

Abstract
Establishment of neuronal diversity is a central topic in developmental neurobiology. Prior studies implicated Pitx2, a paired-like homeodomain transcription factor, in mouse subthalamic nucleus neuronal development, but precise stages of neuronal differentiation affected (migration, axon outgrowth, fate specification) and underlying mechanisms were unknown. Here we report lineage tracing experiments using Pitx2cre/+, Pitx2cre/null, and conditional nuclear lacZ reporter mice to track embryonic Pitx2 expressing neurons. Migration of subthalamic nucleus and hypothalamic neurons was severely arrested in Pitx2cre/null embryos, and subclasses of subthalamic nucleus neurons identified by Lmx1b, Foxp1, and Foxp2-gene expression revealed differing sensitivities to Pitx2 dosage. Interestingly, embryonic subthalamic nucleus development was unaffected in Lmx1b null mice, suggesting that Pitx2 and Lmx1b act via independent genetic pathways. These data provide the first direct evidence for Pitx2-dependent neuronal migration in the developing hypothalamus, and demonstrate that complex transcriptional networks regulate regional specialization of distinct hypothalamic and subthalamic nucleus neurons.
Keywords: development, differentiation, mice, migration, mutant, transcription factor
Introduction
Generation of neuronal diversity in the mammalian brain requires coordinated expression of transcription factors and signaling molecules (Puelles and Rubenstein 2003; Sur and Rubenstein 2005; Lim and Golden 2006). Regional specialization of these complex neuronal populations also requires that differentiating neurons travel long, sometimes circuitous routes to their final destinations in the brain. Central questions in developmental neurobiology include how these distinct neuronal populations are formed and which molecular signals are used to guide their terminal differentiation. Recent advances in genetic fate mapping of cells with restricted gene expression have made it possible to explore cellular fates and phenotypes of specific neuronal lineages in the developing mouse brain (Branda and Dymecki 2004; Joyner and Zervas 2006).
Restricted expression of Pitx2, a paired-like homeodomain transcription factor, in the developing mouse hypothalamus occurs concomitant with or soon after terminal mitosis of neural progenitors around E9.5–E10.5 (Martin et al., 2002), and is necessary for normal development of neurons in the subthalamic nucleus (Martin et al., 2004). Due to complex central nervous system phenotypes and a lack of reporter alleles, earlier studies could not distinguish the effects of Pitx2 deficiency on neural cell migration, axon outgrowth, or cell fate specification. Moreover, it was not possible to exclude altered Pitx2 gene expression as an explanation for defects seen in Pitx2 null embryos (Martin et al., 2004).
Pitx2 exhibits pleiotropic, tissue specific effects during development, with a short (30 minutes) mRNA half-life that is regulated by Wnt/Dvl/β-catenin signaling (Kioussi et al., 2002). Pitx2 promotes migration of cultured HeLa cells (Wei and Adelstein 2002) and cells that give rise to craniofacial and cardiac structures (Liu et al., 2002; Liu et al., 2003), and Pitx2 function is critical for survival of pituitary hormone-producing cells (Charles et al., 2005). Based on these observations, we hypothesized that Pitx2 may be required for one or more aspects of neuronal differentiation including maintenance of gene expression, cell identity, neuronal migration, and axonal outgrowth. To explore the fates of Pitx2 deficient neurons during mid-gestation, we designed a lineage tracing strategy using previously characterized Pitx2null (Gage et al., 1999) and Pitx2cre knock-in (Liu et al., 2003) alleles and transgenic mice containing a Cre conditional nuclear localized lacZ reporter under the control of the chicken β-actin promoter (N-lacZ) (Zinyk et al., 1998). Our analysis revealed that Pitx2 is essential for lineage specific neuronal migration in the developing hypothalamus and subthalamic nucleus.
Results
Nuclear localized β-galactosidase labels Pitx2 mutant neurons
PITX2 protein and mRNA are expressed in differentiating neurons of the E9.5–E14.5 mouse brain around the time of terminal mitosis–(Martin et al., 2002) and unpublished data. Prior studies showed that homozygous Pitx2 null embryos require Pitx2 for normal gene expression and formation of neuronal projections in the developing subthalamic nucleus, but the lack of a permanent reporter made it impossible to track the fates of Pitx2 mutant neurons (Martin et al., 2004). To evaluate for region specific defects in Pitx2 gene expression or localized defects in cell migration or fate, we generated a system for indelibly marking Pitx2 heterozygous mutant and compound heterozygous mutant cells with nuclear localized β-galactosidase. Matings between Pitx2+/null; N-lacZ/N-lacZ and Pitx2cre/+ mice were used to generate littermate embryos of genotype Pitx2cre/+;N-lacZ and Pitx2cre/null;N-lacZ (Fig. 1A). This approach allowed for direct comparison ofβ-galactosidase activity in embryos with one normal copy of Pitx2 to those with complete (compound heterozygous) Pitx2 deficiency.
Figure 1.
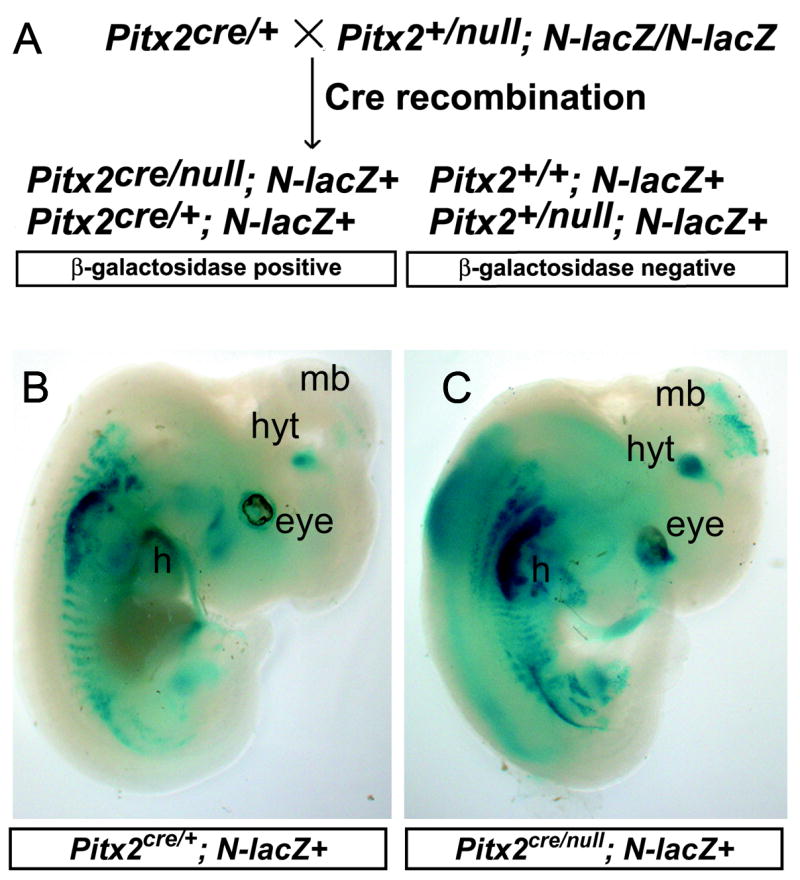
Pitx2cre; N-lacZ lineage tracing approach labels Pitx2 mutant neurons. (A) Pitx2cre/+ embryos were mated with Pitx2+/null; N-lacZ/N-lacZ mice to yield embryos of four possible genotypes. Pitx2cre/+;N-lacZ (B) and Pitx2cre/null:N-lacZ (C) embryos express β-galactosidase in Pitx2-expressing tissues at E12.5, including the eye, hypothalamus (hyt), midbrain (mb), heart (h), craniofacial structures, and somites. Pitx2cre/null;N-lacZ embryos exhibit lethality after E14.5, likely due to cardiac and abdominal defects that are not well visualized in this sagittally oriented embryo (C).
Whole mount X-gal staining of E12.5 Pitx2cre/+;N-lacZ and Pitx2cre/null;N-lacZ embryos revealed β-galactosidase activity in known sites of Pitx2 expression in the brain, eye, heart, proximal limb, and craniofacial regions (Fig. 1B), consistent with prior observations showing reporter expression in the developing brain, spinal cord, heart, and some skeletal muscles (Zinyk et al., 1998). Close analysis of sectioned E14.5 Pitx2cre/+;N-lacZ+ embryos demonstrated β-galactosidase activity in brain regions known to express Pitx2, including the superior colliculus, subthalamic nucleus, mammillary region, posterior hypothalamus, zona limitans intrathalamica and first rhombomere. The pattern of β-galactosidase activity in the postnatal hypothalamus and subthalamic nucleus of Pitx2cre/+;N-lacZ embryos matched previously reported patterns of Pitx2 mRNA and protein expression in wildtype and Pitx2+/− embryos, as illustrated with double immunofluorescence for β-gal and PITX2 (Suppl. Fig. 1) (Martin et al., 2002). There were no ectopic regions of β-galactosidase activity in E12.5–E14.5 Pitx2cre/+;N-lacZ brains (data not shown), indicating that Pitx2 expression perdures in the mid-gestation developing mouse brain.
