

Abstract
Background
The emergence of multi- and extensively-drug resistant Mycobacterium tuberculosis strains has created an urgent need for new agents to treat tuberculosis (TB). The enzymes of shikimate pathway are attractive targets to the development of antitubercular agents because it is essential for M. tuberculosis and is absent from humans. Chorismate synthase (CS) is the seventh enzyme of this route and catalyzes the NADH- and FMN-dependent synthesis of chorismate, a precursor of aromatic amino acids, naphthoquinones, menaquinones, and mycobactins. Although the M. tuberculosis Rv2540c (aroF) sequence has been annotated to encode a chorismate synthase, there has been no report on its correct assignment and functional characterization of its protein product.
Results
In the present work, we describe DNA amplification of aroF-encoded CS from M. tuberculosis (MtCS), molecular cloning, protein expression, and purification to homogeneity. N-terminal amino acid sequencing, mass spectrometry and gel filtration chromatography were employed to determine identity, subunit molecular weight and oligomeric state in solution of homogeneous recombinant MtCS. The bifunctionality of MtCS was determined by measurements of both chorismate synthase and NADH:FMN oxidoreductase activities. The flavin reductase activity was characterized, showing the existence of a complex between FMNox and MtCS. FMNox and NADH equilibrium binding was measured. Primary deuterium, solvent and multiple kinetic isotope effects are described and suggest distinct steps for hydride and proton transfers, with the former being more rate-limiting.
Conclusion
This is the first report showing that a bacterial CS is bifunctional. Primary deuterium kinetic isotope effects show that C4-proS hydrogen is being transferred during the reduction of FMNox by NADH and that hydride transfer contributes significantly to the rate-limiting step of FMN reduction reaction. Solvent kinetic isotope effects and proton inventory results indicate that proton transfer from solvent partially limits the rate of FMN reduction and that a single proton transfer gives rise to the observed solvent isotope effect. Multiple isotope effects suggest a stepwise mechanism for the reduction of FMNox. The results on enzyme kinetics described here provide evidence for the mode of action of MtCS and should thus pave the way for the rational design of antitubercular agents.
Background
Tuberculosis (TB) remains a major global health concern. Its causative agent, Mycobacterium tuberculosis, has been estimated to infect approximately one-third of the world's population [1], and approximately 30 million people have died from the disease in the past decade [2]. The World Health Organization estimated a total of 9 million new cases of TB and approximately 2 million deaths from this disease in 2004, second only to AIDS among infectious diseases [3]. The emergence of drug resistant isolates of M. tuberculosis, particularly of multi drug-resistant TB (MDR-TB), defined as resistant to at least isoniazid and rifampicin [4], imposes a great challenge to public health [5]. Treatment of MDR-TB requires the administration of second-line drugs that are more toxic and less effective [6]. More recently, it was reported cases of extensively drug-resistant (XDR) TB, which are defined as cases in persons with TB whose isolates were resistant to isoniazid and rifampicin and at least three of the six main classes of second-line drugs (aminoglycosides, polypeptides, fluoroquinolones, thioamides, cycloserine, and para-aminosalicylic acid) [7]. XDR-TB has a wide geographic distribution and it raises the bleak prospect of a future epidemic of virtually untreatable TB. New antimycobacterial agents are thus needed to improve the treatment of MDR- and XDR-TB, to shorten the treatment course and increase patient compliance, and to provide more effective treatment of latent TB infection.
A valuable approach to the development of selective antimicrobial chemotherapy is to exploit the inhibition of targets unique and vital to the pathogen [8]. The enzymes of the shikimate pathway are attractive examples of these targets because this route is essential in higher plants, fungi, bacteria and algae and is absent in mammals [9,10]. In M. tuberculosis, the shikimate pathway leads to the biosynthesis of a wide range of primary and secondary metabolites, including aromatic amino acids, folate, naphthoquinones, menaquinones and mycobactins [11]. Disruption of aroK-encoding shikimate kinase gene has recently shown that the shikimate pathway is essential for M. tuberculosis viability [12], which establishes the enzymes of this pathway as potential targets for the development of new antimycobacterial agents.
Homologues to the shikimate pathway enzymes were identified in the complete genome sequence of M. tuberculosis H37Rv [13]. Among them, chorismate synthase (CS; EC 4.2.3.5; 5-O-(1-carboxyvinyl)-3-phosphoshikimate phosphate lyase) encoding gene (aroF, Rv2540c) was proposed to be present by sequence homology. CS catalyzes an unusual 1,4-anti-elimination of the 3-phosphate group and the C-(6proR) hydrogen from 5-enolpyruvylshikimate-3-phosphate (EPSP) forming chorismate and phosphate [14]. Although there is no overall change in its redox state, there is an absolute requirement for a reduced flavin mononucleotide (FMNred) [15,16]. Another interesting feature of CSs from different organisms is how reduced FMNred is obtained, which divides these enzymes into two classes: monofunctional and bifunctional [17]. The CSs from fungi are bifunctional as they display a second enzymatic activity (Figure 1), an NAD(P)H-dependent (dependent of reduced form of nicotinamide adenine dinucleotide) flavin reductase (described as "diaphorase" activity in a number of papers), which confers them an intrinsic ability to reduce flavin using NAD(P)H [18]. The CSs from plants and Escherichia coli are monofunctional as they do not possess this activity and are active only in anaerobic conditions in the presence of either chemically or enzymatically reduced flavin [16]. However, there has been no report on both confirmation of the correct assignment of Rv2540c (aroF) as the M. tuberculosis chorismate synthase (MtCS) coding sequence and functional characterization of its protein product. Moreover, there has been no report on whether MtCS is bifunctional or monofunctional.
Figure 1.
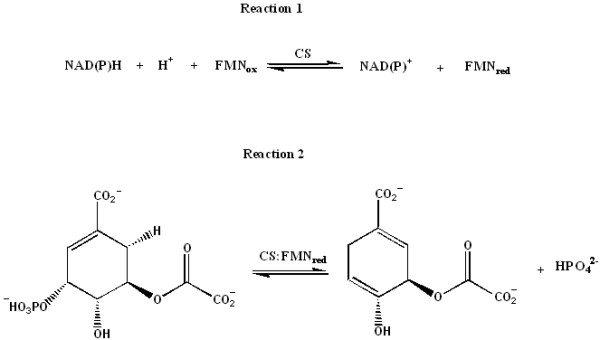
Reactions catalysed by bifunctional CSs.
Here we report the amplification and cloning of Rv2540c DNA sequence (putative aroF gene) from M. tuberculosis, and heterologous overexpression of the MtCS protein. The recombinant protein was purified to homogeneity using a three-step purification protocol and its identity was confirmed by N-terminal sequencing, and electrospray ionization mass spectrometry (ESI-MS). The oligomeric state of MtCS was determined by gel filtration. We also show that CS from M. tuberculosis is bifunctional, as in fungi, based on activity measurements of both chorismate synthase and NADH-dependent flavin reductase activities. Moreover, FMN appears to be bound to MtCS based on ionic exchange chromatography, and FMNox (oxidized flavin mononucleotide) and NADH binding to MtCS were assessed by spectroscopic measurements at equilibrium. The apparent kinetic constants for the holoenzyme MtCS-FMNox for NADH were determined. The NADH-dependent flavin reductase activity was characterized by isotope effects. Measurements of primary deuterium and solvent kinetic isotope effects were carried out to probe the nature of the rate-limiting step of the redox reaction. Multiple isotope effects were also determined showing that a stepwise mechanism is involved in this reaction. Proton inventory on the maximal velocity allowed us to address the number of kinetically important transferred protons. The results presented here are important to understand the reaction mechanism of this enzyme, which should pave the way for the rational-based design of MtCS inhibitors with potential antitubercular activity and low toxicity.
