

Abstract
Degradation of cytoplasmic components by autophagy requires the class III phosphatidylinositol 3 (PI(3))–kinase Vps34, but the mechanisms by which this kinase and its lipid product PI(3) phosphate (PI(3)P) promote autophagy are unclear. In mammalian cells, Vps34, with the proautophagic tumor suppressors Beclin1/Atg6, Bif-1, and UVRAG, forms a multiprotein complex that initiates autophagosome formation. Distinct Vps34 complexes also regulate endocytic processes that are critical for late-stage autophagosome-lysosome fusion. In contrast, Vps34 may also transduce activating nutrient signals to mammalian target of rapamycin (TOR), a negative regulator of autophagy. To determine potential in vivo functions of Vps34, we generated mutations in the single Drosophila melanogaster Vps34 orthologue, causing cell-autonomous disruption of autophagosome/autolysosome formation in larval fat body cells. Endocytosis is also disrupted in Vps34−/− animals, but we demonstrate that this does not account for their autophagy defect. Unexpectedly, TOR signaling is unaffected in Vps34 mutants, indicating that Vps34 does not act upstream of TOR in this system. Instead, we show that TOR/Atg1 signaling regulates the starvation-induced recruitment of PI(3)P to nascent autophagosomes. Our results suggest that Vps34 is regulated by TOR-dependent nutrient signals directly at sites of autophagosome formation.
Introduction
Engulfment of cytoplasmic material into specialized double-membrane vesicles known as autophagosomes is the defining feature of a process referred to as macroautophagy or simply autophagy. Subsequent fusion of autophagosomes with the endolysosomal network leads to hydrolytic degradation of the sequestered material. This process provides eukaryotic cells with a mechanism for cytoplasmic renewal by which they can rid themselves of defective organelles and protein complexes (Yorimitsu and Klionsky, 2005). In addition, nonselective autophagy can be induced to high levels by starvation, providing an internal source of nutrients on which cells can survive extended periods of nutrient deprivation. Conversely, under some circumstances autophagy may be used as a killing mechanism, acting as an alternative or augmentation to apoptotic cell death (Neufeld and Baehrecke, 2008). As autophagy has been implicated in several physiological and pathological conditions, including neurodegeneration, tumorigenesis, and aging (Huang and Klionsky, 2007), better understanding of the molecular mechanisms controlling autophagy and identification of pharmacological regulators of this process are important goals.
Wortmannin and 3-methyladenine are well established inhibitors of autophagy. These compounds are broad-spectrum phosphatidylinositol 3 (PI(3))–kinase inhibitors that disrupt autophagy by inhibiting Vps34 (Petiot et al., 2000), the enzymatic component of a multiprotein complex which also includes Vps15, Beclin1/Atg6, UVRAG, and Bif-1 in mammals and Vps15, Atg6, and Atg14 in yeast (Mari and Reggiori, 2007). Localized production of PI(3) phosphate (PI(3)P) by Vps34 can act to recruit proteins containing FYVE and PX domains to specific membrane compartments (Lindmo and Stenmark, 2006). In yeast, this Vps34 complex is critical for recruiting autophagy-related (Atg) proteins to the preautophagosomal structure, the yeast-specific site of autophagosome formation (Suzuki et al., 2007). The role of PI(3)P in autophagosome biogenesis is less well understood in higher eukaryotes, and whether it functions at the autophagosomal, the donor, or another membrane has not been determined (Pattingre et al., 2007).
Vps34 is also required more broadly for several vesicular trafficking processes that may have indirect impacts on autophagy. These include sorting of hydrolytic enzymes to the lysosome/vacuole and early steps in the endocytic pathway (Lindmo and Stenmark, 2006). In mammalian cells, autophagosomes have been shown to fuse with early or late endosomes before fusion with lysosomes, resulting in intermediate structures known as amphisomes (Eskelinen, 2005). Recently, mutations in components of the endosomal sorting complex required for transport (ESCRT) complex, which is required for the transition from early to late (multivesicular) endosomes, have been shown to block autophagy by inhibiting autophagosome-endosome fusion (Nara et al., 2002; Lee et al., 2007; Rusten et al., 2007). Thus, the effect of PI(3)-kinase inhibitors on autophagy may be due, in part, to these more general trafficking functions of Vps34.
Recent work has shown that Vps34 can also function in a nutrient-sensing pathway upstream of the target of rapamycin (TOR) in several mammalian cell lines (Byfield et al., 2005; Nobukuni et al., 2005). Disruption of Vps34 activity with blocking antibodies or siRNA was found to inhibit activation of TOR by insulin, amino acids, and glucose (Byfield et al., 2005; Nobukuni et al., 2005). As TOR signaling inhibits autophagy, these findings are at odds with the conserved role of Vps34 in promoting autophagy under starvation conditions, suggesting that distinct complexes or pools of Vps34 may be subject to different modes of regulation.
In this paper, we address how these multiple potential roles of Vps34 are coordinated to regulate autophagy in an intact organism, the fruit fly Drosophila melanogaster. Our findings suggest that despite a critical role for Vps34 in endocytic uptake and recycling, its primary function in autophagy in vivo is limited to its direct role at the nascent autophagosome. We further show that Vps34 is not required for TOR activity in this system and that starvation results in a TOR/Atg1-dependent recruitment of Vps34 activity to the autophagosomal membrane.
Results
Generation of null and hypomorphic Vps34 mutants
A single representative of each of the three classes of PI(3)-kinase is present in D. melanogaster. The class III PI(3)-kinase orthologue (referred to previously as Pi3K_59F and herein as Vps34) shows greatest similarity to human Vps34 (hVps34) in the kinase domain, with an overall identity of 37 and 57% to Vps34 proteins in yeast and human (Linassier et al., 1997). Previous work has shown that D. melanogaster Vps34 displays substrate specificity for PtdIns, with high sensitivity to wortmannin and nonionic detergent as is characteristic of this class (Linassier et al., 1997). Vps34 transcripts are expressed widely during development and in adult flies, with expression in the larval fat body and relative enrichment in neuronal and gut tissues (Chintapalli et al., 2007).
To generate loss-of-function mutations in the Vps34 gene, we established isogenic homozygous-viable stocks of the P element line NP1626 (Hayashi et al., 2002) inserted in the Vps34 5′ untranslated region. Transposase-mediated excision of this element generated several lines that displayed hemizygous larval lethality in trans to a deficiency for this region, Df(2R)bw5. By PCR analysis, we identified an excision line that contains a deletion extending 3,349 bases downstream of the P element insertion site (Fig. 1 A). In this line, Δm22, the entire Vps34 coding region is eliminated, thus resulting in a presumptive null allele. In addition, other lines displaying full or partially penetrant lethality were found to contain partial remnants of the excised P element (Fig. 1 A) and likely represent hypomorphic Vps34 alleles, as in the case of ex32. As expected, Vps34 transcripts were undetectable by RT-PCR in Δm22 mutants and were reduced in ex32 animals (Fig. 1 B). To confirm that these lesions specifically affect the Vps34 locus, we used the GAL4–UAS system to ubiquitously express a wild-type Vps34 transgene in mutant animals, resulting in a complete rescue of lethality (unpublished data).
Figure 1.