These observations confirm that labeling of Pitx2-expressing neurons with the βgal reporter in Pitx2cre/+;N-lacZ mice exhibits high fidelity. Moreover, inherent delays in Cre expression, excision of the conditional reporter, and subsequent transcription and translation of β-galactosidase (as long as 24 hours) (Metzger et al., 1995; Zervas et al., 2004), do not appear to interfere with the ability ofβ-galactosidase to label Pitx2 mutant neurons. Loss of only one copy of Pitx2 does not disrupt Pitx2 protein or mRNA expression in the brain relative to wildtype controls (Martin et al., 2004), and heterozygous Pitx2cre/+; N-lacZ embryos exhibit no identifiable defects in neuronal location, thus validating the use of Pitx2cre/+ mice as controls for comparisons with Pitx2cre/null neuronal phenotypes.
Pitx2cre/null embryonic neurons are mislocalized in the developing hypothalamus
Embryos with compound heterozygosity for Pitx2 deficient alleles (Pitx2cre/null; N-lacZ) did not survive beyond E14.5, with externalized heart and abdominal structures and distal turning defects similar to Pitx2null/null and Pitx2cre/cre embryos (Gage et al., 1999; Kitamura et al., 1999; Lin et al., 1999; Lu et al., 1999; Liu et al., 2002; Liu et al., 2003). In order to determine the locations of Pitx2 heterozygous and compound heterozygous Pitx2 mutant neurons, we stained E12.5–E14.5 embryos for β-galactosidase activity and obtained vibratome sections throughout the developing hypothalamus (Fig. 2). In the E14.5 hypothalamus, Pitx2cre/+;N-lacZ embryos expressed high levels of β-galactosidase (Fig. 2) in a pattern similar to Pitx2 mRNA and protein expression (Martin et al., 2002; Martin et al., 2004). In contrast, β-galactosidase positive cells were absent from the Pitx2cre/null;N-lacZ lateral hypothalamus, and the density of β-galactosidase label in the medial hypothalamus was increased (Fig. 2F–H). Similar observations were made in E13.5 Pitx2cre/null;N-lacZ embryos, in which β-galactosidase positive cells were concentrated in medial regions of the hypothalamus, with no visible cells occupying more lateral positions (Fig. 2M, N). At E12.5, there were no visible differences between the locations of β-galactosidase positive cells in the Pitx2cre/null;N-lacZ and Pitx2cre/+;N-lacZ hypothalamus (Fig. 2K, L, O, P). Thus, hypothalamic Pitx2 null neurons became mislocalized after E12.5, and are retained medially at E13.5–E14.5.
Figure 2.

Pitx2cre/null; N-lacZ mutants exhibit defects in hypothalamic neuronal location. Littermate embryos of genotype Pitx2cre/+; N-lacZ or Pitx2cre/null; N-lacZ were fixed, stained for β-galactosidase activity, and vibratome sectioned at 100 μm in the plane of section indicated in A (for C–H) and 150 μm in the plane of section shown in B (for I–P). Anterior is up for each section. Hatched areas depict the lateral hypothalamus where subthalamic nucleus neurons are located. Serial adjacent sections of caudal to rostral hypothalamus from E14.5 embryos (C–H) reveal absence of β-galactosidase positive cells in the lateral hypothalamus of Pitx2cre/null; N-lacZ mutants and a medial abundance of β-galactosidase positive cells. Pitx2cre/null mutants also exhibit increased β-galactosidase expression at the ventral midline (brackets), suggesting a failure of these cells to respect the midline boundary. Serial sections from E13.5 embryos (I, J, M, N) also exhibit medially shifted β-galactosidase positive cells in Pitx2cre/null; N-lacZ embryos, whereas locations of β-galactosidase expressing cells are similar in E12.5 Pitx2cre/+; N-lacZ (K, L) and Pitx2cre/null; N-lacZ (O, P) mutants. Abbreviations: latV, lateral ventricle; STN, subthalamic nucleus; IIIv, third ventricle; Aq, cerebral aqueduct; pit, pituitary.
To test whether lateral hypothalamicβ-galactosidase label represents subthalamic nucleus neurons, we co-stained transverse vibratome E14.5 sections for β-galactosidase activity and Calretinin immunoreactivity (Fig. 3). These studies revealed co-expression of Calretinin and β-galactosidase in the lateral Pitx2cre/+; N-lacZ hypothalamus, consistent with prior reports showing co-localization of PITX2 and Calretinin in subthalamic nucleus neurons (Martin et al., 2004). We observed absence of β-galactosidase and Calretinin in the lateral hypothalamus of Pitx2cre/null; N-lacZ embryos, providing definitive evidence for loss of PITX2-lineage neurons from the subthalamic nucleus region.
Figure 3.
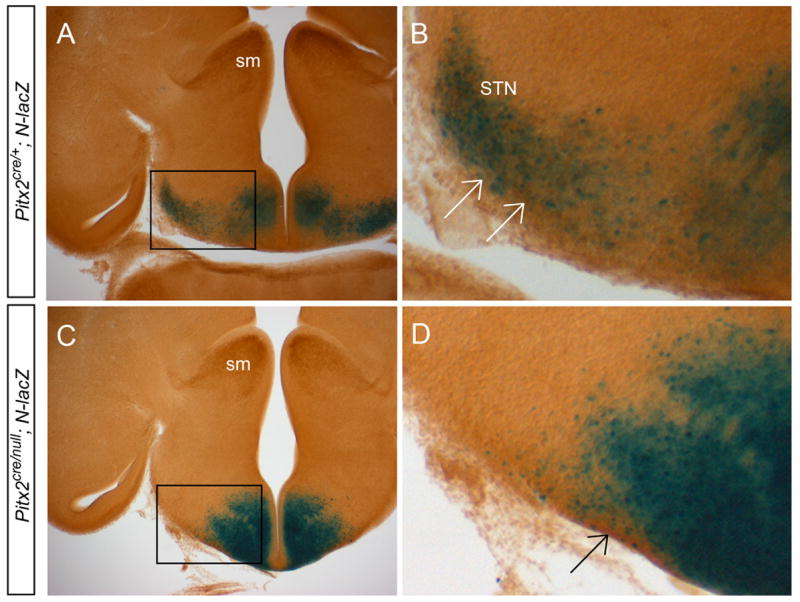
Pitx2cre/+; N-lacZ embryos lack subthalamic nucleus neurons. Vibratome sections of E14.5 Pitx2cre/+; N-lacZ (A, B) and Pitx2cre/null; N-lacZ (C, D) littermate embryos were processed for X-gal immunohistochemistry and Calretinin immunoreactivity using DAB. Single X-gal stained images are shown in figure 3 (D, G). Images in B and D are enlarged from boxes shown in A and C. Calretinin immunolabel is abundant in the stria medullaris (sm), subthalamic nucleus (STN), and hypothalamus of Pitx2cre/+; N-lacZ embryos, but is missing from the Pitx2cre/null; N-lacZ subthalamic nucleus. These data show that PITX2-lineage cells (labeled by Calretinin; arrows in B, D) are absent from the Pitx2cre/null; N-lacZ mutant subthalamic nucleus.
In addition to the medial shift in β-galactosidase, we observed increased β-galactosidase expression at the ventral hypothalamic midline in E14.5 Pitx2cre/null; N-lacZ mutants, suggesting aberrant midline positioning or crossing of these cells (brackets in Fig. 2, C–H). Wildtype E12.5–E14.5 mouse hypothalamic neurons migrate from medial to lateral, ventral to dorsal, and caudal to rostral as they progress toward their final destinations in the subthalamic nucleus (Suppl. Fig. 2) (Altman and Bayer 1986; Marchand 1987; Martin et al., 2004). This 3-dimensional route of subthalamic nucleus neuronal migration includes mixed radial and tangential components, and may also result from distinct waves of neurogenesis (Altman and Bayer 1986). The increased medial and aberrant midlineβ-gal label in Pitx2cre/null; N-lacZ mutants suggests that neuronal migration might become arrested or disrupted shortly after E12.5.