Results and Discussion
Molecular cloning, expression, and purification of MtCS
PCR amplification of Rv2540c DNA sequence (putative aroF gene) from genomic M. tuberculosis DNA yielded a fragment with the expected length (1206 bp). The fragment was cloned into pET23a(+) expression vector, and the aroF gene was sequenced, which confirmed its identity and the absence of PCR (Polymerase Chain Reaction)-introduced mutations. A number of E. coli host strains were employed to express MtCS, including BL21(DE3), Origami(DE3), and BL21trxB(DE3), but no expression could be obtained. Different cultivating temperatures (25°, 30° and 37°C) and presence or absence of isopropyl β-D-thiogalactoside (IPTG) were also employed to no avail (data not shown). MtCS overexpression could only be achieved in E. coli Rosetta(DE3) host cells, which provide tRNAs for codons that are rarely used by E. coli. In agreement, ten rare codons were identified on M. tuberculosis aroF sequence (7 × CCC, AUA, CUA, GGA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis revealed a cell extract containing a significant amount of a soluble protein with an apparent molecular weight in agreement with the predicted value based on amino acid sequence for MtCS (41.8 kDa) (Figure 2A).
Figure 2.
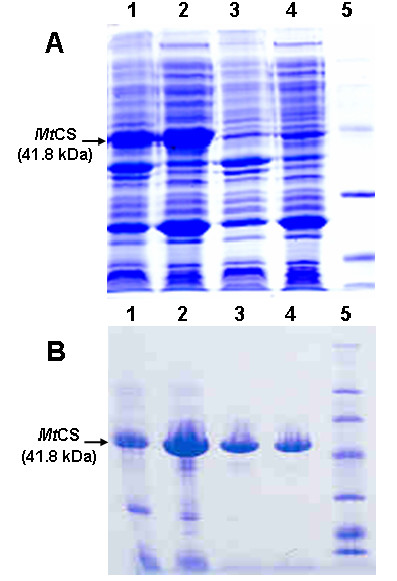
Recombinant MtCS protein expression and purification. (A) Analysis of MtCS expression using E. coli Rosetta(DE3) host cells by SDS-poliacrilamide gel 12%. Lanes: 1 – pellet Rosetta(DE3)+pET23a(+)::aroF; 2 – soluble extract Rosetta(DE3)+pET23a(+)::aroF; 3 – pellet Rosetta(DE3)+pET23a(+) (negative control); 4 – soluble extract Rosetta(DE3)+pET23a(+) (negative control); 5 – protein molecular weight standards, high range (Gibco) (43 kDa; 29 kDa; 18 kDa; 14 kDa) (B) Analysis of purification steps of MtCS by SDS-polyacrylamide gel 12%. Lanes: 1 – soluble extract after dialysis; 2 – soluble extract after Q Sepharose Fast Flow column; 3 – CS after Phenyl-Sepharose column; 4 – CS after MonoQ column; 5 – protein molecular weight standards, high range (Gibco) (200 kDa; 97 kDa; 68 kDa; 43 kDa; 29 kDa; 18 kDa; 14 kDa).
Interestingly, recombinant protein expression was achieved in E. coli Rosetta(DE3) host cells grown for 24 h at 37°C in the absence of IPTG induction. It has been previously shown that the lac-controlled systems, including the pET system, could have a high-level protein expression, in the absence of inducer. This phenomenon occurs in the cell stationary phase, when there is the presence of a complex medium, cyclic AMP, acetate and low pH [19]. Recently, another shikimate pathway enzyme from M. tuberculosis, 3-Deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) synthase, was reported to be expressed only in the absence of IPTG [20]. The three-step purification protocol yielded approximately 55 mg of MtCS from 6 liters of cell culture. No other protein can be observed on SDS-PAGE, which indicates the high level of MtCS purity (Figure 2B). The MtCS activity before the purification can hardly be measured, probably due to interfering compounds, and thus only the amount of total recombinant protein could be assessed. The ammonium sulfate precipitation of purified MtCS resulted in total inactivation of the enzyme (data not shown). Homogeneous recombinant protein was thus instantaneously frozen in liquid nitrogen in the absence of ammonium sulfate, which proved to be a convenient protocol for active homogeneous recombinant MtCS protein storage.
Analysis of purified MtCS
The ESI-MS of homogeneous MtCS revealed a subunit molecular mass of 41,804 Da, consistent with the theoretical molecular mass (41,792 Da). The first 16 N-terminal amino acid residues were identified as MLRWITAGESHGRALV, which confirmed the identity of homogeneous MtCS and presence of N-terminal methionine residue. Analytical gel filtration chromatography revealed a single peak of approximately 104 kDa, suggesting that MtCS is a dimer in solution, which is in agreement with the hydrodynamic properties of the recombinant protein assessed by sedimentation velocity and sedimentation equilibrium [18].
The CS activity of MtCS
The synthesis of EPSP was carried out in a vial containing the enzymes MtSK and MtEPSPS, which convert shikimate, ATP and phosphoenolpyruvate (PEP) to 5-enolpyruvylshikimate-3-phosphate (EPSP), ADP and Pi. The equilibrium of the forward reaction was displaced using Purine Nucleoside Phosphorylase (PNP), which consumes Pi, increasing the final concentration of EPSP in the reaction mixture. The enzymes were removed by ultrafiltration to avoid any residual MtEPSPS enzyme activity that could release Pi in solution, which would thus interfere with specific measurements of MtCS enzyme activity. Conversion of EPSP to chorismate and Pi catalyzed by MtCS enzyme activity (Figure 1) was determined by measuring the release of Pi using PNP, which converts 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG) and Pi to ribose-1-phosphate and 7-methyl-6-thio-guanine base, monitoring absorbance of the latter at 360 nm. The enzymatic activity of homogenous MtCS was dependent on enzyme volume added to the reaction mixture (data not shown), showing that the initial velocity is proportional to total enzyme concentration and that true initial velocities are being measured. The conversion of EPSP to chorismate in aerobic conditions by the addition of FMNox and NADH is the first solid evidence that MtCS is bifunctional, since the monofunctional CSs can only be assayed under strictly anaerobic conditions in the presence of chemically or enzymatically reduced flavin [16]. The specific activity of bifunctional MtCS (0.004 U.mg-1) is approximately 175-fold lower than the specific activity of bifunctional Neurospora crassa CS (NcCS; 0.7 U.mg-1) [21]. In the presence of oxygen the CS activity is limited by the reoxidation of FMNred cofactor; but in bifunctional CSs, NAD(P)H is used in consecutive cycles to maintain the FMN in its reduced form (Figure 1). Moreover, the structures of CS complexed with FMN from Helicobacter pylori [22] and Streptococcus pneumoniae [23] have shown that there are a number of positively charged amino acids in the FMN binding pocket, which could increase the reduction potential of FMNox/FMNred couple, likely permitting the decrease of the rate of reoxidation. Indeed, in bifunctional NcCS the redox potential of the couple FMNred/FMNox was determined to be -167 mV, i.e. 40 mV more positive than that of the free couple in solution (-207 mV) [24].
The NADH:FMN-oxidoreductase activity of MtCS
The NADH-dependent reduction of FMNox was measured in the forward reaction monitoring the decrease in NADH concentration. The NADH:FMN-oxidoreductase activity of homogenous MtCS is dependent on the enzyme volume added to the reaction mixture; therefore the initial velocity is proportional to total enzyme concentration, and true initial velocities are being measured. To verify the stability of the MtCS-FMNox complex, MtCS was incubated with excess FMNox and loaded on an anion exchange column. Two peaks were observed and collected into separated fractions measuring absorbance at 280 nm and 445 nm, and pooled (Figure 3). To roughly estimate the presence of protein in the two pools, coomassie-blue was added to them and only the second pool was shown to contain protein (indicating presence of recombinant MtCS), while the first was not stained (indicating absence of recombinant MtCS). The UV-Vis spectrum for the second pool containing recombinant MtCS protein showed that all the characteristic absorbance peaks of FMNox (267, 373, and 445 nm) could be observed (Figure 3 inset). These results suggest that MtCS and FMNox form a stable binary complex.
Figure 3.