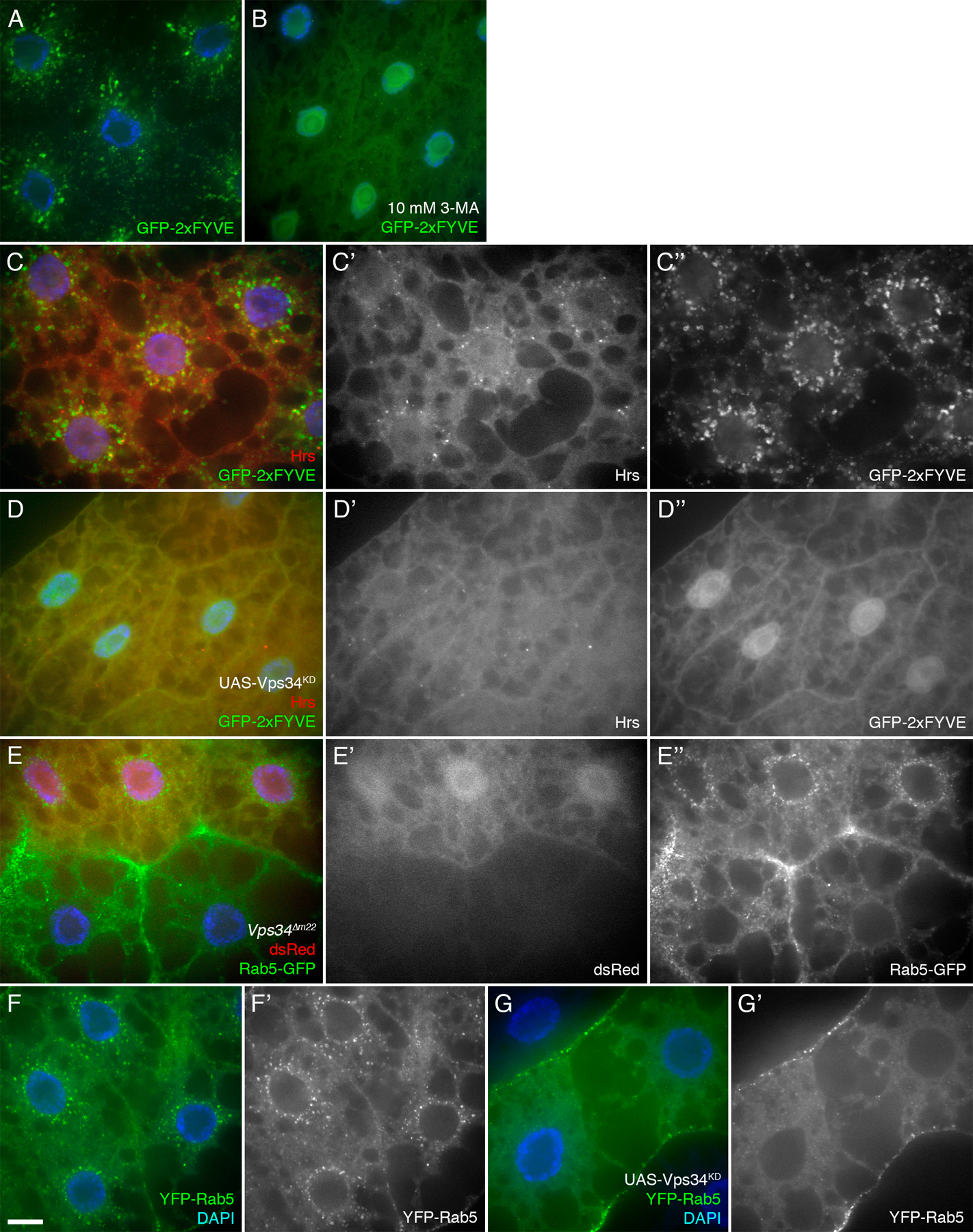
Reagents to disrupt and monitor Vps34 function in D. melanogaster. (A) Map of the Vps34 locus, showing the location and extent of the Δm22 deletion. (B) RT-PCR analysis of Vps34 mRNA expression in control and mutant third instar larvae. Vps34 transcript levels are strongly reduced in ex32 animals and undetectable in the Δm22 mutant. (C–E) Disruption of Vps34 function causes mislocalization of the PI(3)P reporter GFP-2xFYVE. In wild-type fat body cells (C and D), GFP-2xFYVE accumulates at the perinuclear endosomal compartment. This pattern of localization is lost in Vps34Δm22 clones (C; mutant cell marked by lack of dsRed) or in response to expression of Vps34KD (E). Bar, 10 μm. Genotypes: (C) hsflp; FRT42D UAS-GFP-2xFYVE Vps34Δm22/Cg-GAL4 FRT42D UAS-GFP-2xFYVE UAS-dsRed, (D) hsflp; UAS-GFP-2xFYVE/+; Act>CD2>GAL4/+, and (E) hsflp; UAS-GFP-2xFYVE/+ ; Act>CD2>GAL4 / UAS-Vps34KD.
The activity of Vps34 can be monitored in vivo by examining the intracellular localization of FYVE domain–containing proteins (Gillooly et al., 2000). To assay Vps34 function in mutant cells, we used a transgenic UAS line expressing EGFP fused to two tandem copies of the FYVE domain from the early endosomal protein Hrs (GFP-2xFYVE) (Wucherpfennig et al., 2003). For these experiments, we assayed GFP-2xFYVE distribution in FLP-FRT–generated clones of Vps34 mutant cells, which are marked by the loss of a UAS-driven dsRed marker. This system allows side-by-side comparison of mutant and wild-type cells within the same animal. In wild-type fat body cells, GFP-2xFYVE localized primarily to vesicular structures in the perinuclear region, with additional punctate staining occurring at low levels throughout the cytoplasm and at the cell periphery (Fig. 1, C and D). In contrast, GFP-2xFYVE staining was diffuse in Δm22 mutant cells, with no perinuclear localization and few or no apparent punctate structures (Fig. 1 C). Collectively, these results indicate that the Δm22 deletion specifically eliminates Vps34 expression, resulting in a cell-autonomous loss of function.
As a complement to these mutations in the endogenous Vps34 gene, we also generated UAS-regulated transgenic lines that express D. melanogaster Vps34 containing point mutations in conserved residues of the kinase domain (referred to as Vps34KD). Analogous mutations in yeast and mammalian Vps34 result in a kinase-defective protein with dominant-inhibitory properties (Row et al., 2001). Coexpression of Vps34KD with GFP-2xFYVE in FLP-FRT–generated fat body clones (Britton et al., 2002) resulted in diffuse GFP-2xFYVE localization similar to the deletion mutant (Fig. 1 E), which is consistent with a disruption of Vps34 activity in Vps34KD-expressing cells. Incubation of dissected fat body with the PI(3)K inhibitor 3-methyladenine also caused a similar defect in GFP-2xFYVE localization (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200712051/DC1).
Although Vps34 has been localized to several distinct subcellular compartments in different cell types and experimental systems, its highest levels of activity are typically found in the early endosome, and GFP-2xFYVE is commonly used as an early endosomal marker (Gillooly et al., 2000). To test whether the perinuclear localization of GFP-2xFYVE in the fat body reflects the early endosomal compartment in this cell type, we examined the localization of other early endosomal markers. Both Hrs and Rab5-GFP displayed a similar perinuclear pattern which overlapped with GFP-2xFYVE or myc-2xFYVE and which was disrupted in Δm22 mutant cells and in cells expressing Vps34KD (Fig. S1). We also found that the GFP-2xFYVE–labeled compartment was readily accessible to the endocytic tracer Texas Red (TR) avidin (see subsequent sections). In contrast, no colocalization was observed between GFP-2xFYVE and the Golgi marker p120 nor the late endosomal/lysosomal marker Lamp1 (Fig. S2). We conclude that Vps34 activity is concentrated in a perinuclear endosomal compartment in larval fat body cells. Disruption of GFP-2xFYVE, Hrs, and Rab5 localization in Vps34 mutant cells indicates either that Vps34 activity is necessary for the recruitment or stable association of these markers or that the perinuclear endosomal compartment itself is disrupted in the mutant cells.
Vps34 is required for early steps of autophagosome formation
The fat body is specialized for a robust autophagic response to starvation, acting as a reserve source of nutrients for other critical tissues (Neufeld, 2004). Starvation results in an autophagy-dependent expansion and acidification of the lysosomal system, and thus the fluorescent Lysotracker dyes can be used to assay progression to late stages of autophagy in this tissue (Rusten et al., 2004; Scott et al., 2004). Starvation-induced formation of Lysotracker-positive structures was severely inhibited in clones of Vps34 mutant cells and in cells expressing Vps34KD (Fig. 2, A and B), which is consistent with a requirement for Vps34 in autophagy induction or progression. Overexpression of wild-type Vps34 was not sufficient to induce Lysotracker-positive structures under fed conditions (Fig. 2 C) but did result in a more robust response to starvation (Fig. 2 D). To examine earlier stages of autophagy, we determined the localization of the autophagosomal marker GFP-Atg8/LC3 in wild-type and Vps34 mutant cells. Starvation-induced accumulation of GFP-Atg8 punctae was strongly curtailed in both Vps34 mutant and Vps34KD-expressing cells (Fig. 2, E–G), indicating an essential cell-autonomous role for Vps34 in autophagosome biogenesis. Expression of Vps34KD also blocked the starvation-induced accumulation of preautophagosomal membranes labeled with GFP-Atg5 (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200712051/DC1). Disruption of autophagosome and autolysosome formation was also evident by ultrastructural analysis of starved Vps34 loss-of-function larvae (Fig. 2, H–J). Morphometric analysis revealed that the percent volume of cytoplasm occupied by autophagosomes and autolysosomes was reduced by more than 10-fold in both Vps34Δm22 mutants and in animals overexpressing kinase-dead Vps34 in the fat body, relative to controls (P < 0.0001; Fig. 2 K).