Pitx2cre/null neurons exhibit defects in migration
To test for changes in hypothalamic cell number with loss of Pitx2, we counted β-galactosidase positive cells in the medial vs. lateral hypothalamus of transverse sections from Pitx2cre/+;N-lacZ and Pitx2cre/null;N-lacZ embryos (Fig. 4). Slightly increased numbers of β-galactosidase positive cells were located medially in the ventral hypothalamus of Pitx2cre/null;N-lacZ embryos compared to Pitx2cre/+;N-lacZ littermates, but this difference was not statistically significant (Pitx2cre/+;N-lacZ = 590 vs. Pitx2cre/null;N-lacZ = 746 in representative embryos from N = 3 pairs) (Fig. 4A, B). This trend toward increased Pitx2-expressing cells in the Pitx2cre/null hypothalamus might be due to extrinsic effects on neighboring cells. We also detected a shift in β-galactosidase positive cells toward locations farther away from the floor plate in Pitx2cre/null;N-lacZ embryos (Fig. 4C, D).
Figure 4.
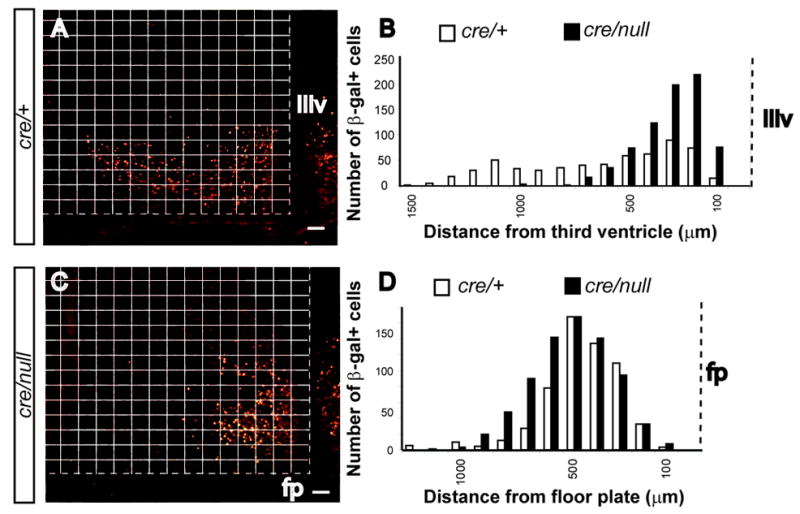
Hypothalamic Pitx2cre/null neurons are shifted medially and away from the floor plate. β-galactosidase positive cells were counted in three different transverse sections from Pitx2cre/+; N-lacZ (A) and Pitx2cre/null; N-lacZ (C) littermate embryos at the level of the hypothalamus (same sections as in figure 7 I, M). β-galactosidase positive cells were more abundant in medial regions of the hypothalamus toward the third ventricle (IIIv) of Pitx2cre/null; N-lacZ embryos compared to Pitx2cre/null; N-lacZ embryos. The overall number of β-galactosidase positive cells was slightly increased in the mutant (N = 590 cells for Pitx2cre/+; N = 746 cells for Pitx2cre/null). There was also a minimal shift of βgal+ cells away from the floor plate (fp) in Pitx2cre/null; N-lacZ cells compared to Pitx2cre/+; N-lacZ embryos (C, D). Scale bar = 100 μm.
To test for defects in neuronal migration, we performed BrdU immunohistochemistry on E14.5 embryos exposed to BrdU by maternal intraperitoneal BrdU injection at 1 hour, 1 day, or 2 days prior to embryo collection (Fig. 5). In embryos injected with BrdU 1 hour or 1 day prior to sacrifice, there were no differences in the locations of BrdU-positive cells in the developing hypothalamus of Pitx2cre/null;N-lacZ mutants compared with Pitx2cre/+;N-lacZ embryos (Fig. 5, A–B, E–F). However, in Pitx2cre/+;N-lacZ embryos injected 2 days prior to sacrifice, BrdU-positive cells were distributed throughout the hypothalamus (including the laterally located subthalamic nucleus) (Fig. 5I), whereas in Pitx2cre/null;N-lacZ mutants, BrdU-positive cells were restricted to more medial locations (Fig. 5J).
Figure 5.

Hypothalamic Pitx2cre/null; N-lacZ neurons fail to migrate. E14.5 Pitx2cre/+; N-lacZ (A, C, E, G, I, K) and Pitx2cre/null; N-lacZ (B, D, F, H, J, L) embryos were exposed to BrdU by intraperitoneal injection 1 hour (A–D), 1 day (E–H), or 2 days (I–L) prior to collection, and transverse sections processed for anti-BrdU immunohistochemistry (A, B, E, F, I, J) or double-label immunofluorescence with anti-BrdU and anti-β-galactosidase (C, D, G, H, K, L). There were no differences in BrdU labeling between Pitx2cre/+ and Pitx2cre/null mutants injected with BrdU 1 hour (A–D) or 1 day (E–H) prior to analysis. Pitx2cre/null mutants injected with BrdU 2 days prior to analysis (I–L) exhibited fewer BrdU positive cells in the lateral hypothalamus (subthalamic nucleus region) compared with Pitx2cre/+ embryos (bracketed areas). There was no cellular colocalization between BrdU and β-gal in embryos injected at E14.5 (C, D) or E13.5 (G, H), whereas double-labeled cells were present in embryos injected at E12.5 (white arrows in K and L). Scale bars = 100 μm.
To identify Pitx2-expressing, BrdU-positive cells, we co-labeled transverse sections with anti-BrdU and anti-β-gal. These studies showed no cellular colocalization of β-gal and BrdU in embryos injected at E13.5 or E14.5 (Fig. 5C–D, G–H), consistent with prior evidence that Pitx2 expression is restricted to postmitotic neurons (Martin et al., 2002). In contrast, there were numerous β-gal+/BrdU+ cells in Pitx2cre/+;N-lacZ and Pitx2cre/null;N-lacZ embryos injected at E12.5, indicating that some normally migrating and arrested cells are of the Pitx2 lineage. Co-label of β-gal and BrdU in a subset of cells in Pitx2cre/null;N-lacZ embryos injected at E12.5 is consistent with prior studies showing that subthalamic nucleus neurons are born over a five day period between E10.5 and E14.5 in the mouse (Altman and Bayer 1986; Marchand 1987; Martin et al., 2004). These data also provide evidence that Pitx2cre/null neurons undergo terminal mitosis normally but become arrested after E12.5 during their migration away from the third ventricle.
To test whether defects in Pitx2cre/null neuronal migration are associated with changes in cell survival, we performed TUNEL studies on E10.5 and E12.5 embryos. We found very few TUNEL-positive cells in the hypothalamus of both Pitx2cre/+;N-lacZ and Pitx2cre/null;N-lacZ embryos (Suppl. Fig. 3), suggesting normal cell survival. This observation also corroborates previous data showing normal cell survival in E14.5 Pitx2 null mutants (Martin et al., 2004). Altogether, these data demonstrate that loss of Pitx2 disrupts migration of hypothalamic neurons after E12.5, but does not impair neuronal survival or cell cycle exit.
As an initial step toward identifying a mechanism for arrested or delayed migration of Pitx2 mutant neurons, we tested expression of candidate molecules implicated in neuronal migration based on their known expression in the embryonic mouse hypothalamus and midbrain. Immunohistochemistry for N-cadherin, neural cell adhesion molecule (NCAM), and doublecortin-like protein kinase DCAMKL1/DCLK (Lin et al., 2000) were unchanged in Pitx2cre/null; N-lacZ+ mutant embryos compared to Pitx2cre/+; N-lacZ+ littermates (data not shown), indicating that altered expression of these molecules does not likely explain the observed neuronal migration defects. Additional studies are necessary to determine the molecules and/or signaling factors that mediate PITX2-dependent hypothalamic neuronal migration.