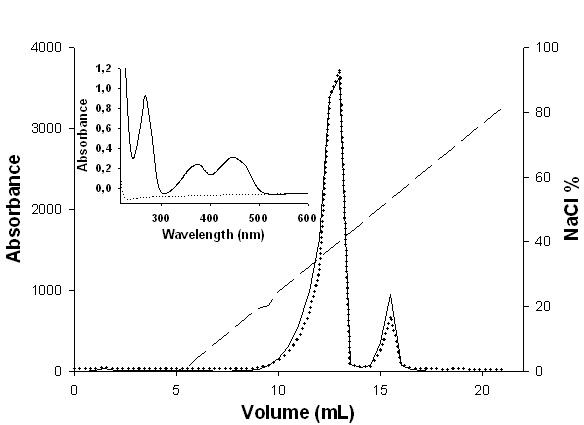
Elution profile of incubated MtCS and FMNox, and UV-Vis spectrum showing stable binary complex formation. Elution of MtCS and FMNoxincubation in a HiTrapQ HP column upon a linear gradient from 0 to 1 M NaCl (dashed line). The absorbance was monitored at 445 nm (dotted line) and 280 nm (solid line). The first peak corresponds to free FMNox, and the second peak corresponds to MtCS-FMNox complex. Inset: UV-Vis spectrum ranging from 210 nm to 600 nm of the second pool (solid line) and Tris-HCl (dotted line). All characteristic peaks of FMNox can be observed (267 nm, 373 nm, and 445 nm).
Although the FMNox usually appears as a prosthetic group in NAD(P)H:FMN oxidoreductases from other organisms [25], it was not clear whether in MtCS the FMN is covalently bound. CSs from other organisms, such as that from E. coli, showed low affinity for FMNox; but the value of Kd for FMNox depends strongly on EPSP binding, decreasing ca. 1000-fold in the presence of this substrate (from 30 μM to 20 nM) [26]. On the other hand, EPSP has a much smaller effect on the affinity of bifunctional NcCS to FMNox [21]. Interestingly, the purified MtCS is FMN-free, which could be an effect of the insufficient intracellular concentration of FMN, probably due to MtCS overexpression into the host cell. As described below, FMN appears to have a value lower than 20 μM for the overall dissociation constant for MtCS-FMN binary complex formation at equilibrium. Accordingly, it appears to be more likely that FMN is lost during the protein purification protocol because of a rather weak affinity of MtCS for FMN. Accordingly, the kinetic data were collected considering the holoenzyme as the MtCS-FMNox complex brought about by incubating MtCS and near-saturating FMNox concentration.
The oxidoreductase activity is functionally independent of CS activity, since the consumption of NADH occurs in the absence of EPSP. Considering the low specific activity of NADH:FMN-oxidoreductase in MtCS, all measurements were made in triplicate, including the controls. Accordingly, the apparent kinetic parameters for the oxidoreductase activity were determined in the absence of EPSP in steady-state kinetic experiments. No activity was observed when NADPH was used at concentrations ranging from 100 to 500 μM (data not shown). Kinetics of reduction of the MtCS-bound FMNox by NADH was determined using fixed amount of holoenzyme and varying levels of NADH (Figure 4). The data were fitted to equation 1, and yielded the following values: KNADH = 36 ± 4 μM, kcat = 8.3 (± 0.3) × 10-3 s-1 and kcat/KNADH = 231 M-1 s-1. The KNADH observed for MtCS is similar to the KNADPH determined for NcCS [24] and ScCS [27] (43 μM and 70 μM, respectively). In other NAD(P)H:FMN oxidoreductases (EC 1.5.1.30), which are present in various organisms, the apparent kinetic constants show significant variation. For instance, the KNAD(P)H can vary from 208 μM in Rhodococcus erythropolis [28] to 0.85 μM in Bacillus subtilis [29].
Figure 4.
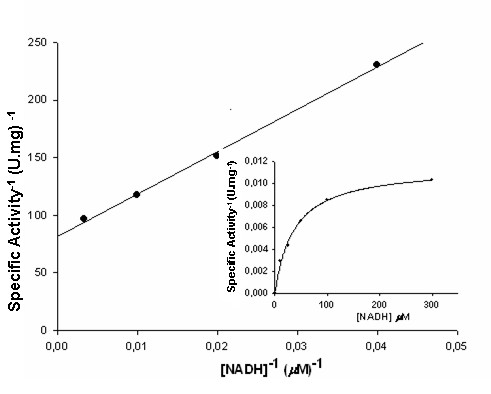
Determination of steady-state kinetic parameters. Double-reciprocal plot of initial velocity data for holoenzyme MtCS-FMNox in the presence of varying concentration of NADH (10, 25, 50, 75, 100, 200, 300 μM). Inset: Michaelis-Menten representation. The data were fitted to equation 1, yielding the following values: KNADH = 36 ± 4 μM, kcat = 8.3 (± 0.3) × 10-3 s-1 and kcat/KNADH = 231 M-1 s-1.
Equilibrium measurements of FMNox and NADH binding to MtCS
The binding of FMNox to MtCS was assessed by UV-visible difference spectra as described by Kitzing et al. [21]. The observed spectral changes are characterized by hypochromic effects on the flavin absorbance at both 378 nm and 450 nm up to 20 μM FMNox (Figure 5). At FMNox concentrations larger than 30 μM there is a hyperchromic effect that is probably due to free oxidized FMN in solution (Figure 5 – inset). The spectral changes at low FMNox concentrations yielded low values for the difference spectra and thus no reliable data could be obtained at concentrations lower than 10 μM. Accordingly, the data presented here allow only to estimate an upper limit value of 20 μM for the FMNox equilibrium dissociation constant. We have also tried to determine the dissociation constant of FMNox from protein fluorescence titration as recently described by Broco et al [30], which could allow reliable measurement of oxidized FMN binding to MtCS at low concentrations of FMNox. However, there was no quench/enhancement in protein fluorescence upon FMNox binding to MtCS (data not shown). At any rate, the UV-visible difference spectra show MtCS-FMNox binary complex formation.
Figure 5.
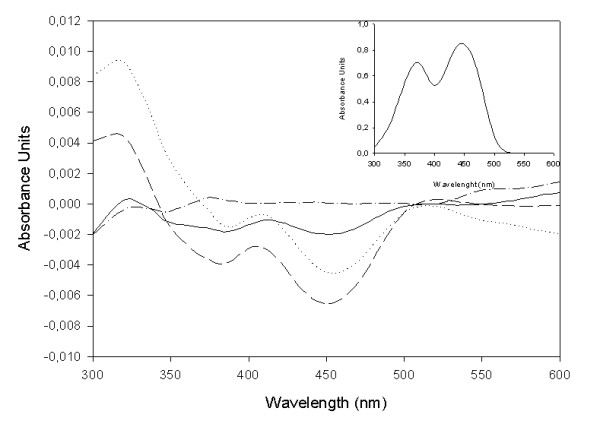
UV-visible difference spectroscopy. Difference absorbance spectra were recorded from 300 to 600 nm. The difference spectra are as follows: dash-dotted line (-·-), 35 μM MtCS; solid line (--), 35 μM MtCS in the presence of 10 μM FMNox; dashed line (- -), 35 μM MtCS in the presence of 20 μM FMNox; and dotted line (·····), 35 μM MtCS in the presence of 30 μM FMNox. The inset shows absorbance spectrum of 100 μM free FMNox.
The equilibrium binding of NADH to MtCS was assessed by fluorescence spectroscopy. There is a quench in protein fluorescence upon NADH binding to MtCS (Figure 6 – inset). A titration curve showing the quench in protein fluorescence upon the binding of NADH to MtCS is given in Figure 6. The data were fitted to a hyperbolic equation yielding a dissociation constant value of 156 ± 12 μM. Even though the NADH-binding site remains unknown, the results presented here clearly indicate that there is indeed an NADH binding site on MtCS enzyme.
Figure 6.