Figure 2.

Starvation-induced autophagy in the larval fat body requires Vps34. All images depict live fat body tissue dissected from fed (C) or 4-h starved animals (all other panels). (A) Vps34Δm22 loss-of-function clones (marked by lack of GFP) fail to accumulate autolysosomes (labeled by punctate Lysotracker Red staining) after starvation. (B) Vps34KD-expressing cells (GFP positive) fail to accumulate punctate Lysotracker Red staining under starvation conditions. (C and D) Clonal expression of wild-type Vps34 (GFP-positive cells) does not induce autophagy under fed conditions (C) but increases the response to starvation (D). (E) Vps34Δm22 loss-of-function clones (marked by lack of myrRFP) fail to accumulate autophagosomes (marked by punctate GFP-Atg8) in response to starvation. (F and G) Starvation-induced accumulation of GFP-Atg8a punctae is observed in controls (F) but not in animals expressing Vps34KD (G). (H–K) TEM images reveal abundant autophagosomes (AP) and autolysosomes (AL) in control animals (H) but not Vps34Δm22 mutants (I) nor animals expressing Vps34KD (J). LD, lipid droplet. The graph in K shows autophagosome and autolysosome area ratios calculated from electron micrographs of five animals per genotype. Error bars show SD from the mean. P-values (Mann-Whitney U test): control versus Vps34Δm22, AP, 1.45e-11 and AL, 3.9e-9; and control versus Vps34KD, AP, 2.9e-11 and AL, 1.5 e-11. Bars: (A–G) 10 μm; (H–J) 1 μm. Genotypes: (A) hsflp; FRT42D Vps34Δm22/UAS-2xeGFP FRT42D fb-GAL4, (B) hsflp; Act>CD2>GAL4 UAS-GFP/UAS-Vps34KD, (C and D) hsflp; Act>CD2>GAL4 UAS-GFP/UAS-Vps34WT, (E) hsflp; FRT42D Vps34Δm22/Cg-GAL4 UAS-GFP-Atg8a FRT42D UAS-myrRFP, (F) hsflp; Act>CD2>GAL4 UAS-GFP-Atg8a/+, (G) hsflp; Act>CD2>GAL4 UAS-GFP-Atg8a/UAS-Vps34KD, (H) Vps34Δm22/CyO-GFP, (I) Vps34Δm22/Δm22, (J) Cg-GAL4 UAS-Vps34KD/Cg-GAL4 UAS-Vps34KD, and (K) as in H–J.
We previously found that a hypomorphic P element allele of D. melanogaster Atg6/beclin 1 results in a moderate defect in starvation-induced autophagy (Scott et al., 2004), and we also find that mutations in the Vps15 homologue Ird1 result in a severe autophagy block (Fig. S3). To determine whether Vps34 functions in a complex containing these proteins, we first tested for physical association between epitope-tagged versions of Vps34, Vps15, and Atg6. We found that Atg6 could readily be precipitated with anti-Vps34 antibodies from larval extracts containing overexpressed myc-Atg6, Vps15-FLAG, and Vps34-TAP (Fig. 3 A). Consistent with findings in mammalian cells (Byfield et al., 2005), this association was observed under both fed and starved conditions, although starvation usually led to increased coprecipitation. Similar interactions were observed between Vps34-TAP and Vps15-FLAG (unpublished data).
Figure 3.

Physical and genetic interactions between D. melanogaster Vps34, Vps15, and Atg6. (A) Epitope-tagged Atg6 and Vps34 coimmunoprecipitate from larval extracts. myc-Atg6 and Vps15-FLAG were coexpressed with and without Vps34-TAP during larval development by heat shock–dependent GAL4 induction of UAS transgenes under either fed or starved conditions. Immunoprecipitation of larval extracts with hVps34 antibodies coprecipitates myc-Atg6. Levels of coprecipitated myc-Atg6 are higher in extracts from larvae overexpressing Vps34-TAP (lanes 2 and 6) than from larvae expressing only endogenous Vps34 (lanes 4 and 8). (B) Coexpression of Vps34-TAP with Atg6 or Vps15 results in cooperative induction of GFP-Atg8a–labeled autophagosomes under fed conditions. Number of GFP-Atg8a–positive spots per field in fat body of fed (F) and starved (S) control animals and in fed transgenic animals is indicated. Data represent mean ± SE of 10 animals per genotype. P-values were determined by two-tailed Student's t test.
To determine whether these physical associations are functionally relevant, we tested for genetic interactions between Vps34, Vps15, and Atg6. GAL4/UAS-mediated expression of Vps34-TAP resulted in a moderate statistically insignificant induction of GFP-Atg8a punctae formation under fed conditions (Fig. 3 B). Although expression of Atg6 or Vps15 also had little effect on their own, coexpression of these proteins with Vps34-TAP resulted in a significant increase in GFP-Atg8a punctae formation (Fig. 3 B). Thus, overexpression of Vps34 complexes promotes formation of GFP-Atg8a–labeled autophagosomes, but not Lysotracker-labeled autolysosomes, under fed conditions (Fig. 3 B and Fig. S3), suggesting that Vps34 activity is sufficient to drive early, but not later, steps of autophagy. Collectively, our findings suggest that D. melanogaster Vps34 is required for early events in autophagosome formation, at least in part via a conserved complex containing Vps34, Vps15, and Atg6.
Vps34 function in the endocytic pathway is distinct from its role in autophagy
Given the localization of Vps34 activity to the endosomal compartment and its role in formation of and localization to multivesicular bodies in mammalian cells (Gillooly et al., 2000; Futter et al., 2001), we next asked whether the loss of autophagy in Vps34 mutants could be attributed in part to its role in the endocytic pathway. Consistent with recent studies (Rusten et al., 2007), we found that mutations in components of ESCRT-I (Vps25), -II (Vps28), or -III (Vps32) disrupted starvation-induced lysotracker staining in the larval fat body (Fig. 4 A; Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200712051/DC1; and not depicted). In contrast to the lack of autophagosomes in Vps34 mutants (Fig. 4 C), mutations in these ESCRT subunits led to an accumulation of GFP-Atg8a–labeled autophagosomes under fed conditions (Fig. 4 B and Fig. S4), which is consistent with a block in autophagosome-endosome fusion, as previously reported (Lee et al., 2007; Rusten et al., 2007). This difference between Vps34 and ESCRT phenotypes suggests that these mutations disrupt distinct steps of autophagy progression.
Figure 4.

Vps34 and ESCRT components are required for distinct steps in autophagy. (A) Mutation of the ESCRT-II component Vps28 (mutant clone marked by lack of GFP) disrupts formation of Lysotracker Red punctae in response to 4-h starvation. Bar, 10 μm. (B–D) GFP-Atg8a–marked autophagosomes accumulate in clones of Vps28 mutant (B) and Vps34 Vps28 double mutant (D) cells but not in Vps34 mutant cells (C). Mutant clones are marked by lack of myrRFP. (E–G) Mosaic eye imaginal discs showing accumulation of ubiquitin-positive punctae in Vps25 (E) and Vps25 Vps34 (G) mutant clones but not in Vps34 mutant clones (F). Lack of GFP marks mutant clones. Bar, 10 μm (B–G). Genotypes: (A) hsflp; FRT42D Vps28k15603/UAS-2xeGFP FRT42D fb-GAL4, (B) hsflp; FRT42D Vps28k15603/Cg-GAL4 UAS-GFP-Atg8a FRT42D UAS-myrRFP, (C) hsflp; FRT42D Vps34Δm22/Cg-GAL4 UAS-GFP-Atg8a FRT42D UAS-myrRFP, (D) hsflp; FRT42D Vps28k15603 Vps34Δm22/Cg-GAL4 UAS-GFP-Atg8a FRT42D UAS-myrRFP, (E) hsflp; FRT42D Vps25A3/FRT42D Ubi-GFPnls, (F) hsflp; FRT42D Vps34Δm22/FRT42D Ubi-GFPnls, and (G) hsflp; FRT42D Vps25A3 Vps34Δm22/FRT42D Ubi-GFPnls.