Pitx2 mutant neurons retain some of their molecular fates
Pitx2 acts as a transcriptional regulator in other tissues including the heart, pituitary, craniofacial structures, skeletal muscle, and ocular tissues (Hjalt et al., 2001; Ganga et al., 2003; Charles et al., 2005; Berry et al., 2006; Diehl et al., 2006; Shih et al., 2007). We hypothesized that hypothalamic neuronal migration defects arise due to disrupted gene expression in hypothalamic neurons. To test this hypothesis, we analyzed expression of Lmx1b, a LIM homeodomain transcription factor expressed in the subthalamic nucleus and posterior hypothalamus which regulates development of serotonergic and dopaminergic neurons (Smidt et al., 2000; Asbreuk et al., 2002; Ding et al., 2003; Guo et al., 2007). We found, by double immunofluorescence, that PITX2 and LMX1B colocalize extensively in the E14.5 subthalamic nucleus and medial hypothalamus (data not shown). LMX1B expression in the hypothalamus is disrupted with loss of Pitx2, based on double immunofluorescence with antibodies againstβ-galactosidase and LMX1B (Fig. 6, A–H). Complete absence of LMX1B immunofluorescence was seen in the subthalamic nucleus region of E14.5 Pitx2cre/null;N-lacZ mutants (Fig. 6F), whereas double-labeled (β-gal+/LMX1B+) and single-labeled LMX1B+ neurons were preserved in the medial hypothalamus (Fig. 6F, G). Thus, LMX1B-expressing neurons destined to occupy the subthalamic nucleus require Pitx2 for normal migration, whereas medial hypothalamic neurons express Lmx1b independent of Pitx2 function.
Figure 6.

Subclasses of subthalamic nucleus neurons require Pitx2 for normal migration. Sections of E14.5 Pitx2cre/+; N-lacZ (A–D, I–L, Q–T) and Pitx2cre/null; N-lacZ (E–H, M–P, U–X) embryos in the coronal (A–H) or transverse plane (I–X) at the level of the subthalamic nucleus were double labeled with anti-β-galactosidase and anti-LMX1B, anti-FOXP1, or anti-FOXP2. Merged images are in C, G, K, O, S, and W, with enlarged subthalamic nucleus regions shown in D, H, L, P, T, and X. Hatched ovals show the lateral hypothalamic region where the subthalamic nucleus normally resides. Loss of β-galactosidase (E, M, and U), and LMX1B (F) in the lateral hypothalamus of Pitx2cre/null; N-lacZ mutants is consistent with absence of these subthalamic nucleus neurons. In contrast, some FOXP1+/β-gal-negative and FOXP2+/β-gal-negative cells remain in the lateral Pitx2cre/null; N-lacZ hypothalamus (P and X), suggesting that these cells escape the migration defect. Some medially located neurons co-expressβ-galactosidase and LMX1B, FOXP1, or FOXP2, indicating that Pitx2 is not globally required for expression of these genes. Scale bars = 100μm (A–C, E–G, I–K, M–O, Q–R, and U–W) or 50 μm (D, H, L, P, T, and X).
We also tested expression of several members of the forkhead family of transcription factors, since they are highly expressed in the developing hypothalamus and subthalamic nucleus (Ferland et al., 2003; Teramitsu et al., 2004). Expression of Foxb1 mRNA, which encodes a forkhead transcription factor required for formation of ventral diencephalon and mammillary bodies (Alvarez-Bolado et al., 2000), was expressed throughout the basal hypothalamus of E14.5 Pitx2+/− and Pitx2−/− embryos (Suppl. Fig. 3). In Pitx2cre/+;N-lacZ embryos, some hypothalamic cells were double-labeled with anti-βgal and anti-FOXP1 (Fig. 6, K) or anti-FOXP2 (Fig. 6, S). In Pitx2cre/null;N-lacZ mutants, FOXP1 and FOXP2 immunoreactivity were reduced in, but not absent from, the subthalamic nucleus region, suggesting that some FOXP1+/β-gal-negative and FOXP2+/β-gal-negative subthalamic nucleus neurons escape the Pitx2 mutant migration defect (Fig. 6, O, W). FOXP1+/β-gal+ and FOXP2+/β-gal+ cells were also present in the medial Pitx2cre/null;N-lacZ hypothalamus, indicating that Pitx2 is not required for FOXP1 or FOXP2 gene expression in these cells (Fig. 6, O, W).
Lmx1b is not required for subthalamic nucleus neuronal development
Certain Lmx1b neuronal lineages (subthalamic nucleus neurons) are sensitive to Pitx2 deficiency, whereas others (dorsal midbrain and medial thalamic/hypothalamic neurons) are not. To test whether Lmx1b acts genetically upstream of Pitx2 in the developing subthalamic nucleus, perhaps through coordinated activity with other transcription factors, we analyzed Lmx1bn/n embryos (Chen et al., 1998) at E14.5 for defects in Pitx2 mRNA and protein and FOXP1/FOXP2 expression (Fig. 7) (Ferland et al., 2003; Martin et al., 2004; Tamura et al., 2004). Expression of each of these markers in the hypothalamus of E14.5 Lmx1b embryos remained intact, indicating that loss of Lmx1b does not recapitulate the Pitx2 mutant phenotype in the subthalamic nucleus. Since Lmx1b expression is preserved in a subset of Pitx2 mutant neurons of the developing hypothalamus, expression of Pitx2 and Lmx1b are likely regulated by independent mechanisms in subthalamic nucleus neuronal development.
Figure 7.

Lmx1b is dispensable for subthalamic nucleus development. Coronal sections of E14.5 Lmx1b+/n (A–E) and Lmx1bn/n (F–J) paraffin embedded embryos were labeled for anti-LMX1B (A, F), Pitx2 mRNA (B, G), anti-PITX2 (C, H), anti-FOXP1 (D, I), or anti-FOXP2 (E, J). Absence of LMX1B confirms the null status of Lmx1bn/n embryos. Pitx2 mRNA and immunofluorescence and FOXP1/FOXP2 immunofluorescence are unchanged in Lmx1bn/n embryos, indicating Lmx1b is not required for subthalamic nucleus development.
These observations suggested that specific populations of developing hypothalamic neurons (with unique PITX2/LMX1B/FOXP1/FOXP2 transcription factor expression profiles) exhibit variable sensitivities to Pitx2 dosage. LMX1B-positive subthalamic nucleus neurons are highly disrupted by loss of Pitx2, whereas some FOXP1-positive and FOXP2-positive cells migrate normally. To test whether this reflects differences in Pitx2-lineage, we performed a Pitx2-lineage tracing experiment in postnatal day 1 Pitx2cre/+;N-lacZ hypothalamus and subthalamic nucleus (Fig. 8). Double label experiments revealed extensive but differential colocalization between β-gal and the following subthalamic nucleus markers: PITX2, Calretinin, LMX1B, FOXP1, and FOXP2. LMX1B+/β-gal-negative cells were present in the caudomedial subthalamic nucleus, whereas FOXP1+/β-gal-negative and FOXP2+/β-gal-negative cells were abundant in the rostral and caudomedial subthalamic nucleus (Fig. 8).
Figure 8.