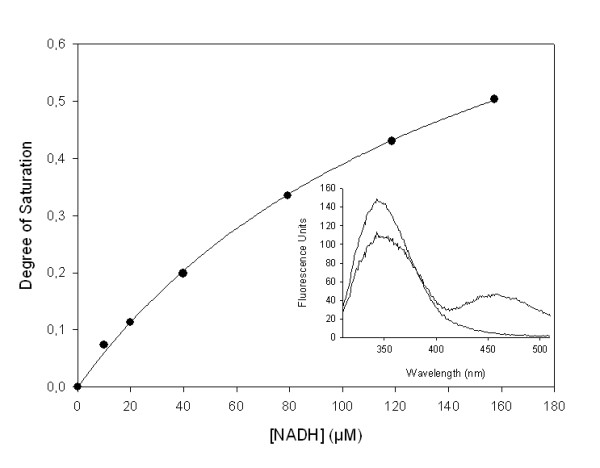
Equilibrium binding of NADH to MtCS assessed by monitoring the protein fluorescence quench upon binary complex formation. The binding of NADH to MtCS causes a quench in protein fluorescence (λexc = 290 nm; 310 ≤ λem ≤ 510 nm; with a maximum at 345.5 nm). The MtCS enzyme solution (1 μM) was titrated with increasing concentrations of NADH, and the data points (fluorescence intensities at 345.5 nm) were fitted to a hyperbolic equation (solid line). Inset: Emission spectra of free MtCS (1 μM) and enzyme in the presence of 80 μM NADH. Emission spectrum of free enzyme shows a peak at 345.5 nm and no emission at ~450 nm. Emission spectrum of MtCS in the presence of NADH shows a quench in protein fluorescence concomitant to an increase in nucleotide fluorescence at ~450 nm.
Primary deuterium kinetic isotope effects
To probe for rate-limiting steps and determine the stereospecificity of hydride transfer, primary deuterium kinetic isotope effects were determined. Initial velocity data were collected using either [4S-1H]NADH or [4S-2H]NADH (Figure 7), and values of 3.5 ± 0.2 and 3.0 ± 0.4 were determined for, respectively, DV and DV/K. The magnitude of the primary isotope effects when the [4S-2H]NADH substrate is used indicates that C4-proS hydrogen is being transferred during the reduction of FMNox catalyzed by MtCS. Isotope effects on V are related to events following the formation of the complex capable of undergoing catalysis (MtCS-FMNox-NADH in the case studied here), including chemical steps, possible enzyme conformational changes, and product release. Isotope effects on V/K report on steps in the reaction mechanism from the binding of the labeled substrate to the first irreversible step, usually product release [31]. The values for primary deuterium isotope effects of biochemical interest usually range from 3 to 7 [32], and steps such as conformational changes accompanying hydride transfer and product release may account for the lower values. In particular, primary deuterium kinetic isotope effects typically range from 1 to 3 for enzyme reactions involving NAD(P)H oxidation [33]. The DV value obtained for MtCS-FMNox (3.5) suggests that the hydride transfer contributes significantly to the rate-limiting step of the FMN reduction reaction. The value of 3.0 for the DV/K indicates that NADH is not sticky, since DV/K values for sticky substrates are around unity [34]. It should be remembered that a substrate is sticky if it reacts to give products as fast as, or faster than, it dissociates from the enzyme. The stickiness of a substrate depends on the external part of its commitment: the larger the external commitment, the sticker the substrate [34].
Figure 7.
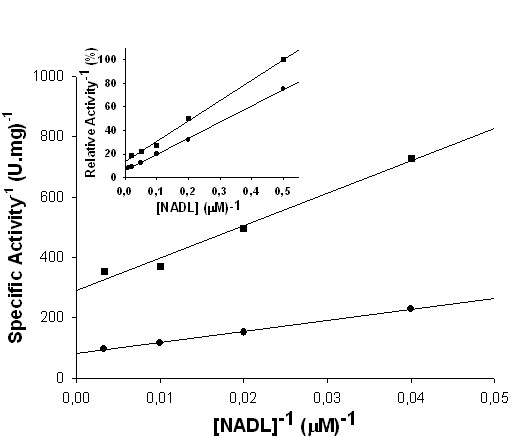
Primary and multiple deuterium kinetic isotope effects. Measurements of primary deuterium kinetic isotope effects for MtCS yielded values of 3.5 ± 0.2 and 3.0 ± 0.4 for, respectively, DV and DV/K. Inset: Measurements of primary isotope effect in 90 atom % D2O (multiple) yielded values of 1.3 ± 0.1 and 2.2 ± 0.1 for D2OV[4S-2H]NADH and D2OV/K[4S-2H]NADH, respectively. In both [4S-1H]NADH (●) or [4S-2H]NADH (■) is varied in the reaction mixture. L represents either [4S-1H]NADH or [4S-2H]NADH.
Solvent isotope effects and proton inventory
Solvent isotope effects were employed to assess the contribution of solvent proton transfer to the rate of flavin reduction. Initial velocity data were collected in either H2O or 91 atom % D2O (Figure 8), and values of 1.7 ± 0.3 and 1.3 ± 0.1 were obtained for D2OV and D2OV/K, respectively. The magnitude of the solvent isotope effects indicate that proton transfer partially limit the rate of FMN reduction reaction. In order to determine the number of kinetically important protons transferred during the reaction, a proton inventory on V was carried out. The mole fraction of D2O was varied from 0 to 80%, and a linear relationship between V and the mole fraction of D2O was observed (Figure 8, inset). This result indicates that a single proton transfer gives rise to the observed solvent isotope effect [35].
Figure 8.
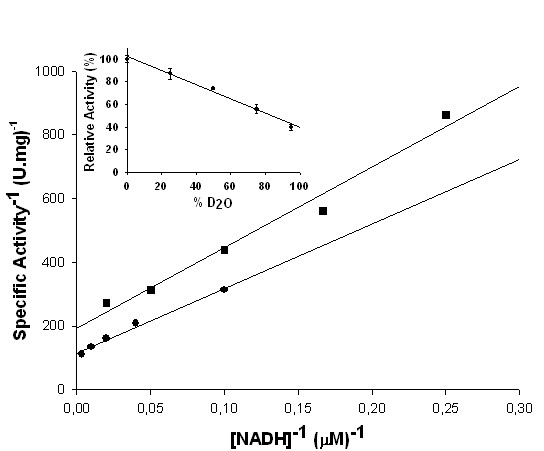
Solvent deuterium kinetic isotope effects and proton inventory. Solvent isotope effects for MtCS. NADH is varied in the reaction mixture containing either 0 (●) or 91 (■) atom % D2O. Values of 1.7 ± 0.3 and 1.3 ± 0.1 were obtained for D2OV and D2OV/K, respectively. Inset: Proton inventory at saturating concentration of NADH, indicating that a single proton transfer from solvent contribute to the rate of flavin reduction.