To investigate this further, we generated clones of fat body cells mutant for both Vps34 and 25 or Vps34 and 28. In these double mutant cells, GFP-Atg8a punctae still accumulated under fed conditions, albeit at somewhat reduced levels (Fig. 4 D and Fig. S4). Accumulation of autophagosomes was also evident in transmission electron micrographs of eye imaginal discs doubly mutant for Vps34 and 25 (Fig. S5). This finding that autophagosomes accumulate in Vps34-Vps25 and Vps34-Vps28 double mutants, but not in Vps34 single mutants, indicates that deletion of Vps34 has a more severe kinetic effect on autophagosome formation than on autophagosome-lysosome fusion, such that the early steps in autophagosome formation become rate limiting. With additional mutation of Vps25 or Vps28, autophagosome-lysosome fusion becomes rate limiting and autophagosomes accumulate. Thus, the ESCRT-dependent step required for autophagy is at least partially functional in cells lacking Vps34.
As reported previously (Vaccari and Bilder, 2005), loss of Vps25 leads to accumulation of ubiquitin-positive punctae in cells of the developing eye (Fig. 4 E). Similar ubiquitin accumulation was observed in Vps34-Vps25 mutant cells, but not in Vps34 single mutants (Fig. 4, F and G), further indicating that the ESCRT pathway is not disrupted in Vps34 mutants. The ubiquitin-containing structures in Vps34-Vps25 mutant cells largely colocalized with the endosomal marker Hrs and were often adjacent to GFP-Atg8a–labeled punctae (Fig. S5). Similarly, immuno-EM analysis of Vps25 and Vps34-Vps25 mutant eye discs revealed accumulation of ubiquitin in tubulovesicular compartments but not in autophagosomes (Fig. S5). Thus, the ubiquitinated structures in Vps25 and Vps34-Vps25 mutant cells likely represent stalled intermediates in the endocytic pathway.
Despite the relatively normal progression of ESCRT-dependent processes in Vps34 mutants, we found that they were severely defective in fluid-phase endocytosis. Vps34 mutant clones in the larval fat body failed to incorporate the endocytic tracer TR-avidin beyond the cortical region of the cell, where it often accumulated to high levels (Fig. 5 A). A similar disruption was observed in Vps34-Vps25 double mutant fat body cells (Fig. S4). Mutation of Vps34 or expression of Vps34KD also inhibited TR-avidin uptake in the highly endocytic Garland cells (Fig. 5, B–E). In addition, we observed accumulation of Notch in Vps34 mutant cells of the eye imaginal disc (Fig. 5 F), which is consistent with defective endocytic recycling (Vaccari and Bilder, 2005). Finally, loss of Vps34 dramatically altered the distribution of GFP-Lamp1 and Spin-GFP (Fig. S2), markers which localize to or transit through late endosomes and lysosomes. Collectively, these findings indicate that D. melanogaster Vps34 has an essential role in the endocytic pathway but that this role is distinct from ESCRT-dependent endocytic processes and does not constitute a rate-limiting step for its function in autophagy.
Figure 5.

Vps34 is required for endocytosis. (A) Vps34Δm22 loss-of-function fat body clones (marked by lack of GFP) fail to incorporate the endocytic tracer TR avidin into the perinuclear early endosome (20-min pulse followed by 10-min chase). (B and C) TR avidin accumulates in Garland cells of heterozygous controls (B) but not Vps34Δm22 homozygous mutants (C; 2-min pulse followed by 20-min chase). (D and E) Mosaic larvae expressing Rab5-GFP (D) or Rab5-GFP and Vps34KD (E) in a subset of Garland cells. TR avidin uptake and cortical localization of Rab5-GFP are disrupted in Vps34KD-expressing cells (15-min pulse, no chase). Bar, 10 μm (B–E). (F) Clonal loss of Vps34 (marked by lack of GFP) in the eye imaginal disc results in accumulation of Notch-positive punctae. Bar, 10 μm (A and F). Genotypes: (A) hsflp; FRT42D Vps34Δm22/UAS-2xeGFP FRT42D fb-GAL4, (B) Vps34Δm22/CyO-GFP, (C) Vps34Δm22/Δm22, (D) hsflp; Act>CD2>GAL4 UAS-Rab5-GFP/+, (E) hsflp; Act>CD2>GAL4 UAS-Rab5-GFP/UAS-Vps34KD, and (F) hsflp; FRT42D Vps34Δm22/FRT42D Ubi-GFPnls.
Vps34 is not required for nutrient-dependent signaling to TOR
In D. melanogaster and other systems, a hallmark of TOR signaling is its stimulation of cell growth and proliferation (Arsham and Neufeld, 2006). Down-regulation of this pathway in fat body cells through Tor mutation (Scott et al., 2007), RNAi-mediated inhibition of Tor expression (Fig. 6, A and L), or overexpression of the TOR inhibitors Tsc1 and 2 (Fig. 6, B and L) results in a significant reduction in cell size. In contrast, cells mutant for Vps34 or expressing Vps34KD were similar in size to wild-type cells (Fig. 6, C, D, and L). Similarly, in the eye imaginal disc, which is comprised of mitotically active diploid cells, mutations in Tor cause a strong proliferative disadvantage (Zhang et al., 2000), whereas Vps34 mutant cells proliferate at a rate similar to that of control cells (Fig. 6, E and F). The normal rate of growth and proliferation suggests that TOR signaling is not appreciably compromised in Vps34 mutant cells.
Figure 6.

Vps34 does not influence TOR signaling or function. Transgene-expressing cells are marked by coexpression of GFP in A, B, D, and G–J. Loss-of-function mutant clones are marked by lack of GFP in C, E, and F. All images are from fed larvae except G and H, which are from 48-h starved animals. (A and B) Inhibition of TOR signaling by expression of Tor double-stranded RNA (dsRNA; A) or overexpression of Tsc1 and 2 (B) results in a reduction in fat body cell size. (C and D) Fat body cells lacking Vps34 (C) or expressing Vps34KD (D) are normal in size. (E and F) In the eye imaginal disc, cells mutant for Tor (E; GFP negative) proliferate more slowly than their wild-type “twin spot” control cells (two copies of GFP). Vps34Δm22 homozygous mutant cells (F) proliferate at a rate similar to that of twin spot controls. (G and H) Overexpression of Rheb activates TOR and increases cell growth both in the absence (G) or presence (H) of coexpressed kinase-defective Vps34. (I and J) Inhibition of TOR signaling by cooverexpression of Tsc1 and 2 reduces cell growth in control (I) and Vps34Δm22 (J) animals. (K) Ubiquitous hsGAL4-driven overexpression of wild-type Vps34 (lane 2) or kinase-defective Vps34 (lane 3) does not affect TOR-dependent phosphorylation of S6K-Thr398 or Akt-Ser505 relative to control extracts (lane 1). (L) Quantitation of the area of mutant or transgene-expressing cells in A–D relative to wild-type cells from the same tissue. Data represent mean ± SEM of 7–10 animals per genotype. Bar: 10 μm. Genotypes: (A) hsflp; Act>CD2>GAL4 UAS-GFP/UAS-TORdsRNA5092R-2, (B) hsflp; Act>CD2>GAL4 UAS-GFP/UAS-Tsc1 UAS-Tsc2, (C) hsflp; FRT42D Vps34Δm22/UAS-2xeGFP FRT42D fb-GAL4, (D) hsflp; Act>CD2>GAL4 UAS-GFP/UAS-Vps34KD, (E) hsflp; TorΔP FRT40A/Ubi-GFPnls FRT40A, (F) hsflp; FRT42D Vps34Δm22/FRT42D Ubi-GFPnls, (G) hsflp; Act>CD2>GAL4 UAS-GFP UAS-RhebEP50.084/+, (H) hsflp; Act>CD2>GAL4 UAS-GFP UAS-RhebEP50.084/UAS-Vps34KD, (I) hsflp; Act>CD2>GAL4 UAS-GFP/UAS-Tsc1 UAS-Tsc2, and (J) hsflp; Vps34Δm22/Vps34Δm22; Act>CD2>GAL4 UAS-GFP/UAS-Tsc1 UAS-Tsc2.
