NaCl, 0.2% NP-40, and protease inhibitors (Sigma-Aldrich); repelleted; incubated (1 h at 37°C) with shaking in NEB buffer for BamHI (New England Biolabs, Inc.) + 0.3% SDS; and subsequently incubated in 1.8% Triton-X (1 h at 37°C). 106 nuclei were digested (16 h at 37°C) with 600 U BamHI and incubated (20 min at 65°C) in a final concentration of 1.6% SDS. Samples containing 2 μg DNA were incubated (1 h at 37°C) in 1% Triton X-100 and the volume was adjusted to 2 ml, then incubated (4 h at 16°C) in 2,000 U T4 DNA ligase (New England Biolabs, Inc.), incubated (16 h at 65°C) with 100 μg/ml proteinase K (Sigma-Aldrich), and treated (30 min at 37°C) with 0.4 μg/ml RNase A (Roche). DNA was then purified by phenol extraction and ethanol precipitation. PCR (30 cycles; controls showed this was in the linear range) was performed with serially diluted samples using oligo pairs 14–17, and PCR products were sized by gel electrophoresis. Expected ligation products were generally prepared in vitro from each pair of templates and used to confirm that amplification efficiencies were equivalent. Plasmid pTP18 (Pavelitz et al., 1995) and a BAC (RP-11-114A14; BACPAC Resources Center) were used as sources of host U2 and host ALDOA DNA, respectively. The relative cross-linking frequency is the intensity of the DsRed–EGFP or plasmid–host U2 band divided by the product of the intensities of the ALDOA–ALDOA and PCR control bands, normalized relative to equivalent values given by the reference experiment (i.e., II + III or CMV in Fig. 5).
For Fig. 5, transcriptionally active chromatin fractions were selected before 3C by incubation (16 h at 4°C) with antibodies against trimethyl K4 in histone H3 (Abcam). After immunoselection (Cai et al., 2006) on protein G–Sepharose beads, beads were washed once (at 4°C for 5 min) with low-stringency buffer (10 mM Tris-HCl, 150 mM NaCl, and 10 mM MgCl2, pH 7.5), twice (at 4°C for 5 min) with high-stringency buffer (10 mM Tris-HCl, 500 mM NaCl, and 10 mM MgCl2, pH 7.5), and once (at 4°C for 5 min) with low-stringency buffer. Beads were then incubated (37°C for 16 h) with 600 U of BamHI or PstI and the 3C ligation was performed. Plasmid copy numbers in experimental and reference samples were shown to be equivalent by PCR amplification of Neor using oligo pair 18 (Fig. 5 B, row 5).
Statistics
The Mann-Whitney test, one-way analysis of variance (ANOVA) and Student's t tests were performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, Inc.). Sample sizes were such that the last five samples analyzed yielded values that lay within 5% of the progressive mean.
Online supplemental material
Fig. S1 shows that nuclear foci containing high concentrations of minichromosomal transcripts colocalize with the active form of RNA polymerase II. Fig. S2 shows that intron-containing RNA colocalizes with faint SC-35 foci. Fig. S3 shows that transcripts made from a polymerase I promoter colocalize with UBF. Table S1 lists oligonucleotide sequences of primers, linkers, and probes. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710053/DC1.
Supplementary Material
Acknowledgments
We thank S. Murphy and N.J. Proudfoot for constructs.
We thank the University of Oxford (Clarendon Award), the UK Government (Overseas Research Student Award), and the E.P. Abraham Research Fund for support.
Abbreviations used in this paper: 3C, chromosome conformation capture; ChIP, chromatin immunoprecipitation; CMV, cytomegalovirus; DIG, digoxigenin; LCR, locus control region; qRT-PCR, quantitative RT-PCR; UBF, upstream binding factor.