Multiple isotope effects
Significantly different magnitudes between primary and solvent kinetic isotope effects may indicate that they are reporting on distinct steps of a reaction, as reported for the NADPH-dependent ketoacyl-ACP reductase from S. pneumoniae [36]. The primary isotope effect values (DV = 3.5 and DV/K = 3.0) are larger than those for solvent isotope effects (D2OV = 1.7 and D2OV/K = 1.3), suggesting that proton and hydride transfer may take place in two distinct transition-states, with the latter being more rate-limiting for the MtCS-catalyzed reaction. Multiple isotope effects studies are able to distinguish whether two different isotopic substitutions affect the same or different chemical steps. Theory predicts that if protonation and hydride transfer occur in the same transition state, the primary isotope effects will be larger or unchanged with D2O as compared to H2O. On the other hand, if hydride transfer and protonation occur in distinct steps, the primary isotope effects will be smaller with D2O as solvent, as proton transfer will become more rate limiting [37,38]. Multiple isotope effects were thus evaluated to distinguish whether two different isotopic substitutions affect the same or different chemical steps, i.e., if the reduction of FMNox catalyzed by MtCS occurs, respectively, in a concerted or in a stepwise mechanism. Initial velocity data were collected and values of 1.3 ± 0.1 and 2.2 ± 0.1 were obtained for D2OV[4S-2H]NADH and D2OV/K[4S-2H]NADH, respectively (Figure 7, inset). The values for the primary kinetic isotope effects on V and V/K in D2O were smaller than the effects in H2O, thereby suggesting a stepwise mechanism for the reduction of FMNox. A mechanism for bifunctional NcCS has been proposed and involves an electron transfer from FMNred to C1 EPSP and C-O bond cleavage, protonation of the leaving phosphate group by His17, tautomerization of the resulting C4(a)-neutral flavin semiquinone to a radical species with concomitant abstraction of the C-(6proR) hydrogen of the dephosphorylated substrate intermediate, and reduced flavin deprotonation restores the initial state of the cofactor [24]. More recently, it has been shown for NcCS that the carboxylate group of Asp367 participates in the water-mediated deprotonation of the N(5) atom of the isoalloxazine ring system of FMNred that is involved in abstraction of C-(6proR) hydrogen of EPSP [39]. A comparison of the primary sequences of CSs from different sources has shown that these residues (His11 and Asp343, M. tuberculosis numbering) are conserved [18]. Interestingly, His11 and Asp343 are present in monofunctinal and bifunctional CSs and across all known species [40], and these residues are part of the characteristic CS signature sequence [23]. The role of His11 and Asp343 in the mode of action of bifunctional MtCS should await site-directed mutagenesis studies that are currently underway in our laboratory.
Conclusion
It has recently been pointed out that we are currently unable to predict bifuncionality based on sequence information and three-dimensional structures available at present because of difficulties in identifying the NAD(P)H-binding site of bifunctional CSs [40]. Incidentally, the three-dimensional structural model [41] and X-ray diffraction structure [18] of MtCS employed the crystal structure of CS from Streptococcus pneumoniae [23] as template, which is a monofunctional CS. It is thus necessary to measure CS enzyme activity to show whether it depends on a source of free reduced FMN (monofunctional) or can directly reduce FMN cofactor at the expense of NAD(P)H (bifunctional). Here we present experimental evidence that recombinant MtCS is bifunctional. To the best of our knowledge, this is the first report showing that a bacterial CS is bifunctional. We also show that FMN and NADH bind to free MtCS. Primary deuterium kinetic isotope effects of the NADH-dependent flavin reductase activity of MtCS showed that C4-proS hydrogen is transferred during the reduction of FMNox, and that hydride transfer contributes significantly to the rate-limiting step of FMN reduction. Solvent deuterium kinetic isotope effects suggest that proton transfer from solvent partially limits the rate of FMN reduction and that a single proton is transferred from solvent. Multiple isotope effects indicate that a stepwise mechanism for the reduction of FMNox. Expression of functional proteins in soluble form has been identified as an important bottleneck in efforts to determine biological activity and crystal structure of M. tuberculosis proteins [42]. Moreover, protein purification has become an important asset in any structural genomic effort as there has been increasing demand for homogeneous proteins [43]. Accordingly, the work presented here should contribute to overcome these obstacles for, at least, MtCS and allow functional and structural efforts to be pursued. We provide experimental evidence for the correct assignment of the Rv2540c DNA sequence as a CS-coding aroF gene in M. tuberculosis, which is a pivotal step for the rational design of inhibitors of MtCS enzyme activity with potential antitubercular action. The availability of MtCS and the experimental evidence given here for an NADH-binding site warrant further efforts to obtain the crystal structure of the binary complex to try to elucidate the structural features of this interaction and the mechanism of action for bifunctional CSs. The results reported here should thus pave the way for further functional and structural studies to guide the rational design of antitubercular agents.
Methods
Chemical, reagents, enzymes and bacterial strains
Pfu DNA polymerase was from Stratagene. The pET23a(+) expression vector and E. coli Rosetta(DE3) host cell were from Novagen. All chromatographic supports, including the molecular weight calibration kits, were purchased from GE Healthcare. The protease inhibitor cocktail was from Roche. Purine nucleotide phosphorylase (PNP, EC 2.4.2.1) and 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG) were purchased from Molecular Probes. NADH, NAD+, oxidized FMN (FMNox), ATP, phosphoenolpyruvate (PEP), Leuconostoc mesenteroides glucose-6-phosphate dehydrogenase (type XXIII) and yeast hexokinase (type C-300) were from Sigma Chemical Co. D-glucose-1-d (97 atom % D) was from Aldrich and deuterium oxide (99.9 atom % D) was from Cambridge Isotope Laboratories.
Molecular cloning, and overexpression of M. tuberculosis Rv2540c (aroF) DNA sequence
Synthetic oligonucleotide primers complementary to amino-terminal coding and carboxy-terminal noncoding strands of aroF (Rv2540c) gene (5' ggtcatatgttgcgctggatcaccgcgg 3' and 5' cggatcctcaaccggagacccgcgcggc 3', respectively) were designed based on the complete genome sequence of M. tuberculosis [13]. These primers, containing 5'NdeI and 3'BamHI restriction sites, in bold, were used to amplify aroF-CS encoding gene (1,206 bp) from M. tuberculosis genomic DNA, using Pfu DNA polymerase and standard PCR conditions. The amplified fragment was purified by low melting agarose electrophoresis, digested with NdeI and BamHI, and cloned into pET23a(+) expression vector previously digested with the same restriction enzymes. The aroF gene identity and the absence of PCR-introduced mutations were confirmed by DNA sequencing. The pET23a (+)::aroF was transformed by electroporation into E. coli Rosetta(DE3) host cells and selected on LB agar plates containing 50 μg.mL-1 carbenicillin and 34 μg.mL-1 chloramphenicol. Single colonies were used to inoculate 1 L of Luria-Bertani medium containing the same antibiotics and grown for 24 h at 37°C at 180 rpm with no isopropyl β-D-thiogalactoside (IPTG) induction. The cells (4.5 g) were harvested by centrifugation at 3,000 g for 30 min at 4°C.
Recombinant MtCS protein purification
All purification procedures were performed at 4°C. Cells (25 g) were suspended in 75 mL of 50 mM Tris-HCl, pH 7.8 (buffer A) containing proteinase inhibitor cocktail and 0.2 mg.mL-1 lisozyme; and the mixture was stirred for 30 min. The cells were disrupted by sonication and centrifuged at 48,000 g for 30 min to remove cell debris. Streptomycin sulfate was added to the supernantant to a final concentration of 1% (w/v) and the mixture was stirred for 30 min. The soluble fraction was collected by centrifugation at 48,000 g for 30 min and dialyzed 3 times against 2 L of buffer A. This sample was clarified by centrifugation (48,000 g for 30 min), loaded on a Q-Sepharose Fast Flow (2.6 cm × 8.2 cm) anion exchange column previously equilibrated with buffer A and the proteins eluted using a linear gradient from 0.0 to 0.5 M NaCl. The fractions containing MtCS were pooled and ammonium sulfate was added to a final concentration of 0.8 M. This sample was loaded on a High Load Phenyl-Sepharose (1.6 cm × 10 cm) hydrophobic interaction column pre-equilibrated with 50 mM Tris-HCl, pH 7.8, 0.8 M (NH4)2SO4(buffer B). The proteins were fractionated using a linear gradient from 0.8 to 0.0 M (NH4)2SO4. The active fractions were pooled and dialyzed 3 times against 2 L of buffer A. The sample was loaded on a MonoQ (1.6 cm × 10 cm) anion exchange column previously equilibrated with buffer A, and the proteins eluted using a linear gradient of NaCl (0.0 – 0.5 M). The fractions containing homogeneous MtCS were pooled, quickly frozen in liquid nitrogen and stored at -80°C. Samples of each purification step were analyzed by SDS-PAGE [44]. Protein concentration was determined by Bradford method [45], using Bio-Rad protein assay kit (Bio-Rad) and bovine serum albumin as standard.