To ask whether a role for Vps34 in TOR signaling might be apparent only at higher or lower than normal levels of TOR activity, we induced clones of cells with altered expression of Rheb (a TOR activator) or Tsc1 and 2 in control or Vps34-deficient backgrounds. Cells overexpressing Rheb reached a larger size than neighboring cells, as previously reported (Saucedo et al., 2003), and coexpression of Rheb and Vps34KD resulted in a similar cell enlargement (Fig. 6, G and H). Conversely, overexpression of Tsc1 and 2 inhibited cell growth to a similar extent in Vps34 mutant and control animals (Fig. 6, I and J). Expression of wild-type or kinase-defective Vps34 also had no effect on cell size in starved animals or animals that were allowed to recover on normal media after 48 h of starvation (unpublished data). We also tested for loss-of-function genetic interactions between Vps34 and components of the TOR pathway. Heterozygous mutation of Tor or S6k dominantly suppresses the lethality of Tsc1 homozygous mutant animals (Radimerski et al., 2002; Zhang et al., 2006), presumably by restraining the abnormally high levels of TOR signaling caused by loss of Tsc1. In contrast, Vps34 mutants had no effect on the development or viability of Tsc1 mutant animals (unpublished data).
To more directly assess the requirement for Vps34 in TOR activation, we examined the phosphorylation of S6K-Thr398, a direct substrate of the rapamycin-sensitive TOR complex I, as well as Akt-Ser505, which is phosphorylated by TOR complex II. Although the phosphorylation status of these sites is highly sensitive to the levels of Tsc1 or Rheb (Hennig et al., 2006; Scott et al., 2007), ubiquitous overexpression of UAS-driven wild-type or kinase-deficient Vps34 had no effect on the phosphorylation of these sites in whole larval extracts (Fig. 6 K). Fat body–specific expression of these transgenes also failed to alter the phosphorylation of these sites in extracts from dissected fat body tissue (unpublished data). Collectively, these results indicate that in contrast to the case in cultured mammalian cells, D. melanogaster Vps34 does not play a critical role in TOR activation in vivo.
Recruitment of Vps34 activity to autophagosomal membranes is regulated by TOR/Atg1-dependent nutrient signaling
Although Vps34 is required for starvation-induced autophagy, it remains uncertain whether and how the PI(3)-kinase enzymatic activity of autophagy-specific Vps34 complexes is regulated by nutrient conditions. For example, some studies have reported an increase in Beclin 1–associated PI(3)-kinase activity in response to starvation (Byfield et al., 2005; Nobukuni et al., 2005), whereas others report a decrease (Tassa et al., 2003; Takahashi et al., 2007). Comparison of GFP-2xFYVE localization in fed and starved animals revealed that starvation induced a relative shift from a perinuclear concentration of this marker to a more widely dispersed pattern (Fig. 7, A–D and F). This did not reflect a disruption of the early endosome itself, as the TR-avidin–accessible compartment remained in the perinuclear region in starved larvae (Fig. 7, A and B). Rab5-GFP also remained associated with the perinuclear early endosome under starvation conditions (Fig. 7, C and D), and the accumulation of peripheral GFP-2xFYVE punctae was not associated with decreased perinuclear localization (Fig. 7 F).
Figure 7.

Recruitment of PI(3)P from early endosomes to autophagosomes in response to TOR/Atg1-dependent nutrient signaling. (A and B) Internalized TR avidin colocalizes with GFP-2xFYVE under fed conditions (A) but not after 4-h starvation (B). (C and D) Starvation causes dispersion of myc-2xFYVE but not Rab5-GFP. (E) Under starvation conditions, myc-2xFYVE displays extensive colocalization with GFP-Atg8a. Yellow circles in E′ and E′′ show overlap between GFP-Atg8a and myc-2xFYVE label. Bar, 10 μm. (F) Quantitation of the myc-2xFYVE–labeled compartment area in a 2-μm ring surrounding the nucleus (perinuclear) and the remaining peripheral cell area (cytoplasmic). Genotypes: (A and B) Cg-GAL4/UAS-GFP-2xFYVE, (C and D) hsflp; Act>CD2>GAL4 UAS-Rab5-GFP/UAS-myc-2xFYVE, and (E) Cg-GAL4 UAS-GFP-Atg8a/UAS-myc-2xFYVE.
These findings suggest that starvation causes recruitment of Vps34 activity to peripheral locations throughout the cell, presumably nascent autophagosomes. This was confirmed by the observation that the nonendosomal myc-2xFYVE punctae induced by starvation displayed a nearly complete overlap with the autophagosomal marker GFP-Atg8a (Fig. 7 E). Thus, autophagy-inducing signals result in mobilization of Vps34 activity directly to the autophagosomal membrane. Although we were unable to detect endogenous Vps34 protein in vivo, UAS-driven Vps34 did not appear to be concentrated at the early endosome nor on autophagosomes, and its localization was not affected by starvation (unpublished data). Thus, the mobilization of Vps34 activity may reflect activation of preexisting Vps34 protein at distinct cellular locations. Alternatively, the high levels of UAS-driven expression may mask more subtle changes in the distribution of endogenous protein.
TOR signaling plays a critical role in conveying nutrient status to the autophagic machinery and may directly regulate the activity of the Ser-Thr kinase Atg1 (Kamada et al., 2000; Scott et al., 2007). To determine whether this pathway is involved in regulation of Vps34, we examined its effect on nutrient-dependent relocalization of GFP-2xFYVE. In clones of cells mutant for Tsc2 or overexpressing Rheb, both of which result in constitutive TOR activation, formation of GFP-2xFYVE cytoplasmic punctae in response to starvation was greatly diminished (Fig. 8 A and not depicted). Similar results were observed in cells lacking Atg1 (Fig. 8 B). Thus, signaling through TOR and Atg1 is necessary for proper nutrient-responsive recruitment of Vps34 activity to autophagosomes. Accordingly, down-regulation of TOR signaling by cooverexpression of Tsc1 and 2 led to autophagy induction in control animals, but not in Vps34 mutants (Fig. 8, C and D), which is consistent with Vps34 functioning downstream of TOR-dependent nutrient signaling.
Figure 8.

Vps34 functions downstream of TOR signaling. (A and B) Starvation-induced redistribution of Vps34 activity requires TOR-Atg1 signaling. 4-h starvation causes relocalization of GFP-2xFYVE in control cells but not in Tsc2 homozygous mutant cells (A) nor in cells mutant for Atg1 (B). Mutant clones are marked by lack of dsRed. Note also the changes in GFP-2xFYVE levels in the mutant cells. (C and D) Cooverexpression of Tsc1 and 2 in GFP-marked clones induces formation of autolysosomes under fed conditions in control (C) but not Vps34 mutant (D) animals. Bar: (A and B) 10 μm; (C and D) 25 μm. Genotypes: (A) hsflp; Cg-GAL4 UAS-GFP-2xFYVE/+; Tsc2192 FRT80B/UAS-dsRed FRT80B, (B) hsflp; Cg-GAL4 UAS-GFP-2xFYVE/+; Atg1Δ3D FRT80B/UAS-dsRed FRT80B, (C) hsflp; Vps34Δm22/+; Act>CD2>GAL4 UAS-GFP/UAS-Tsc1 UAS-Tsc2, and (D) hsflp; Vps34Δm22/Vps34Δm22 ; Act>CD2>GAL4 UAS-GFP/UAS-Tsc1 UAS-Tsc2.