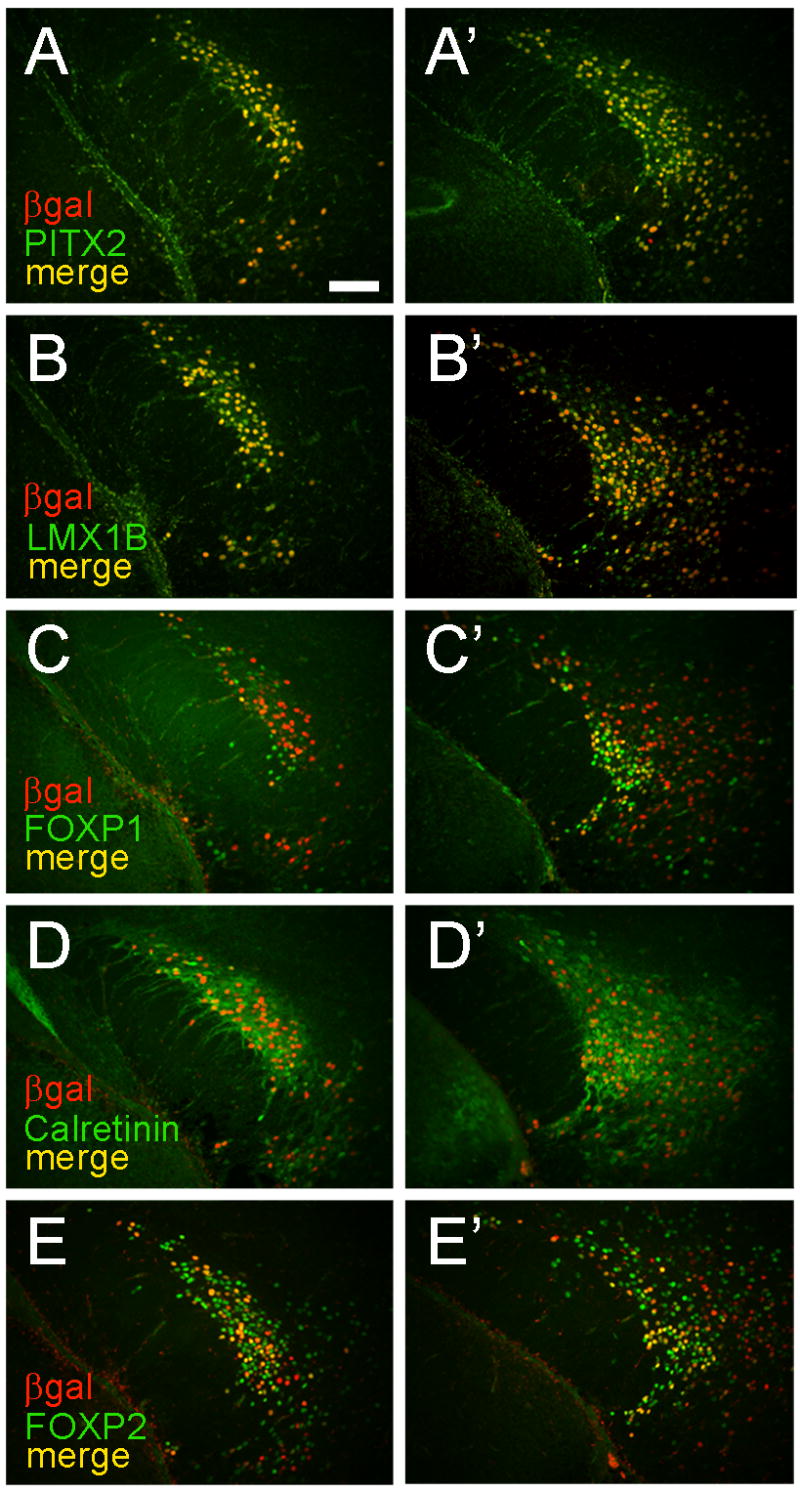
Neurons in the postnatal subthalamic nucleus exhibit heterogenous lineages. Adjacent coronal sections from rostral (A–E) or caudal (A″–E″) hypothalamus of postnatal day 1 Pitx2cre/+; N-lacZ embryos were co-labeled with anti-β-gal and anti-PITX2 (A, A″), anti-LMX1B (B, B″), anti-FOXP1 (C, C″), anti-Calretinin (D, D″), or anti-FOXP2 (E, E″). Medial is to the right for each section. Extensive colocalization of β-gal is seen with all five markers; however, anti-FOXP1 and anti-FOXP2 both label numerousβ-gal negative subthalamic nucleus cells, suggesting they derive from non Pitx2-expressing lineages. Scale bar in A (50 μm) applies to all panels.
Discussion
We demonstrate, through the use of a Cre lineage tracing Pitx2 deficiency allele, neuronal migration defects in the developing mouse hypothalamus with loss of Pitx2 function. Our studies provide direct evidence of a genetic requirement for Pitx2 in normal neuronal migration in the developing hypothalamus, which, unlike the cortex and cerebellum, has not been extensively explored (Ayala et al., 2007; Lim and Golden 2007).
A primary role for Pitx2 in region specific neuronal migration
Prior studies demonstrated a requirement for Pitx2 in neuronal differentiation of the developing diencephalon, but the precise stages of differentiation (i.e. migration, axon outgrowth, cell specification) and causative mechanisms were not clear (Martin et al., 2004). A detailed understanding of the stages of neuronal differentiation that require Pitx2 is critical, since these stages may involve separate molecular genetic mechanisms that are differentially susceptible to injury or developmental insult. Pitx2 expression is high in postmitotic neurons of the hypothalamus (Martin et al., 2002), and data presented here demonstrate that defective neuronal migration is a primary phenotype in this brain region of Pitx2 null mutants (Suppl. Fig. 2). Initial patterning of hypothalamic regions appears intact in embryos with complete loss of Pitx2, yet refined formation of subgroups of PITX2-positive neurons or neurons derived from PITX2-positive cells is impaired. Moreover, migration appears to be an early aspect of neuronal differentiation that is sensitive to Pitx2 deficiency, but these defects in neuronal migration may influence later cell identity or axon outgrowth.
The primary neuronal migration defect in Pitx2 deficient mice disrupts organization of the subthalamic nucleus. Pitx2 mutant cells accumulate medially in the hypothalamus, and appear to be retained medially or aberrantly cross the ventral midline. These data are consistent with previous results showing midline crossing of cells in the chick diencephalon, where retroviral labeling produced bilateral clones on either side of the third ventricle (Golden and Cepko 1996).
Prior studies also raised the possibility that anatomical shifts in expression or stability of Pitx2 mRNA might occur with Pitx2 deficiency (Martin et al., 2004), but data presented here make that unlikely. In mouse pituitary cell lines, Pitx2 mRNA is highly unstable, with an estimated half-life of 30 minutes (Briata et al., 2003). Wnt/β-catenin signaling in pituitary cells stabilizes Pitx2 mRNA, via changes in interactions between 3′ UTR sequences in the Pitx2 gene and other proteins (Briata et al., 2003). DNA sequences in the 3′ UTR of the Pitx2 gene that regulate Pitx2 mRNA stability are also present in the Pitx2null and Pitx2cre alleles; hence, any inherent difference in Pitx2 mRNA stability between these alleles is not related to these 3′ mRNA sequences. Moreover, the Pitx2cre allele has the advantage of expressing Cre from IRES mediated bicistronic cassettes, allowing for distinction between indelibly marked Pitx2 expressing mutant neurons and those Pitx2 mutant neurons that are actively expressing Pitx2 mRNA. Since our Cre lineage tracing approach uses a β-gal reporter that is expressed in trans (i.e. a different genomic locus) to Pitx2 and permanently marks Pitx2-expressing neurons (Zinyk et al., 1998), the data provide definitive evidence for defects in migration of Pitx2cre/null neurons that cannot be explained by intrinsic differences in Pitx2 mRNA expression or stability.
Pitx2 function in neurogenesis and cell fate
We found no evidence to support a major role for Pitx2 in neural progenitor proliferation or cell cycle exit, indicating that neurogenesis likely proceeds independent of Pitx2. These results are similar to those obtained in earlier studies (Martin et al., 2004), and suggest that Pitx2 may have tissue and cell type specific roles in progenitor proliferation. Canonical Wnt β-catenin signaling regulates Pitx2 expression in cellular proliferation and survival of the developing pituitary gland and heart (Kioussi et al., 2002); however, our data show that mouse brain neurons are born and survive independent of Pitx2 function. Developing mouse hypothalamic neurons seem to exhibit highly regionally specific requirements for Pitx2 for normal migration. Knowledge about specific roles for Pitx2 in neuronal cell fate are limited to a study in C. elegans showing that an orthologue of Pitx2 (unc-30), is critical for formation of GABAergic neurons (Westmoreland et al., 2001). Identifying altered cell fates in the mouse brain with Pitx2 loss of function remains a major challenge.
Our observations in neurons are also supported by results obtained in craniofacial tissues, where Pitx2 mutant cells exhibit misdirected or arrested migration (Liu et al., 2003). In cultured cells, Pitx2 may regulate one or more aspects of migration through changes in the cytoskeleton, as suggested by studies showing that Pitx2 controls Rho GTPase activity of HeLa cells through regulation of Trio, a guanine nucleotide exchange factor (Wei and Adelstein 2002). Since Rho GTPases are implicated in non-canonical planar cell polarity (PCP) (Winter et al., 2001; Habas et al., 2003), these observations also support the possibility of a more generalized role for Pitx2 in brain non-canonical Wnt signaling.
Genetic interactions in the developing hypothalamus
Expression of LMX1B and PITX2 in the developing subthalamic nucleus implied that these two proteins might actively cooperate or repress each other during neuronal development (Asbreuk et al., 2002). Lmx1b is required for proper expression of Pitx3 in tyrosine hydroxylase positive neurons (Smidt et al., 2000) and for development of the tectum and cerebellum via regulation of Wnt1 and Fgf8 (Guo et al., 2007). Our studies identified colocalization of LMX1B and PITX2 in the subthalamic nucleus. We also found that Lmx1b expression is absent in the Pitx2 mutant subthalamic nucleus, consistent with a failure in migration of subthalamic nucleus neurons (Martin et al., 2004). Lmx1b expression is retained in medially located hypothalamic Pitx2 mutant neurons, suggesting that Lmx1b positive neurons require Pitx2 for proper migration to the subthalamic nucleus but not for Lmx1b gene expression. Dorsal midline LMX1B-positive midbrain neurons are also unaffected by loss of Pitx2.