Mass spectrometry analysis and N-terminal amino acid sequencing
Recombinant MtCS was analyzed using mass spectrometry in an adaptation of Chassaigne and Lobinski systems [46]. Samples were analyzed on a triple quadrupole mass spectrometer, model QUATRO II, equipped with a standard electrospray (ESI) probe (Micromass, Altrinchan) adjusted to ca. 250 μL.min-1. The source temperature (80°C) and needle voltage (3.6 kV) were maintained constant throughout the data collection, applying a drying gas flow (nitrogen) of 200 L.h-1 and a nebulizer gas flow of 20 L.h-1. The equipment was calibrated with intact horse heart myoglobin. Approximately 50 pmol of sample was injected into electrospray transport solvent to determine the subunit molecular mass of MtCS. The N-terminal amino acid residues of purified recombinant MtCS were identified by automated Edman degradation sequencing using a PPSQ 21A gas-phase sequencer (Shimadzu).
Determination of native MtCS molecular mass
The molecular mass of native MtCS was determined by gel filtration using Superdex S-200 (10 mm × 30 cm) column eluted with buffer A containing 200 μM NaCl at 0.4 mL.min-1. The protein elution was monitored at 280 nm. The protein molecular weight standards were from Low Molecular Weight and High Molecular Weight Calibration kits (GE Healthcare).
EPSP synthesis
Since there is no ESPS commercially available, EPSP was synthesized using the enzymes shikimate kinase (MtSK) and EPSP synthase (MtEPSPS) from M. tuberculosis [47,48], and PNP [49,50]. The synthesis was carried out using 9.6 mM shikimate, 2.4 mM ATP, 3 mM PEP, 0.4 mM MESG. This reaction mixture was pre-incubated in 50 mM Tris-HCl, 2.5 mM MgCl2, 2.5 mM KCl pH 7.6 for 3 min at 25°C. Then the three enzymes were added (2.2 U MtSK, 0.7 U MtEPSPS, 2 U PNP) and the reaction mixture was incubated further at 25°C for 30 min. The enzymes were then removed by ultrafiltration using a Centricon 3000 Da cut-off (Amicon). The filtrated solution, containing the substrate EPSP, was collected and used to measure chorismate synthase activity. The reaction catalyzed by EPSP synthase releases an inorganic phosphate molecule, which is consumed by PNP. This coupled reaction changes the equilibrium constant of ESPS synthase reaction, allowing synthesis of higher concentration of EPSP. One unit (U) of enzyme activity for all enzymes cited in this work is defined as the amount of enzyme that catalyzes the conversion of 1 μmol of substrate per minute at 25°C in an optical path of 1 cm.
MtCS enzymatic assay for CS activity
The MtCS assay was performed in the forward direction using 15 μL of ESPS-containing solution (see above), 0.04 mM FMNox, 0.3 mM NADH, 0.2 mM MESG, 1 U PNP in buffer A. The reaction mixture was incubated for 4 min at 25°C for total consumption of Pi by PNP, and MtCS was then added. The CS reaction was monitored using a coupled reaction with the PNP enzyme [50]. This enzyme catalyses the cleavage of MESG by the inorganic phosphate molecule which is released by MtCS, and formation of 2-amine-6-mercapto-7-methylpurine is monitored at 360 nm (ε = 11.0 × 103M-1 cm-1). The assay was performed at 25°C and monitored using a Shimadzu UV-2550 spectrophotometer. All measurements, including the blanks, were carried out in triplicate to ensure that reliable data were being collected.
MtCS and FMNox interaction
The stability of the interaction between FMNox and MtCS was tested by incubating MtCS with excess FMNox in buffer A for 12 hours at 4°C in the dark. The mixture was injected into a HiTrapQ HP anion exchange column previously equilibrated with buffer A. The sample was eluted with a linear gradient from 0.0 to 1 M NaCl. Absorption spectrum of the pool of fractions containing FMNox and MtCS was measured at wavelengths values ranging from 210 to 600 nm by a Shimadzu UV-2550 spectrophotometer.
FMNox and NADH binding to MtCS measured by, respectively, spectrophotometry and spectrofluorimetry
The binding of FMNox to MtCS (35 μM) was assessed as described by Kitzing et al. [21]. UV-visible difference absorbance spectra were monitored at 25°C in a double beam spectrophotometer UV-2550 (Shimadzu) with photometric accuracy of ± 0.002 absorbance units in the range 0 – 0.5 absorbance. The difference spectra were recorded using quartz cells with two chambers (Hellma GmbH & Co) placing identical arrangements in both the sample (sample cuvette) and reference beam (reference cuvette). Spectra were recorded from 300 to 600 nm. Differences between solutions were minimized by filling both sides (sample and reference cuvettes) from single stock solutions of FMNox. Microliter additions of FMNox to both one chamber of the reference cuvette and to the sample cuvette, followed by mixing of enzyme and FMNox solutions for the latter, allowed direct measurements of the difference spectra. This procedure is almost mandatory when change in absorbance signal is relatively small on interaction. All solutions were in 50 mM Tris-HCl, pH 7.8 (buffer A).
Fluorescence titration was performed at equilibrium in a RF-5301PC Spectrofluorophotometer (Shimadzu) at 25°C by making microliter additions of NADH stock solution (10 mM) to 2 mL of 1 μM MtCS (active site concentration) in 50 mM Tris-HCl, pH 7.8 (buffer A). Controls were determined, following exactly the same procedure, by microliter additions of buffer A to account for changes in protein fluorescence due to dilution. In order to assess the NADH inner-filter effect in the fluorimeter, two cuvettes were placed in series so that the contents of the first cuvette acted as a filter of the excitation light and the light emitted from the second cuvette detected. To the first cuvette, NADH was added, while the second cuvette contained MtCS. In this manner, NADH inner-filter effects upon the protein fluorescence could be assessed. The binding of NADH to MtCS causes a quench in protein fluorescence (λexc = 290 nm; 310 ≤ λem ≤ 510 nm), and fluorescence values at the maximum emission wavelength (345.5 nm) were plotted against increasing NADH concentrations. The data were fitted to a hyperbolic function, yielding a value of 156 (± 12) μM for the dissociation constant of MtCS-NADH binary complex formation.
MtCS enzymatic assay for NADH:FMN-oxidoreductase activity
FMN-reductase activity of MtCS was monitored for the forward reaction 25°C in buffer A. The activity measurements were based on the decrease of NADH concentration upon FMN reduction. Owing to the high absorption coefficient of FMN at 360 nm, the consumption of NADH was monitored at 380 nm (ε = 0.893 × 103 M-1 cm-1). The apparent steady state kinetic parameters Km and Vmax of FMN reductase activity were determined for holoenzyme MtCS-FMNox in the presence of varying concentrations of NADH (10, 25, 50, 75, 100, 200, 300 μM). The reaction was started with the addition of 60 nmol of homogeneous MtCS previously incubated with 40 μM FMNox.
Kinetic isotope effects and proton inventory
[4S-2H]NADH was synthesized as described by Ottolina et al. [51]. The substrates, [4S-4-1H]- or [4S-4-2H]-NADH, were purified on a MonoQ column as previously reported [52], and the fractions with absorbance ratios A260 nm/A340 nm ≤ 2.3 were pooled. Both kinetic isotope effects and proton inventory were determined in buffer A with homogeneous MtCS previously incubated with 40 μM FMNox. Primary deuterium kinetic isotope effects were determined from measurements of initial rates in the presence of varying concentrations of either [4S-4-1H]- or [4S-4-2H]-NADH, and solvent kinetic isotope effects from measurements of initial rates in the presence of varying concentrations of NADH in either H2O or 91 atom % D2O. Multiple isotope effects were determined by measuring initial velocities in the presence of varying concentrations of either [4S-4-1H]- or [4S-4-2H]-NADH in 90 atom % D2O. The pD of buffers used on solvent and multiple isotope effects were measured on a pH meter considering pD = pH + 0.4. Proton inventory was carried out at saturating NADH concentrations at various mole fractions of D2O. All measurements were performed in triplicate. The nomenclature proposed by Northrop [31] and Cook and Cleland [34] was used to express isotope effects.