Discussion
Previous work in mammalian and yeast systems has identified a wide range of vesicle trafficking processes regulated by Vps34, including autophagy, endocytosis, endosome maturation, and both anterograde and retrograde trafficking between the Golgi and lysosome (Lindmo and Stenmark, 2006). Our findings indicate that despite these activities, the in vivo role of Vps34 in autophagy is largely limited to its function at the autophagosome. Although fluid-phase endocytosis, endocytic recycling of Notch, and trafficking of lysosomal proteins were disrupted by mutation of Vps34, our results suggest that events subsequent to autophagosome formation, including fusion between autophagosomes and endosomes or lysosomes and subsequent lysosomal degradation, are not rate limiting in the absence of Vps34. Why does endocytic disruption lead to autophagosome fusion defects in ESCRT mutants but not in Vps34 mutants? Accumulation of endocytic tracer at the periphery of Vps34 mutant cells suggests that Vps34 functions at an early step of endocytosis, and apparently this event, as well as normal endocytic flux is not essential for fusion of autophagosomes with elements of the endosomal-lysosomal compartment. Interestingly, the accumulation of autophagosomes in ESCRT/Vps34 double mutants indicates that loss of Vps34 does not completely prevent autophagosome formation. Similarly, the lack of autophagosome accumulation in Vps34 single mutants indicates that ESCRT complexes are at least partially functional in the absence of Vps34. Thus, PI(3)P may not be absolutely essential for these processes, or perhaps sufficient levels of PI(3)P are generated independently of Vps34 by the class II PI(3) kinase or by PI(3,4)P or PI(3,5)P phosphatases. As ESCRT components are required for multivesicular body formation but not autophagy in yeast (Reggiori et al., 2004), it will be interesting to determine whether the requirement for ESCRT complexes in autophagy in higher eukaryotes reflects their role in multivesicular body formation or an alternate function.
The cellular compartment in which Vps34 acts to promote autophagy and the mechanisms by which it is regulated by nutrient signals have remained unresolved. In mammalian cells, Beclin1/Atg6 has been reported to localize to the trans-Golgi network, ER, and mitochondria (Kihara et al., 2001; Pattingre et al., 2005). It was recently shown that Beclin1–Vps34 complexes can be inhibited by the antiapoptotic factor Bcl-2 in a nutrient-dependent manner (Pattingre et al., 2005). Bcl-2 mutants that are targeted to the ER, but not to mitochondria, retain their capacity to inhibit starvation-induced autophagy, suggesting that the ER is an important site of Beclin1-Vps34 regulation. However, it is unknown how these organelles contribute to the formation of autophagosomes, and recent studies suggest that rather than budding off a preexisting compartment, the autophagosomal membrane is likely to form de novo from small lipid transport vesicles or lipoprotein complexes (Juhasz and Neufeld, 2006; Kovacs et al., 2007). Our finding that myc-2xFYVE is recruited to GFP-Atg8a–positive structures under starvation conditions indicates that Vps34 activity is targeted directly to autophagosomes in a TOR/Atg1-dependent manner. Although these results do not distinguish between a role for TOR/Atg1 signaling in regulating Vps34 activity versus providing a platform on which Vps34 complexes can assemble, together with these previous studies they indicate that Vps34 is likely to promote autophagy by different mechanisms from multiple cellular locations.
How the TOR signaling pathway senses intracellular levels of nutrients, such as amino acids, has been poorly understood despite considerable work in yeast, mammalian, and other model systems. The recent identification of Vps34 as a transducer of this signal in mammalian cells thus represents a significant new insight into this issue (Byfield et al., 2005; Nobukuni et al., 2005). However, further work is necessary to determine the extent to which this mechanism is generally conserved, as starvation appears to have opposing effects on Vps34 activity in different cell types (Tassa et al., 2003; Nobukuni et al., 2005; Takahashi et al., 2007). The results presented here fail to support this model in D. melanogaster, as mutation of Vps34 did not appear to influence TOR-dependent phenotypes nor to disrupt TOR-dependent signaling. This may reflect a fundamental difference in signaling mechanisms between the fly and mammalian systems. The makeup of Vps34 complexes has diverged significantly between yeast and metazoans, and perhaps components of this complex, such as Ambra1 (Fimia et al., 2007), that appear to be unique to vertebrates may confer functions not found in flies. It is also possible that production of PI(3)P by Vps34-independent mechanisms is more efficient in D. melanogaster than in mammalian cells and, thus, a role for Vps34 in TOR signaling may be obscured by these other sources. The continued ESCRT function and basal level of autophagy in Vps34 null mutants are consistent with this possibility. Alternatively, our findings may reflect important differences between the roles of TOR and Vps34 in vivo versus in cultured cells as well as the experimental paradigms of these systems. For example, although complete starvation is commonly used to inactivate TOR in cell culture studies, such experiments may not accurately mimic physiologically relevant events, given the inherent capacity of intact organisms to buffer changes in nutrient levels. Additional studies in an in vivo mammalian system will be helpful to clarify these issues.
Materials and methods
Fly stocks and culture
Flies were raised at 25°C on standard cornmeal/molasses/agar media. The following D. melanogaster stocks were used: P[GawB]NP1626 (Kyoto stock center), Df(2R)bw5 and UAS-YFP-Rab5 (Bloomington stock center), UAS-GFP-2xFYVE, UAS-myc-2xFYVE, and UAS-Rab5-GFP (gifts of M. Gonzalez-Gaitan, Max Planck Institute, Dresden, Germany), UAS-HRP-Lamp1, UAS-GFP-Lamp1, and FRT42D Vps28[k16503] (gifts of H. Kramer, University of Texas Southwestern Medical Center, Dallas, TX), UAS-spin-GFP (gift of G. Davis, University of California, San Francisco, San Francisco, CA), Ird1[3] and UAS-Vps15 (gifts of L. Wu, University of Maryland, College Park, MD), FRT42D Vps25[A3] (gift of D. Bilder, University of California, Berkeley, Berkeley, CA), FRTG13 Vps32/shrb[4] (gift of F.-B. Gao, Gladstone Institute, San Francisco, CA), UAS-Tor dsRNA line 5092R-2 (National Institute of Genetics), UAS-Tsc1 and UAS-Tsc2 (gifts of N. Tapon, London Research Institute, London, UK), Rheb[EP50.084] (gift of E. Hafen, Institute for Molecular Systems Biology, Zürich, Switzerland), Tsc2[192] FRT80B (gift of N. Ito, Massachussetts General Hospital, Charlestown, MA), Tor[ΔP] (Zhang et al., 2000), Tor[R97C] (Zhang et al., 2006), and Atg1[Δ3D] (Scott et al., 2007).
Null and hypomorphic alleles of Vps34 were generated as previously described (Scott et al., 2004) through imprecise excision of P[GawB]NP1626 (Hayashi et al., 2002), inserted after base 13 of the annotated PI3K_59F transcript (Flybase ID FBtr0072064). Excision events were characterized by PCR amplification of the Vps34 locus using the primers 5′-GCTGGAGGAGCACAGATCCGAGACG-3′ and 5′-CAGCAGCTCCATCATCTTGAACAGGAACC-3′.
RT-PCR of wild-type and mutant animals
Total RNA was isolated from 5 mg of larvae per genotype using RevertAid Total RNA Isolation kit (Fermentas). cDNA was synthetized using a First Strand cDNA Synthesis kit (Fermentas) with random primers and used in PCR reactions with the Vps34-specific primers 5′-GGTGTTCAACCAGGGCAGAC-3′ and 5′-GTCCGACATACGCTTTTCGC-3′, with primers 5′-GTCGCTTAGCTCAGCCTCG-3′ and 5′-TAACCCTCGTAGATGGGCAC-3′ specific to actin as control.
Construction of transgenic D. melanogaster lines
All P element constructs were injected into w− embryos according to standard procedures.
UAS-GFP-Atg8a.
A NotI–XbaI fragment containing the GFP-Atg8a coding region was excised from pCasper-HS-GFP-Atg8a (Scott et al., 2004) and ligated into NotI–XbaI-digested pUAST.
UAS-Atg6.
Atg6/CG5429 EST LD35669 was cloned from the vector pOT2 into pUAST using XhoI and EcoRI restriction sites.
UAS-Myc-Atg6.