Lmx1b mutant embryos exhibited normal expression of multiple subthalamic nucleus markers in E14.5 embryos, ruling out an early developmental requirement for Lmx1b in Pitx2 lineage neurons. Combinatorial actions between Pitx genes and Lmx1b may therefore be limited to Pitx3 in mesencephalic dopaminergic neurons (Smidt et al., 2000). Our results are similar to data obtained in developing eye tissues, which demonstrate minimal or unchanged Lmx1b and Pitx2 expression in developing mutant mice (Lu et al., 1999; Pressman et al., 2000). We also analyzed for differences in hypothalamic expression of Ldb1, a cofactor of Lmx1b, and found no changes between Pitx2 mutant and wildtype sections (data not shown). These data indicate that subthalamic nucleus neurons are highly sensitive to Pitx2 but not Lmx1b dosage for normal migration.
Expression of the transcription factors FOXP1 and FOXP2 in specific subthalamic nucleus neurons with unique sensitivities to PITX2 dosage indicates that complex transcriptional networks are required for subthalamic nucleus neuronal differentiation. Mutations in human FOXP2 cause a severe language disorder (Balter 2001), and similar expression of FOXP1 and FOXP2 in humans and birds suggests these two proteins may cooperate to regulate development of specific neuronal populations (Teramitsu et al., 2004). Expression of FOXP1/2 in the mouse cortex, basal ganglia, thalamus, and cerebellum is consistent with roles in postmigratory neurons (Ferland et al., 2003). Some FOXP1-positive and FOXP2-positive subthalamic nucleus neurons require Pitx2 for proper migration, whereas others do not. These observations suggest that Pitx2-expressing subthalamic nucleus neurons exhibit cell intrinsic defects that do not influence the migratory capacities of neighboring neurons. As in other brain regions, functions of FOXP1/2 in the hypothalamus are not well understood, including whether FOXP1 and FOXP2 regulate transcription independently or in cooperation with PITX2 and LMX1B. The differential cellular overlap in FOXP1/2 expression with PITX2 and LMX1B suggests that complex and cell specific combinatorial codes between these four transcriptional regulators are critical for hypothalamic regional specificity and neuronal migration.
Our studies provide indirect evidence about the cell autonomous nature of Pitx2 defects in the developing hypothalamus. Normal migration of some FOXP1-positive and FOXP2-positive subthalamic nucleus suggests cell autonomous Pitx2 defects in migration, whereas the slight increase in hypothalamic Pitx2-expressing cells suggests cell extrinsic influences on gene expression. Further experiments using transplants, chimeras, or reporter tagged, tissue specific Pitx2 knockouts will be helpful in addressing these questions.
Experimental Methods
Generation of mutant mice
We used a previously characterized Pitx2 null allele, Pitx2creneo, that exhibits Cre activity in areas of known Pitx2 expression in the heart and craniofacial structures (Liu et al., 2002; Liu et al., 2003). Pitx2creneo/+ mice are heterozygous for a Pitx2 deficiency allele in which the homeodomain is replaced with an IRES-Cre-neomycin cassette (Liu et al., 2003). Since brain Cre activity patterns in these mice had not been explored in detail, we mated Pitx2creneo/+ mice with FLPe recombinase expressing transgenic mice (Jackson Laboratory, Bar Harbor, Maine; stock #003946) to remove intervening neomycin sequences that might interfere with normal Pitx2 gene expression. Offspring were PCR genotyped for Pitx2cre sequences using Chromo Taq (Denville, Metuchen, NJ) with primers GGTGGGGTGGGGGTGTCTGTAAAA, GCTAGGCGCGAAGGTTCTCCAGTG, and AGATATGGCCCGCGCTGGAGTTTC (Invitrogen, Carlsbad, CA). The resulting PCR product was verified by sequencing (University of Michigan DNA Sequencing Core) to confirm excision of the neomycin cassette. PCR reactions were performed with an initial denaturation at 94°C for 2 min, followed by 35 cycles of 94°C × 30 sec, 59°C × 30 sec, 72°C × 2 min 30 sec, and 10 min incubation at 72°C. In situ hybridization with a Cre antisense RNA probe on Pitx2cre/+ embryos (E10.5 and E12.5) showed Cre mRNA in sites of known Pitx2 expression in the brain, including the developing hypothalamus, midbrain, and zona limitans (Mucchielli et al., 1996) (data not shown). Initial experiments for this study were performed by crossing Pitx2cre/+ mice with Rosa26R mice that are homozygous for a Cre conditional lacZ reporter (Soriano 1999). This approach resulted in preferential β-galactosidase label in the cytoplasm, complicating anatomic definition within the histologically uniform neuroepithelium. To overcome this, we used a strain of mice (N-lacZ) that are transgenic for a Cre conditional, nuclear localized lacZ reporter (Zinyk et al., 1998). Pitx2null/+ mice were as previously described (Gage et al., 1999), and were maintained to generation N11 on a C57BL/6J background. Lmx1b+/n mice were obtained from Iain McIntosh (Johns Hopkins University) and were genotyped as previously described (Chen et al., 1998).
N-lacZ doubly hemizygous mice (Jackson Laboratory, Bar Harbor, Maine; stock #002982) (Zinyk et al., 1998) were mated with Pitx2+/null mice (maintained at generation N9-11 on a C57BL/6J background). Pitx2+/null; N-lacZ/+ mice were mated with N-lacZ doubly hemizygous mice, and Pitx2+/null; N-lacZ/N-lacZ offspring identified by genotyping for Pitx2 wildtype and null alleles as previously described (Gage et al., 1999), and for the N-lacZ transgene using quantitative PCR. In each Q-PCR reaction, doubly hemizygous and hemizygous genomic DNA samples were used for reference. DNA samples were assayed in triplicate at (100 ng/μl, 10 ng/μl, and 2 ng/μl). Each sample was assayed with primers 5′GCCCATCTACACCAACGTAACC3′ and 5′AGTAACAACCCGTCGGATTCTC3′ and probe 6FAM-CGGTCAATCCGCCGTTTGTTCCTA-MRA (Applied Biosystems, Foster City, CA) for detecting LacZ and Assay ID: Mm99999915-g1 (Applied Biosystems, Foster City, CA) for detecting GAPDH with Platinum qPCR SuperMix-UDG with Rox (Invitrogen, Carlsbad, CA). Samples were amplified and detected using an ABI Prism 7000.
Embryo preparation
Timed pregnancies were established using Pitx2cre/+, Pitx2+/null;N-lacZ/N-lacZ, Pitx2+/null, or Lmx1b+/neo mice. The morning of plug identification was designated as E0.5. Litters of embryos were dissected into PBS from pregnant females following cervical dislocation and hysterectomy. From each embryo, an amniotic sac, tail, or limb was retained for genotyping. Embryos were fixed in 4.0% formaldehyde (Fisher, Waltham, MA) for 30 min to 4 hours depending on age, embedded in paraffin and sectioned at 7 μm thickness.
β-galactosidase assay
Litters of E10.5–E14.5 embryos were dissected and fixed in 4% formaldehyde for 20–30 minutes. Embryos were then washed in PBS and X-gal Wash Buffer, as described (Sclafani et al., 2006). Whole embryos were cleared in a glycerol gradient and photographed on a Leica MZ10F dissection microscope. For preparation of vibratome sections, embryos were then post-fixed in 4% formaldehyde overnight at 4° C, craniofacial tissues removed, and brains embedded in 4% low melt Gene Pure agarose (BioExpress, Kaysville, UT) and sectioned at 100–150 μm. Some sections were then double labeled with rabbit anti-Calretinin (Chemicon) at 1:2000 and detected with diaminobenzidine (DAB), as previously described (Martin et al., 2004), then cleared in glycerol and photographed.
Immunohistochemistry, in situ hybridization, and TUNEL staining
Cell proliferation studies with 5-bromo-2′-deoxyuridine (BrdU, Sigma, St. Louis, MO) were done as previously described (Martin et al., 2002; Martin et al., 2004), with intraperitoneal injections of BrdU (300 μl of 1 mg/ml in PBS) at 1 hour, 1 day or 2 days prior to embryo collection. Immunohistochemistry or immunofluorescence on paraffin embedded tissues was also done as described (Martin et al., 2002; Martin et al., 2004), with rat anti-β-galactosidase at 1:1000 (from Tom Glaser), guinea pig anti-LMX1B at 1:10,000 (from Tom Jessell), rabbit anti-FOXP1 at 1:250 (Ferland et al., 2003), rabbit anti-FOXP2 at 1:2000 (Abcam, Cambridge, MA), rabbit anti-DCAMKL1(DCLK) at 1:50 (Lin et al., 2000), rabbit anti-NCAM at 1:500 (Millipore, Billerica, MA), rabbit anti-N-cadherin (Abcam, Cambridge, MA) at 1:100, rabbit anti-PITX2 at 1:400 (Hjalt et al., 2000), rat anti-BrdU at 1:200 (GeneTex, San Antonio, TX), and rabbit anti-Calretinin (Chemicon) at 1:2000. Secondary antibodies were from Vector Laboratories (Burlingame, CA) or Invitrogen (Carlsbad, CA). In situ hybridization was performed as previously described (Martin et al., 2002; Martin et al., 2004), using cRNA probes for Pitx2, Cre, Ldb1, and Foxb1. Cell death assays were done by TUNEL labeling (Oncogene Research Products, Boston, MA) with methyl green counterstain.