Data analysis
Values for the apparent kinetic parameters and their standard errors were obtained by fitting the data to the appropriate equations using the non-linear regression function of SigmaPlot 2000 (SPSS, Inc.). The initial rate measured at seven different NADH concentrations were fitted to eq. 1.
| v = VA/(K + A) | (1) |
Isotope effect data were fitted to eq 2, which assumes isotope effects on both V/K and V. In equations 1 and 2, V is the maximal velocity, K is the Michaelis constant, A is substrate concentrations, EV/K and EV are the isotope effects minus 1 on V/K and V, respectively, and Fi is the fraction of deuterium label.
| v = VA/[K(1 + FiEV/K) + A(1 + FiEV)] | (2) |
Abbreviations
CS: chorismate synthase; DAHP: 3-deoxy-D-arabino-heptulosonate 7-phosphate; EPSP: 5-enolpyruvylshikimate-3-phosphate; ESI-MS: electrospray ionization-mass spectrometry; FMN: flavin mononucleotide; FMNox: oxidized flavin mononucleotide; FMNred: reduced flavin mononucleotide; FPLC: fast protein liquid chromatography; IPTG: isopropyl β-D-thiogalactoside; MDR-TB: multi drug-resistant strains of M. tuberculosis; MESG: 2-amino-6-mercapto-7-methylpurine ribonucleoside; MtCS: chorismate synthase from M. tuberculosis; MtEPSPS: EPSP synthase from M. tuberculosis; MtSK: shikimate kinase from M. tuberculosis; NADH: reduced form of nicotinamide adenine dinucleotide; NcCS: chorismate synthase from Neurospora crassa; PCR: Polymerase Chain Reaction; PEP: phosphoenolpyruvate; PNP: purine nucleotide phosphorylase; SDS-PAGE: sodium docecyl sulfate polyacrylamide gel electrophoresis; TB: tuberculosis; XDR-TB: extensively drug-resistant strains of M. tuberculosis.
Authors' contributions
FE carried out protein expression and purification, enzyme kinetic studies, and drafted the manuscript. JESN carried out FMN and NADH binding studies. EKS carried out gene cloning. JF assisted in protein expression and participated in manuscript preparation. MSP carried out mass spectrometry analysis and N-terrminal amino acid sequencing. DSS conceived and coordinated this study, and helped draft the manuscript. LAB participated in experimental design, supervision, and helped draft the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We are grateful to Rafael G. Silva for his contribution to the analysis of isotope effects. Financial support for this work was provided by Millennium Initiative Program MCT-CNPq, Ministry of Health-Department of Science and Technology and PRONEX/CNPq/FAPERGS (Brazil) to D.S.S. and L.A.B. D.S.S. (CNPq, 304051/1975-06), L.A.B. (CNPq, 520182/99-5), M.S.P. (CNPq, 500079/90-0), and J.F. (CNPq, 301131/2003-1) are research career awardees from the National Council for Scientific and Technological Development of Brazil.
Contributor Information
Fernanda Ely, Email: fernanda_ely@yahoo.com.br.
José ES Nunes, Email: zehsacconi@yahoo.com.br.
Evelyn K Schroeder, Email: evelyn_ks@yahoo.com.
Jeverson Frazzon, Email: jeverson.frazzon@ufrgs.br.
Mário S Palma, Email: mspalma@rc.unesp.br.
Diógenes S Santos, Email: diogenes@pucrs.br.
Luiz A Basso, Email: luiz.basso@pucrs.br.
References
- Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, Dye C. The growing burden of tuberculosis. Arch Intern Med. 2003;163:1009–1021. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]
- Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- World Health Organization . WHO Report. Geneva, Switzerland; 2006. Global tuberculosis control: surveillance, planning, financing. [Google Scholar]
- Basso LA, Blanchard JS. Resistance to antitubercular drugs. Adv Exp Med Biol. 1998;456:115–144. doi: 10.1007/978-1-4615-4897-3_7. [DOI] [PubMed] [Google Scholar]
- Dooley SW, Jr, Castro KG, Hutton MD, Mullan RJ, Polder JA, Snider DE., Jr Guidelines for preventing the transmission of tuberculosis in health care settings, with special focus on HIV-related issues Rep. Morb Mortal Wkly Rep. 1990;39:1–29. [PubMed] [Google Scholar]
- Plabos-Méndez A, Gowda DK, Frieden TR. Controlling multidrug-resistant tuberculosis and access to expensive drugs: a rational framework. Bulletin of the World Health Organization. 2002;80:489–500. [PMC free article] [PubMed] [Google Scholar]
- CDC (Centers for Disease Control and Prevention) Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs worldwide. Morb Mortal Wkly Rep. 2006;55:301–305. [PubMed] [Google Scholar]
- Coates A, Hu Y, Bax R, Page C. The future challenges facing the development of new antimicrobial drugs. Nat Rev Drug Discov. 2002;1:895–910. doi: 10.1038/nrd940. [DOI] [PubMed] [Google Scholar]
- Bentley R. The shikimate pathway – a metabolic tree with many branches. Crit Rev Biochem Mol Biol. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- Herrmann KM. The shikimate pathway: early steps in the biosynthesis of aromatic compounds. Plant Cell. 1995;7:907–919. doi: 10.1105/tpc.7.7.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratledge C. Nutrition, growth and metabolism. In: Ratledge C, Stanford JL, editor. The biology of the Mycobacteria. Academic Press, London; 1982. pp. 185–271. [Google Scholar]
- Parish T, Stoker NG. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology. 2002;148:3069–3077. doi: 10.1099/00221287-148-10-3069. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Hawkes TR, Lewis T, Coggins JR, Mousdale DM, Lowe DJ, Thorneley RNF. Chorismate synthase: pre-steady-state kinetics of phosphate release from 5-enolpyruvylshikimate 3-phosphate. Biochem J. 1990;265:899–902. doi: 10.1042/bj2650899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Abell C, Coggins JR. Observation of an isotope effect in the chorismate synthase reaction. J Am Chem Soc. 1990;112:8581–8583. doi: 10.1021/ja00179a052. [DOI] [PubMed] [Google Scholar]
- White PJ, Millar G, Coggins JR. The overexpression, purification and complete amino acid sequence of chorismate synthase from Escherichia coli K12 and its comparison with the enzyme from Neurospora crassa. Biochem J. 1988;251:313–322. doi: 10.1042/bj2510313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias MVB, Ely F, Palma MS, De Azevedo WF, Jr, Basso LA, Santos DS. Chorismate Synthase: An attractive target for drug development against orphan diseases. Curr Drug Targets. 2007;8:437–444. doi: 10.2174/138945007780058924. [DOI] [PubMed] [Google Scholar]
- Dias MVB, Borges JC, Ely F, Pereira JH, Canduri F, Ramos CH, Frazzon J, Palma MS, Basso LA, Santos DS, de Azevedo WF., Jr Structure of chorismate synthase from Mycobacterium tuberculosis. J Struct Biol. 2006;154:130–143. doi: 10.1016/j.jsb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Grossman TH, Kawaski ES, Punreddy SR, Osburne MS. Spontaneous cAMP-dependent derepression of gene expression in stationary phase plays a role in recombinant expression instability. Gene. 1998;209:95–103. doi: 10.1016/S0378-1119(98)00020-1. [DOI] [PubMed] [Google Scholar]