The Atg6 coding region was PCR amplified from EST LD35669 using the primers 5′-CACCATGAGTGAGGCGGAA-3′ and 5′-TCACGGTGACACAAACTGTG-3′, cloned into pENTR/D-TOPO, and then recombined into the D. melanogaster Gateway vector pTMW using LR clonase.
UAS-Vps34.
The D. melanogaster Vps34/PI3K_59F ORF was PCR amplified from EST GH13170 and ligated into the pUAST vector. A kinase-defective version of this construct (UAS-Vps34KD) with D805→A and N810→I substitutions was generated by site-directed mutagenesis of pUAST-Vps34 and confirmed by sequencing.
UAS-Vps34-TAP.
TOPO-TA cloning was used to clone the D. melanogaster Vps34/PI3K_59F ORF using the primers 5′-GGGGTACCCCAAAAATGGACC-3′ (introducing a 5′ KpnI site) and 5′-CTAGGATTACTCGCTCCTCC-3′ for N-terminal fusion and 5′-GCAGATCTGATATCATCGCC-3′ and 5′-GGGTACCCTTCCGCCAGTAT-3′ (introducing a 3′ KpnI site) for C-terminal fusion. To generate N-terminal UAS-TAP-Vps34, Vps34 was cloned from TOPO-Vps34(N) into pUAST-NTAP (obtained from A. Veraksa, University of Massachussetts, Boston, MA) using KpnI and XbaI restriction sites. C-terminal UAS-Vps34-TAP was generated by cloning Vps34 from TOPO-Vps34(C) into pUAST-CTAP using EcoRI and KpnI restriction sites.
Histology and imaging
Clonal analysis of fat body tissue and lysotracker staining were performed essentially as previously described (Scott et al., 2007), except that LysoTracker Red DND-99 (Invitrogen) was used at 100 nM. TR avidin uptake was performed as previously described (Hennig et al., 2006), with pulse and chase times as indicated. For immunohistochemistry, the following antibodies were used: mouse monoclonal anti–myc (9E10; 1:1,000; gift of M. Stewart, North Dakota State University, Fargo, ND); mouse anti–D. melanogaster Golgi (p120; 1:150; EMD); rabbit anti-hVps34 (1:300; Siddhanta et al., 1998); chicken anti-Avl (1:1,000; gift of D. Bilder); chicken anti-ubiquitin (1:200; eBioscience); and mouse monoclonal anti-Notch extracellular domain (F461.3B; 1:10; Developmental Studies Hybridoma Bank). Images of live lysotracker-stained fat bodies were obtained on an epifluorescence microscope (Axioscope-2; Carl Zeiss, Inc.) with a Plan-Neofluar 40× 0.75 NA objective, coupled to a digital camera (DXM1200; Nikon) controlled by ACT-1 acquisition software (Nikon). Confocal images were acquired on a microscope (Axioplan-2; Carl Zeiss, Inc.) with a Plan-Apochromat 63× 1.4 NA objective, equipped with a CARV spinning disc confocal system and a digital camera (ORCA-ER; Hamamatsu). Axiovision software (Carl Zeiss, Inc.) was used for acquisition, and images were further processed using Photoshop CS2 (Adobe).
GFP-Atg8 quantification
24-h collections of larvae carrying hs-GFP-Atg8a, Lsp2-GAL4, and UAS transgenes were raised to third instar, heat shocked for 1 h at 37°C, and incubated at 25°C for 3 h under feeding or starvation conditions. For starvation experiments, incubations were done in a dish with a wet kimwipe. For feeding experiments, feeding larvae were scooped, along with food, into a dish for incubations. Larval fat body was dissected in phosphate-buffered saline 3 h after completion of heat shock and mounted in SlowFade Gold antifade reagent (Invitrogen). Fat body samples were imaged with a confocal laser scanning microscope (LSM510; Carl Zeiss, Inc.) using a 40× objective. For each genotype, three fields in each of 10 animals were imaged and GFP-positive spots were counted.
Transmission EM
Tissues were dissected and fixed overnight at 4°C in 3.2% PFA, 1% glutaraldehyde, 1% sucrose, and 0.028% CaCl2 in 0.1 N sodium cacodylate, pH 7.4, thoroughly washed in 0.1 N sodium cacodylate, pH 7.4, postfixed in 0.5% osmium tetroxide for 1 h, and embedded in Durcupan resin (Sigma-Aldrich) according to the manufacturer's recommendations. 70-nm sections were cut, stained in Reynolds lead citrate, and viewed on a transmission electron microscope (CM-II 100; JEOL). Immuno-EM was performed as described in Juhasz et al. (2007).
TEM morphometry and statistics
Five images per section were taken randomly at a magnification of 2,700 from five larvae per genotype (25 pictures). The area of autophagosomes, autolysosomes, and total cytoplasm was calculated using Photoshop by manually encircling relevant structures. Values are given as percent of total cytoplasm ± SD. Significance values were calculated by STATISTICA software (StatSoft) using Mann-Whitney U probes.
Immunoprecipitation and Western blot
After 1-h heat shock and 3-h 25°C recovery incubation, larvae were homogenized in lysis buffer (50 mM Hepes, pH 7.4, 150 mM KCl, 6.5% glycerol, 0.5 mM DTT, 0.1% Triton X-100, and complete protease inhibitor tablet [Roche]) and incubated on ice for 15 min. Samples were spun at 10,000 g for 10 min at 4°C to collect protein, which was stored at −80°C. For each IP, 300 μg of protein was precleared with 50 μl of protein A Sepharose 4B 50% slurry (Invitrogen) on ice for 1 h. Precleared lysate was incubated with 2 μg of antibody (rabbit anti-hVps34 #243 [Siddhanta et al., 1998]) at 4°C for 1 h. 50 μl of protein A Sepharose was added, followed by an additional 1-h incubation at 4°C. Beads were collected by centrifugation at 10,000 g for 30 s, washed with lysis buffer, and resuspended in 25 μl of Laemmli buffer. Antigen–antibody complexes were released from beads with a 10-min incubation at 95°C, followed by centrifugation at 10,000 g for 5 min at 4°C. Protein was separated by SDS-PAGE on a 7.5% acrylamide gel and transferred to an Immobilon-P PVDF membrane (Millipore). Membranes were blocked in 3% milk/TBST for 1 h at room temperature and washed three times for 10 min each in TBST. Blots were incubated with primary antibody (anti–c-Myc [1:200] in 2% BSA/TBST) for 1 h at room temperature, followed by three 10-min washes in TBST. Blots were incubated in goat anti–rabbit-HRP secondary antibody and diluted 1:2,000 in 3% milk/TBST for 1 h at room temperature. Blots were washed in TBST and then incubated with ECL reagent (GE Healthcare) and exposed with Hyperfilm ECL (GE Healthcare).
Online supplemental material
Fig. S1 displays the localization of early endosomal markers in wild-type and Vps34 mutant cells. Fig. S2 shows the effect of Vps34 on additional cellular markers. Fig. S3 shows the effects of Vps34 and 15 on markers of autophagy. Fig. S4 displays endocytic and autophagic phenotypes of Vps25-Vps34 mutants. Fig. S5 includes TEM and confocal analysis of Vps25-Vps34 single and double mutants. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200712051/DC1.
Supplementary Material
Acknowledgments
We thank David Bilder, Graeme Davis, Marcos Gonzalez-Gaitan, Fen-Biao Gao, Ernst Hafen, Naoto Ito, Helmut Kramer, Mary Stewart, Nicolas Tapon, Louisa Wu, and Jun Zhang for gifts of fly strains and antibodies, Laura Muller for cell size quantitation, Balázs Érdi for preparing RNAs and cDNAs, and Sarolta Pálfia, Gáborné Komjáti, and Mariann Saródy for technical assistance with EM.
This work was supported by National Institutes of Health grants GM59136 (E.H. Baehrecke), DK070679 (J.M. Backer), and GM62509 (T.P. Neufeld). G. Juhász is a Bólyai Fellow.
Abbreviations used in this paper: dsRNA, double-stranded RNA; ESCRT, endosomal sorting complex required for transport; PI(3), phosphatidylinositol 3; PI(3)P, PI(3) phosphate; TOR, target of rapamycin; TR, Texas red.