Cell counts
For quantification of hypothalamic neurons, we performed β-galactosidase immunofluorescence on at least three transverse sections from Pitx2cre/+; N-lacZ and Pitx2cre/null; N-lacZ E14.5 embryos. Sections were photographed on a Leica DMRB microscope and with Photoshop vCS2 software. β-galactosidase positive cells were counted by superimposing an orthogonal grid with lines spaced 100 μm apart, scoring cells for nuclear β-galactosidase fluorescence, and tallying the cumulative total number of cells in each column or row over three sections spaced 300 μm apart.
Supplementary Material
Supplemental Figure 1. β-galactosidase recapitulates PITX2 expression in the Pitx2cre/+; N-lacZ hypothalamus and subthalamic nucleus. Coronal sections of postnatal day 1 Pitx2cre/+; N-lacZ embryos at the level of the rostral (A–C) or caudal (D–F) subthalamic nucleus (rSTN and cSTN) and were co-labeled with anti-PITX2 (A, D) and β-gal (B, E), with merged images shown in (C, F). All PITX2-immunopositive cells also labeled with anti-β-gal, indicating that the lineage tracing reliably identifies PITX2-lineage cells. There were no β-gal-positive/PITX2-negative cells in the developing hypothalamus, suggesting that PITX2 expression is not transient.
Supplemental Figure 2. Cartoon model depicting neuronal migration routes in the developing Pitx2cre/+ (left) and Pitx2cre/null mouse E14.5 hypothalamus. Shown is a transverse slice at the level of the subthalamic nucleus in an orientation similar to that shown in Fig. 2. Neurons migrate from ventral (V) to dorsal (D), medial (grey represents the wall of the third ventricle) to lateral, and caudal to rostral (R). Note that Pitx2cre/null neurons at E14.5 occupy positions that are more medial and ventral than in their Pitx2cre/+ littermates.
Supplemental Figure 3. Normal cell death and Foxb1 mRNA expression in Pitx2 null mutants. TUNEL labeling of transverse hypothalamic sections from E10.5 (A, B) and E12.5 (C, D) Pitx2cre/+; N-lacZ (A, C) and Pitx2cre/null; N-lacZ (B, D) embryos showed minimal numbers of apoptotic cells. Insert in B″ shows TUNEL positive labeled cells from E10.5 oral ectoderm. Foxb1 mRNA expression (E, F) is unaltered in E14.5 transverse hypothalamic sections from Pitx2null/null embryos (F) compared to Pitx2+/− embryos (E) at the level of the subthalamic nucleus. Pitx2 mRNA in adjacent sections is shown in G and H.
Acknowledgments
We thank Ben Novitch and Yasushi Nakagawa for critical comments on the manuscript. The following people provided reagents: Iain McIntosh (Lmx1b mice), Tord Hjalt (PITX2 antibody), Tom Glaser (β-galactosidase antibody), Tom Jessell (LMX1B antibody), Chris Walsh (CAMKL1 antibody), and Edward Morrissey (FOXP1 antibody). Margaret Lomax and Bob Lyons assisted with Q-PCR for genotyping N-lacZ mice. These data were presented in part at the 2006 annual meetings of the Society for Developmental Biology and the Society for Neuroscience. This work was supported by funds to DMM (NIH KO8 HD40288, R01 NS054784, a Children’s Health Research Center Award, and an award from Janette Ferrantino) and NIH RO1 DE16329 to JFM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman J, Bayer SA. The development of the rat hypothalamus. Adv Anat Embryol Cell Biol. 1986;100:1–178. [PubMed] [Google Scholar]
- Alvarez-Bolado G, Zhou X, Voss AK, Thomas T, Gruss P. Winged helix transcription factor Foxb1 is essential for access of mammillothalamic axons to the thalamus. Development. 2000;127(5):1029–38. doi: 10.1242/dev.127.5.1029. [DOI] [PubMed] [Google Scholar]
- Asbreuk CH, Vogelaar CF, Hellemons A, Smidt MP, Burbach JP. CNS expression pattern of lmx1b and coexpression with ptx genes suggest functional cooperativity in the development of forebrain motor control systems. Mol Cell Neurosci. 2002;21(3):410–20. doi: 10.1006/mcne.2002.1182. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128(1):29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Balter M. Genetics. First gene linked to speech identified. Science. 2001;294(5540):32. doi: 10.1126/science.294.5540.32a. [DOI] [PubMed] [Google Scholar]
- Berry FB, Lines MA, Oas JM, Footz T, Underhill DA, Gage PJ, Walter MA. Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum Mol Genet. 2006;15(6):905–19. doi: 10.1093/hmg/ddl008. [DOI] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6(1):7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R. The Wnt/beta-catenin-->Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell. 2003;12(5):1201–11. doi: 10.1016/s1097-2765(03)00407-6. [DOI] [PubMed] [Google Scholar]
- Charles MA, Suh H, Hjalt TA, Drouin J, Camper SA, Gage PJ. PITX genes are required for cell survival and Lhx3 activation. Mol Endocrinol. 2005 doi: 10.1210/me.2005-0052. [DOI] [PubMed] [Google Scholar]
- Charles MA, Suh H, Hjalt TA, Drouin J, Camper SA, Gage PJ. PITX genes are required for cell survival and Lhx3 activation. Mol Endocrinol. 2005;19(7):1893–903. doi: 10.1210/me.2005-0052. [DOI] [PubMed] [Google Scholar]
- Chen H, Lun Y, Ovchinnikov D, Kokubo H, Oberg KC, Pepicelli CV, Gan L, Lee B, Johnson RL. Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet. 1998;19(1):51–5. doi: 10.1038/ng0598-51. [DOI] [PubMed] [Google Scholar]
- Diehl AG, Zareparsi S, Qian M, Khanna R, Angeles R, Gage PJ. Extraocular muscle morphogenesis and gene expression are regulated by Pitx2 gene dose. Invest Ophthalmol Vis Sci. 2006;47(5):1785–93. doi: 10.1167/iovs.05-1424. [DOI] [PubMed] [Google Scholar]
- Ding YQ, Marklund U, Yuan W, Yin J, Wegman L, Ericson J, Deneris E, Johnson RL, Chen ZF. Lmx1b is essential for the development of serotonergic neurons. Nat Neurosci. 2003;6(9):933–8. doi: 10.1038/nn1104. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460(2):266–79. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126(20):4643–51. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- Ganga M, Espinoza HM, Cox CJ, Morton L, Hjalt TA, Lee Y, Amendt BA. PITX2 Isoform-specific Regulation of Atrial Natriuretic Factor Expression: SYNERGISM AND REPRESSION WITH Nkx2.5. J Biol Chem. 2003;278(25):22437–45. doi: 10.1074/jbc.M210163200. [DOI] [PubMed] [Google Scholar]
- Golden JA, Cepko CL. Clones in the chick diencephalon contain multiple cell types and siblings are widely dispersed. Development. 1996;122(1):65–78. doi: 10.1242/dev.122.1.65. [DOI] [PubMed] [Google Scholar]
- Guo C, Qiu HY, Huang Y, Chen H, Yang RQ, Chen SD, Johnson RL, Chen ZF, Ding YQ. Lmx1b is essential for Fgf8 and Wnt1 expression in the isthmic organizer during tectum and cerebellum development in mice. Development. 2007;134(2):317–25. doi: 10.1242/dev.02745. [DOI] [PubMed] [Google Scholar]
- Habas R, Dawid IB, He X. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 2003;17(2):295–309. doi: 10.1101/gad.1022203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjalt TA, Amendt BA, Murray JC. PITX2 regulates procollagen lysyl hydroxylase (PLOD) gene expression: implications for the pathology of Rieger syndrome. J Cell Biol. 2001;152(3):545–52. doi: 10.1083/jcb.152.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjalt TA, Semina EV, Amendt BA, Murray JC. The Pitx2 protein in mouse development. Dev Dyn. 2000;218(1):195–200. doi: 10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Joyner AL, Zervas M. Genetic inducible fate mapping in mouse: establishing genetic lineages and defining genetic neuroanatomy in the nervous system. Dev Dyn. 2006;235(9):2376–85. doi: 10.1002/dvdy.20884. [DOI] [PubMed] [Google Scholar]
- Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T, Ohgi KA, Lin C, Gleiberman A, Wang J, Brault V, Ruiz-Lozano P, Nguyen HD, Kemler R, Glass CK, Wynshaw-Boris A, Rosenfeld MG. Identification of a Wnt/Dvl/beta-Catenin --> Pitx2 Pathway Mediating Cell-Type-Specific Proliferation during Development. Cell. 2002;111(5):673–85. doi: 10.1016/s0092-8674(02)01084-x. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, Kondo S, Yokoyama M. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126(24):5749–58. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- Lim Y, Golden JA. Patterning the developing diencephalon. Brain Res Brain Res Rev. 2006 doi: 10.1016/j.brainresrev.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Lim Y, Golden JA. Patterning the developing diencephalon. Brain Res Rev. 2007;53(1):17–26. doi: 10.1016/j.brainresrev.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Lin CR, Kioussi C, O’Connell S, Briata P, Szeto D, Liu F, Izpisua-Belmonte JC, Rosenfeld MG. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401(6750):279–82. doi: 10.1038/45803. [DOI] [PubMed] [Google Scholar]
- Lin PT, Gleeson JG, Corbo JC, Flanagan L, Walsh CA. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. J Neurosci. 2000;20(24):9152–61. doi: 10.1523/JNEUROSCI.20-24-09152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129(21):5081–91. doi: 10.1242/dev.129.21.5081. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Lu MF, Martin JF. Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development. 2003;130(25):6375–85. doi: 10.1242/dev.00849. [DOI] [PubMed] [Google Scholar]
- Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature. 1999;401(6750):276–8. doi: 10.1038/45797. [DOI] [PubMed] [Google Scholar]
- Marchand R. Histogenesis of the subthalamic nucleus. Neuroscience. 1987;21(1):183–95. doi: 10.1016/0306-4522(87)90332-0. [DOI] [PubMed] [Google Scholar]
- Martin DM, Skidmore JM, Fox SE, Gage PJ, Camper SA. Pitx2 distinguishes subtypes of terminally differentiated neurons in the developing mouse neuroepithelium. Dev Biol. 2002;252(1):84–99. doi: 10.1006/dbio.2002.0835. [DOI] [PubMed] [Google Scholar]
- Martin DM, Skidmore JM, Philips ST, Vieria C, Gage PJ, Condie BG, Raphael Y, Martinez S, Camper SA. PITX2 is required for normal development of neurons in the mouse subthalamic nucleus and midbrain. Dev Biol. 2004;267(1):93–108. doi: 10.1016/j.ydbio.2003.10.035. [DOI] [PubMed] [Google Scholar]
- Metzger D, Clifford J, Chiba H, Chambon P. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc Natl Acad Sci U S A. 1995;92(15):6991–5. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucchielli ML, Martinez S, Pattyn A, Goridis C, Brunet JF. Otlx2, an Otx-related homeobox gene expressed in the pituitary gland and in a restricted pattern in the forebrain. Mol Cell Neurosci. 1996;8(4):258–71. doi: 10.1006/mcne.1996.0062. [DOI] [PubMed] [Google Scholar]
- Pressman CL, Chen H, Johnson RL. LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis. 2000;26(1):15–25. [PubMed] [Google Scholar]
- Puelles L, Rubenstein JL. Forebrain gene expression domains and the evolving prosomeric model. Trends Neurosci. 2003;26(9):469–76. doi: 10.1016/S0166-2236(03)00234-0. [DOI] [PubMed] [Google Scholar]
- Sclafani A, Skidmore J, Ramaprakash H, Trumpp A, Gage P, Martin D. Nestin-Cre mediated deletion of Pitx2 in the mouse. Genesis. 2006;44:336–344. doi: 10.1002/dvg.20220. [DOI] [PubMed] [Google Scholar]
- Shih HP, Gross MK, Kioussi C. Cranial muscle defects of Pitx2 mutants result from specification defects in the first branchial arch. Proc Natl Acad Sci U S A. 2007;104(14):5907–12. doi: 10.1073/pnas.0701122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, Asbreuk CH, Cox JJ, Chen H, Johnson RL, Burbach JP. A second independent pathway for development of mesencephalic dopaminergic neurons requires Lmx1b. Nat Neurosci. 2000;3(4):337–41. doi: 10.1038/73902. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain [letter] Nature Genetics. 1999;21(1):70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sur M, Rubenstein JL. Patterning and plasticity of the cerebral cortex. Science. 2005;310(5749):805–10. doi: 10.1126/science.1112070. [DOI] [PubMed] [Google Scholar]
- Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E. Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience. 2004;124(2):261–7. doi: 10.1016/j.neuroscience.2003.11.036. [DOI] [PubMed] [Google Scholar]
- Teramitsu I, Kudo LC, London SE, Geschwind DH, White SA. Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J Neurosci. 2004;24(13):3152–63. doi: 10.1523/JNEUROSCI.5589-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Adelstein RS. Pitx2a expression alters actin-myosin cytoskeleton and migration of HeLa cells through Rho GTPase signaling. Mol Biol Cell. 2002;13(2):683–97. doi: 10.1091/mbc.01-07-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland JJ, McEwen J, Moore BA, Jin Y, Condie BG. Conserved function of Caenorhabditis elegans UNC-30 and mouse Pitx2 in controlling GABAergic neuron differentiation. J Neurosci. 2001;21(17):6810–9. doi: 10.1523/JNEUROSCI.21-17-06810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter CG, Wang B, Ballew A, Royou A, Karess R, Axelrod JD, Luo L. Drosophila Rho-associated kinase (Drok) links Frizzled-mediated planar cell polarity signaling to the actin cytoskeleton. Cell. 2001;105(1):81–91. doi: 10.1016/s0092-8674(01)00298-7. [DOI] [PubMed] [Google Scholar]
- Zervas M, Millet S, Ahn S, Joyner AL. Cell behaviors and genetic lineages of the mesencephalon and rhombomere 1. Neuron. 2004;43(3):345–57. doi: 10.1016/j.neuron.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Zinyk DL, Mercer EH, Harris E, Anderson DJ, Joyner AL. Fate mapping of the mouse midbrain-hindbrain constriction using a site-specific recombination system. Curr Biol. 1998;8(11):665–8. doi: 10.1016/s0960-9822(98)70255-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. β-galactosidase recapitulates PITX2 expression in the Pitx2cre/+; N-lacZ hypothalamus and subthalamic nucleus. Coronal sections of postnatal day 1 Pitx2cre/+; N-lacZ embryos at the level of the rostral (A–C) or caudal (D–F) subthalamic nucleus (rSTN and cSTN) and were co-labeled with anti-PITX2 (A, D) and β-gal (B, E), with merged images shown in (C, F). All PITX2-immunopositive cells also labeled with anti-β-gal, indicating that the lineage tracing reliably identifies PITX2-lineage cells. There were no β-gal-positive/PITX2-negative cells in the developing hypothalamus, suggesting that PITX2 expression is not transient.
Supplemental Figure 2. Cartoon model depicting neuronal migration routes in the developing Pitx2cre/+ (left) and Pitx2cre/null mouse E14.5 hypothalamus. Shown is a transverse slice at the level of the subthalamic nucleus in an orientation similar to that shown in Fig. 2. Neurons migrate from ventral (V) to dorsal (D), medial (grey represents the wall of the third ventricle) to lateral, and caudal to rostral (R). Note that Pitx2cre/null neurons at E14.5 occupy positions that are more medial and ventral than in their Pitx2cre/+ littermates.
Supplemental Figure 3. Normal cell death and Foxb1 mRNA expression in Pitx2 null mutants. TUNEL labeling of transverse hypothalamic sections from E10.5 (A, B) and E12.5 (C, D) Pitx2cre/+; N-lacZ (A, C) and Pitx2cre/null; N-lacZ (B, D) embryos showed minimal numbers of apoptotic cells. Insert in B″ shows TUNEL positive labeled cells from E10.5 oral ectoderm. Foxb1 mRNA expression (E, F) is unaltered in E14.5 transverse hypothalamic sections from Pitx2null/null embryos (F) compared to Pitx2+/− embryos (E) at the level of the subthalamic nucleus. Pitx2 mRNA in adjacent sections is shown in G and H.