References
- Barski, A., S. Cuddapah, K. Cui, T.Y. Roh, D.E. Schones, Z. Wang, G. Wei, I. Chepelev, and K. Zhao. 2007. High-resolution profiling of histone methylations in the human genome. Cell. 129:823–837. [DOI] [PubMed] [Google Scholar]
- Bartlett, J., J. Blagojevic, D. Carter, C. Eskiw, M. Fromaget, C. Job, M. Shamsher, I.F. Trindade, M. Xu, and P.R. Cook. 2006. Specialized transcription factories. Biochem. Soc. Symp. 73:67–75. [DOI] [PubMed] [Google Scholar]
- Binnie, A., P. Castelo-Branco, J. Monks, and N.J. Proudfoot. 2006. Homologous gene sequences mediate transcription-domain formation. J. Cell Sci. 119:3876–3887. [DOI] [PubMed] [Google Scholar]
- Boyd, D.C., P.C. Turner, N.J. Watkins, T. Gerster, and S. Murphy. 1995. Functional redundancy of promoter elements ensures efficient transcription of the human 7SK gene in vivo. J. Mol. Biol. 253:677–690. [DOI] [PubMed] [Google Scholar]
- Brinster, R.L., J.M. Allen, R.R. Behringer, R.E. Gelinas, and R.D. Palmiter. 1988. Introns increase transcriptional efficiency in transgenic mice. Proc. Natl. Acad. Sci. USA. 85:836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K. 2002. Visualizing nuclear proteins together with transcribed and inactive genes in structurally preserved cells. Methods. 26:10–18. [DOI] [PubMed] [Google Scholar]
- Cai, S., C.C. Lee, and T. Kohwi-Shigematsu. 2006. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 38:1278–1288. [DOI] [PubMed] [Google Scholar]
- Chakalova, L., E. Debrand, J.A. Mitchell, C.S. Osborne, and P. Fraser. 2005. Replication and transcription: shaping the landscape of the genome. Nat. Rev. Genet. 6:669–677. [DOI] [PubMed] [Google Scholar]
- Cook, P.R. 1997. The transcriptional basis of chromosome pairing. J. Cell Sci. 110:1033–1040. [DOI] [PubMed] [Google Scholar]
- Cook, P.R. 1999. The organization of replication and transcription. Science. 284:1790–1795. [DOI] [PubMed] [Google Scholar]
- Cuello, P., D.C. Boyd, M.J. Dye, N.J. Proudfoot, and S. Murphy. 1999. Transcription of the human U2 snRNA genes continues beyond the 3′ box in vivo. EMBO J. 18:2867–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, D.A. 1997. Import of plasmid DNA into the nucleus is sequence specific. Exp. Cell Res. 230:293–302. [DOI] [PubMed] [Google Scholar]
- Dekker, J., K. Rippe, M. Dekker, and N. Kleckner. 2002. Capturing chromosome conformation. Science. 295:1306–1311. [DOI] [PubMed] [Google Scholar]
- de Laat, W. 2007. Long-range DNA contacts: romance in the nucleus? Curr. Opin. Cell Biol. 19:317–320. [DOI] [PubMed] [Google Scholar]
- Dirks, R.W., F.M. van de Rijke, S. Fujishita, M. van der Ploeg, and A.K. Raap. 1993. Methodologies for specific intron and exon RNA localization in cultured cells by haptenized and fluorochromized probes. J. Cell Sci. 104:1187–1197. [DOI] [PubMed] [Google Scholar]
- Dynan, W.S., and R. Tjian. 1985. Control of eukaryotic messenger RNA synthesis by sequence-specific DNA-binding proteins. Nature. 316:774–778. [DOI] [PubMed] [Google Scholar]
- Frey, M.R., A.D. Bailey, A.M. Weiner, and A.G. Matera. 1999. Association of snRNA genes with coiled bodies is mediated by nascent snRNA transcripts. Curr. Biol. 9:126–135. [DOI] [PubMed] [Google Scholar]
- Gerton, J.L., and R.S. Hawley. 2005. Homologous chromosome interactions in meiosis: diversity amidst conservation. Nat. Rev. Genet. 6:477–487. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1999. Using Antibodies: a Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Springer Harbor, NY. 495 pp.
- Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365–369. [DOI] [PubMed] [Google Scholar]
- Iborra, F.J., D.A. Jackson, and P.R. Cook. 1998. The path of transcripts from extra-nucleolar synthetic sites to nuclear pores: transcripts in transit are concentrated in discrete structures containing SR proteins. J. Cell Sci. 111:2269–2282. [DOI] [PubMed] [Google Scholar]
- Jackson, D.A., and P.R. Cook. 1993. Transcriptionally active minichromosomes are attached transiently in nuclei through transcription units. J. Cell Sci. 105:1143–1150. [DOI] [PubMed] [Google Scholar]
- Jackson, C.E., D. O'Neill, and A. Bank. 1995. Nuclear factor binding sites in human beta globin IVS2. J. Biol. Chem. 270:28448–28456. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., and D.L. Spector. 2003. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4:605–612. [DOI] [PubMed] [Google Scholar]
- Levsky, J.M., S.M. Shenoy, R.C. Pezo, and R.H. Singer. 2002. Single-cell gene expression profiling. Science. 297:836–840. [DOI] [PubMed] [Google Scholar]
- Mais, C., J.E. Wright, J.L. Prieto, S.L. Raggett, and B. McStay. 2005. UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev. 19:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera, A.G., R.M. Terns, and M.P. Terns. 2007. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 8:209–220. [DOI] [PubMed] [Google Scholar]
- McKee, B.D. 2004. Homologous pairing and chromosome dynamics in meiosis and mitosis. Biochim. Biophys. Acta. 1677:165–180. [DOI] [PubMed] [Google Scholar]
- Mearini, G., P.E. Nielsen, and F.O. Fackelmayer. 2004. Localization and dynamics of small circular DNA in live mammalian nuclei. Nucleic Acids Res. 32:2642–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlin, J.E., P. Uguen, A. Taylor, D.L. Bentley, and S. Murphy. 2003. The C-terminal domain of pol II and a DRB-sensitive kinase are required for 3′ processing of U2 snRNA. EMBO J. 22:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon, P., V. Parker, Y. Gluzman, and T. Maniatis. 1981. Identification of DNA-sequences required for transcription of the human alpha-1-globin gene in a new Sv40 host-vector system. Cell. 27:279–288. [DOI] [PubMed] [Google Scholar]
- Mitchell, J.A., and P. Fraser. 2008. Transcription factories are nuclear subcompartments that remain in the absence of transcription. Genes Dev. 22:20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, R.M., D.C. Rio, A.K. Robbins, and R. Tjian. 1981. SV40 gene expression is modulated by the cooperative binding of T antigen to DNA. Cell. 25:373–384. [DOI] [PubMed] [Google Scholar]
- Ohlsson, R., and A. Gondor. 2007. The 4C technique: the ‘Rosetta stone’ for genome biology in 3D? Curr. Opin. Cell Biol. 19:321–325. [DOI] [PubMed] [Google Scholar]
- Osborne, C.S., L. Chakalova, K.E. Brown, D. Carter, A. Horton, E. Debrand, B. Goyenechea, J.A. Mitchell, S. Lopes, W. Reik, et al. 2004. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36:1065–1071. [DOI] [PubMed] [Google Scholar]
- Osborne, C.S., L. Chakalova, J.A. Mitchell, A. Horton, A.L. Wood, D.J. Bolland, A.E. Corcoran, and P. Fraser. 2007. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 5:e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelitz, T., L. Rusche, A.G. Matera, J.M. Scharf, and A.M. Weiner. 1995. Concerted evolution of the tandem array encoding primate U2 snRNA occurs in situ, without changing the cytological context of the RNU2 locus. EMBO J. 14:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleschka, S., R. Jaskunas, O.G. Engelhardt, T. Zurcher, P. Palese, and A. Garcia-Sastre. 1996. A plasmid-based reverse genetics system for influenza A virus. J. Virol. 70:4188–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo, A., J. Ferreira, E. Bridge, and M. Carmo-Fonseca. 1994. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J. 13:5075–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo, A., P. Cuello, W. Schul, J.-B. Yoon, R.G. Roeder, P.R. Cook, and S. Murphy. 1998. Regional and temporal specialization in the nucleus: a transcriptionally-active nuclear domain rich in PTF, Oct1 and PIKA antigens associates with specific chromosomes early in the cell cycle. EMBO J. 17:1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo, A., D.A. Jackson, M. Hollinshead, Z. Wang, R.G. Roeder, and P.R. Cook. 1999. Regional specialization in human nuclei: visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 18:2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., and D.W. Russell. 2001. Molecular Cloning. A Laboratory Manual. Third edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Simonis, M., P. Klous, E. Splinter, Y. Moshkin, R. Willemsen, E. de Wit, B. van Steensel, and W. de Laat. 2006. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 38:1348–1354. [DOI] [PubMed] [Google Scholar]
- Smith, K.P., and J.B. Lawrence. 2000. Interactions of U2 gene loci and their nuclear transcripts with Cajal (coiled) bodies: evidence for PreU2 within Cajal bodies. Mol. Biol. Cell. 11:2987–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, M., R.A. Haeusler, P.D. Good, and D.R. Engelke. 2003. Nucleolar clustering of dispersed tRNA genes. Science. 302:1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, J., S. Huang, H. Rogers, L.A. Dickinson, T. Kohwi-Shigematsu, and C.T. Noguchi. 2005. SATB1 family protein expressed during early erythroid differentiation modifies globin gene expression. Blood. 105:3330–3339. [DOI] [PubMed] [Google Scholar]
- Xie, S.Q., S. Martin, P.V. Guillot, D.L. Bentley, and A. Pombo. 2006. Splicing speckles are not reservoirs of RNA polymerase II, but contain an inactive form, phosphorylated on serine2 residues of the C-terminal domain. Mol. Biol. Cell. 17:1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}