References
- Arsham, A.M., and T.P. Neufeld. 2006. Thinking globally and acting locally with TOR. Curr. Opin. Cell Biol. 18:589–597. [DOI] [PubMed] [Google Scholar]
- Britton, J.S., W.K. Lockwood, L. Li, S.M. Cohen, and B.A. Edgar. 2002. Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev. Cell. 2:239–249. [DOI] [PubMed] [Google Scholar]
- Byfield, M.P., J.T. Murray, and J.M. Backer. 2005. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem. 280:33076–33082. [DOI] [PubMed] [Google Scholar]
- Chintapalli, V.R., J. Wang, and J.A. Dow. 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39:715–720. [DOI] [PubMed] [Google Scholar]
- Eskelinen, E.L. 2005. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 1:1–10. [DOI] [PubMed] [Google Scholar]
- Fimia, G.M., A. Stoykova, A. Romagnoli, L. Giunta, S. Di Bartolomeo, R. Nardacci, M. Corazzari, C. Fuoco, A. Ucar, P. Schwartz, et al. 2007. Ambra1 regulates autophagy and development of the nervous system. Nature. 447:1121–1125. [DOI] [PubMed] [Google Scholar]
- Futter, C.E., L.M. Collinson, J.M. Backer, and C.R. Hopkins. 2001. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol. 155:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly, D.J., I.C. Morrow, M. Lindsay, R. Gould, N.J. Bryant, J.M. Gaullier, R.G. Parton, and H. Stenmark. 2000. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19:4577–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, S., K. Ito, Y. Sado, M. Taniguchi, A. Akimoto, H. Takeuchi, T. Aigaki, F. Matsuzaki, H. Nakagoshi, T. Tanimura, et al. 2002. GETDB, a database compiling expression patterns and molecular locations of a collection of Gal4 enhancer traps. Genesis. 34:58–61. [DOI] [PubMed] [Google Scholar]
- Hennig, K.M., J. Colombani, and T.P. Neufeld. 2006. TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J. Cell Biol. 173:963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J., and D.J. Klionsky. 2007. Autophagy and human disease. Cell Cycle. 6:1837–1849. [DOI] [PubMed] [Google Scholar]
- Juhasz, G., B. Erdi, M. Sass, and T.P. Neufeld. 2007. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 21:3061–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz, G., and T.P. Neufeld. 2006. Autophagy: a forty-year search for a missing membrane source. PLoS Biol. 4:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada, Y., T. Funakoshi, T. Shintani, K. Nagano, M. Ohsumi, and Y. Ohsumi. 2000. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150:1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara, A., Y. Kabeya, Y. Ohsumi, and T. Yoshimori. 2001. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, A.L., Z. Palfia, G. Rez, T. Vellai, and J. Kovacs. 2007. Sequestration revisited: integrating traditional electron microscopy, de novo assembly and new results. Autophagy. 3:655–662. [DOI] [PubMed] [Google Scholar]
- Lee, J.A., A. Beigneux, S.T. Ahmad, S.G. Young, and F.B. Gao. 2007. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 17:1561–1567. [DOI] [PubMed] [Google Scholar]
- Linassier, C., L.K. MacDougall, J. Domin, and M.D. Waterfield. 1997. Molecular cloning and biochemical characterization of a Drosophila phosphatidylinositol-specific phosphoinositide 3-kinase. Biochem. J. 321:849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindmo, K., and H. Stenmark. 2006. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 119:605–614. [DOI] [PubMed] [Google Scholar]
- Mari, M., and F. Reggiori. 2007. Shaping membranes into autophagosomes. Nat. Cell Biol. 9:1125–1127. [DOI] [PubMed] [Google Scholar]
- Nara, A., N. Mizushima, A. Yamamoto, Y. Kabeya, Y. Ohsumi, and T. Yoshimori. 2002. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 27:29–37. [DOI] [PubMed] [Google Scholar]
- Neufeld, T.P. 2004. Role of autophagy in developmental cell growth and death: insights from Drosophila. In Autophagy. D.J. Klionsky, editor. Landes Bioscience, Georgetown. 224-232.
- Neufeld, T.P., and E.H. Baehrecke. 2008. Eating on the fly: function and regulation of autophagy during cell growth, survival and death in Drosophila. Autophagy. http://www.landesbioscience.com/journals/3/article/5782 (accessed April 30, 2008). [DOI] [PMC free article] [PubMed]
- Nobukuni, T., M. Joaquin, M. Roccio, S.G. Dann, S.Y. Kim, P. Gulati, M.P. Byfield, J.M. Backer, F. Natt, J.L. Bos, et al. 2005. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA. 102:14238–14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre, S., A. Tassa, X. Qu, R. Garuti, X.H. Liang, N. Mizushima, M. Packer, M.D. Schneider, and B. Levine. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 122:927–939. [DOI] [PubMed] [Google Scholar]
- Pattingre, S., L. Espert, M. Biard-Piechaczyk, and P. Codogno. 2007. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 90:313–323. [DOI] [PubMed] [Google Scholar]
- Petiot, A., E. Ogier-Denis, E.F. Blommaart, A.J. Meijer, and P. Codogno. 2000. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275:992–998. [DOI] [PubMed] [Google Scholar]
- Radimerski, T., J. Montagne, M. Hemmings-Mieszczak, and G. Thomas. 2002. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev. 16:2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori, F., C.W. Wang, U. Nair, T. Shintani, H. Abeliovich, and D.J. Klionsky. 2004. Early stages of the secretory pathway, but not endosomes, are required for Cvt vesicle and autophagosome assembly in Saccharomyces cerevisiae. Mol. Biol. Cell. 15:2189–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Row, P.E., B.J. Reaves, J. Domin, J.P. Luzio, and H.W. Davidson. 2001. Overexpression of a rat kinase-deficient phosphoinositide 3-kinase, Vps34p, inhibits cathepsin D maturation. Biochem. J. 353:655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusten, T.E., K. Lindmo, G. Juhasz, M. Sass, P.O. Seglen, A. Brech, and H. Stenmark. 2004. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell. 7:179–192. [DOI] [PubMed] [Google Scholar]
- Rusten, T.E., T. Vaccari, K. Lindmo, L.M. Rodahl, I.P. Nezis, C. Sem-Jacobsen, F. Wendler, J.P. Vincent, A. Brech, D. Bilder, and H. Stenmark. 2007. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 17:1817–1825. [DOI] [PubMed] [Google Scholar]
- Saucedo, L.J., X. Gao, D.A. Chiarelli, L. Li, D. Pan, and B.A. Edgar. 2003. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 5:566–571. [DOI] [PubMed] [Google Scholar]
- Scott, R.C., O. Schuldiner, and T.P. Neufeld. 2004. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell. 7:167–178. [DOI] [PubMed] [Google Scholar]
- Scott, R.C., G. Juhasz, and T.P. Neufeld. 2007. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddhanta, U., J. McIlroy, A. Shah, Y. Zhang, and J.M. Backer. 1998. Distinct roles for the p110α and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J. Cell Biol. 143:1647–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K., Y. Kubota, T. Sekito, and Y. Ohsumi. 2007. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 12:209–218. [DOI] [PubMed] [Google Scholar]
- Takahashi, Y., D. Coppola, N. Matsushita, H.D. Cualing, M. Sun, Y. Sato, C. Liang, J.U. Jung, J.Q. Cheng, J.J. Mul, et al. 2007. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9:1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassa, A., M.P. Roux, D. Attaix, and D.M. Bechet. 2003. Class III phosphoinositide 3-kinase–Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 376:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari, T., and D. Bilder. 2005. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev. Cell. 9:687–698. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig, T., M. Wilsch-Brauninger, and M. Gonzalez-Gaitan. 2003. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol. 161:609–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu, T., and D.J. Klionsky. 2005. Autophagy: molecular machinery for self-eating. Cell Death Differ. 12:1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., J.P. Stallock, J.C. Ng, C. Reinhard, and T.P. Neufeld. 2000. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 14:2712–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., C.J. Billington Jr., D. Pan, and T.P. Neufeld. 2006. Drosophila target of rapamycin kinase functions as a multimer. Genetics. 172:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}