- Rizzi C, Frazzon J, Ely F, Weber PG, Fonseca IO, Gallas M, Oliveira JS, Mendes MA, Souza BM, Palma MS, Santos DS, Basso LA. DAHP synthase from Mycobacterium tuberculosis H37Rv: cloning, expression, and purification of functional enzyme. Protein Expr Purif. 2005;40:23–30. doi: 10.1016/j.pep.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Kitzing K, Macheroux P, Amrhein N. Spectroscopic and kinetic characterization of the bifunctional chorismate synthase from Neurospora crassa. J Biol Chem. 2001;276:42658–42666. doi: 10.1074/jbc.M107249200. [DOI] [PubMed] [Google Scholar]
- Ahn HJ, Yoon HJ, II Lee B, Suh SW. Crystal structure of chorismate synthase: A novel FMN-binding protein fold and functional insights. J Mol Biol. 2004;336:903–915. doi: 10.1016/j.jmb.2003.12.072. [DOI] [PubMed] [Google Scholar]
- Maclean J, Ali S. The structure of chorismate synthase reveals a novel flavin binding site fundamental to a unique chemical reaction. Structure. 2003;11:1499–1511. doi: 10.1016/j.str.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Kitzing K, Auweter S, Amerhein N, Macheroux P. Mechanism of chorismate synthase. J Bio Chem. 2004;279:9451–9461. doi: 10.1074/jbc.M312471200. [DOI] [PubMed] [Google Scholar]
- Inouye S. NAD(P)H-flavin oxidoreductase from the bioluminescent bacterium, Vibrio fischeri ATCC is a flavoprotein. FEBS Lett. 7744;347:163–168. doi: 10.1016/0014-5793(94)00528-1. [DOI] [PubMed] [Google Scholar]
- Macheroux P, Petersen J, Bornemann S, Lowe DJ, Thorneley RNF. Binding of the oxidized, reduced, and radical flavin species to chorismate synthase. An investigation by spectrophotometry, fluorimetry, and electron paramagnetic resonance and electron nuclear double resonance spectroscopy. Biochemistry. 1996;35:1643–1652. doi: 10.1021/bi951705u. [DOI] [PubMed] [Google Scholar]
- Henstrand JM, Schaller A, Braun M, Amrhein N, Schmid J. Saccharomyces cerevisiae chorismate synthase has a flavin reductase activity. Mol Microbiol. 1996;22:859–866. doi: 10.1046/j.1365-2958.1996.01534.x. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Ohshiro T, Nishina Y, Izumi Y. Purification, characterization, and overexpression of flavin reductase involved in dibenzothiophene desulfurization by Rhodococcus erythropolis D-1. Appl Environ Microbiol. 2001;67:1179–1184. doi: 10.1128/AEM.67.3.1179-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenno S, Kobori T, Tanokura M, Saigo K. Purification and characterization of NfrA1, a Bacillus subtilis nitro/flavin reductase capable of interacting with the bacterial luciferase. Biosci Biotechnol Biochem. 1998;62:1978–1987. doi: 10.1271/bbb.62.1978. [DOI] [PubMed] [Google Scholar]
- Broco M, Soares CM, Oliveira S, Mayhew SG, Rodrigues-Pousada C. Molecular determinants for FMN-binding in Desulfovibrio gigas flavoredoxin. FEBS Letters. 2007;581:4397–4402. doi: 10.1016/j.febslet.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Northrop DB. Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- Schowen KB, RL Schowen. The use of isotope effects to elucidate enzyme mechanisms. BioScience. 1981;31:826–831. doi: 10.2307/1308680. [DOI] [Google Scholar]
- Cook PF. Kinetic and Regulatory Mechanisms of Enzymes from Isotope Effects. In: Cook PF, editor. Enzyme Mechanism from Isotope Effects. CRC Press, Florida; 1991. pp. 203–228. [Google Scholar]
- Cook PF, Cleland WW. Mechanistic deductions from isotope effects in multireactant enzyme mechanisms. Biochemistry. 1981;20:1790–1796. doi: 10.1021/bi00510a013. [DOI] [PubMed] [Google Scholar]
- Quinn DM, Sutton LD. Theoretical Basis and Mechanistic Utility of Solvent Isotope Effects. In: Cook PF, editor. Enzyme Mechanism from Isotope Effects. CRC Press, Florida; 1991. pp. 73–126. [Google Scholar]
- Patel MP, Liu WS, West J, Tew D, Meek DT, Thrall SH. Kinetic and chemical mechanisms of the fabG-encoded Streptococcus pneumoniae β-ketoacyl-ACP reductase. Biochemistry. 2004;44:16753–16765. doi: 10.1021/bi050947j. [DOI] [PubMed] [Google Scholar]
- Belasco JG, Albery J, Knowles JR. Double isotope fractionation: test for concertedness and for transition-state dominance. J Am Chem Soc. 1983;105:2475–2477. doi: 10.1021/ja00346a062. [DOI] [Google Scholar]
- Hermes JD, Roeske CA, O'Leary MH, Cleland WW. Use of multiple isotope effects to determine enzyme mechanisms and intrinsic isotope effects. Biochemistry. 1984;21:5106–5114. doi: 10.1021/bi00263a040. [DOI] [PubMed] [Google Scholar]
- Rauch G, Ehammer H, Bornemann S, Macheroux P. Mutagenic analysis of an invariant aspartate residue in chorismate synthase supports its role as an active site base. Biochemistry. 2007;46:3768–3774. doi: 10.1021/bi602420u. [DOI] [PubMed] [Google Scholar]
- Ehammer H, Rauch G, Prem A, Kappes B, Macheroux P. Conservation of NADPH utilization by chorismate synthase and its implications for the evolution of the shikimate pathway. Mol Micribiol. 2007;65:1249–1257. doi: 10.1111/j.1365-2958.2007.05861.x. [DOI] [PubMed] [Google Scholar]
- Fernandes CL, Breda A, Santos DS, Basso LA, Souza ON. A structural model for chorismate synthase from Mycobacterium tuberculosis in complex with coenzyme and substrate. Comput Biol Med. 2007;25:434–441. doi: 10.1016/j.compbiomed.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Vincentelli R, Bignon C, Gruez A, Canaan S, Sulzenbacher G, Tegoni M, Campanacci V, Cambillau C. Medium-scale structural genomics: strategies for protein expression and crystallization. Acc Chem Res. 2003;36:165–172. doi: 10.1021/ar010130s. [DOI] [PubMed] [Google Scholar]
- Chapman T. Pure but not simple. Nature. 2005;434:795–798. doi: 10.1038/434795a. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Bradford MM, McRorie RA, Williams WL. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chassaigne H, Lobinski R. Characterization of horse kidney metallothionein isoforms by electrospray MS and reversed-phase HPLC-electrospray MS. Analyst. 1998;123:2125–2130. doi: 10.1039/a804713h. [DOI] [PubMed] [Google Scholar]
- Oliveira JS, Pinto CA, Basso LA, Santos DS. Cloning and overexpression in soluble form of functional shikimate kinase and 5-enolpyruvylshikimate 3-phosphate synthase enzymes from Mycobacterium tuberculosis. Protein Expr Purif. 2001;22:430–435. doi: 10.1006/prep.2001.1457. [DOI] [PubMed] [Google Scholar]
- Oliveira JS, Mendes MA, Palma MS, Basso LA, Santos DS. One-step purification of 5-enolpyruvylshikimate-3-phosphate synthase enzyme from Mycobacterium tuberculosis. Protein Expr Purif. 2003;28:287–292. doi: 10.1016/S1046-5928(02)00708-8. [DOI] [PubMed] [Google Scholar]
- Upson RH, Haugland RP, Malekzadeh MN, Haugland RP. A spectrophotometric method to measure enzymatic activity in reactions that generate inorganic pyrophosphate. Anal Biochem. 1996;243:41–45. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci USA. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolina G, Riva S, Carrea G, Danieli B, Buckmann AF. Enzymatic synthesis of [4R-2H]NAD(P)H and [4S-2H]NAD(P)H and determination of the stereospecificity of 7 alpha- and 12 alpha-hydroxysteroid dehydrogenase. Biochem Biophys Acta. 1989;998:173–178. doi: 10.1016/0167-4838(89)90270-7. [DOI] [PubMed] [Google Scholar]
- Orr GA, Blanchard JS. High-performance ion-exchange separation of oxidized and reduced nicotinamide adenine dinucleotides. Anal Biochem. 1984;142:232–234. doi: 10.1016/0003-2697(84)90544-X. [DOI] [PubMed] [Google Scholar]