

Abstract
The Drosophila fruitless (fru) gene encodes a transcription factor that essentially regulates all aspects of male courtship behavior. The use of alternative 5′-splice sites generates fru isoforms that determine gender-appropriate sexual behaviors. Alternative splicing of fru is regulated by TRA and TRA2 and depends on an exonic splicing enhancer (fruRE) consisting of three 13-nucleotide repeat elements, nearly identical to those that regulate alternative sex-specific 3′-splice site choice in the doublesex (dsx) gene. dsx has provided a useful model system to investigate the mechanisms of enhancer-dependent 3′-splice site choice. However, little is known about enhancer-dependent regulation of alternative 5′-splice sites. The mechanisms of this process were investigated using an in vitro system in which recombinant TRA/TRA2 could activate the female-specific 5′-splice site of fru. Mutational analysis demonstrated that one 13-nucleotide repeat element within the fruRE is required and sufficient to activate the regulated female-specific splice site. As was established for dsx, the fruRE can be replaced by a short element encompassing tandem 13-nucleotide repeat elements, by heterologous splicing enhancers, and by artificially tethering a splicing activator to the pre-mRNA. Complementation experiments showed that Ser/Arg-rich proteins facilitate enhancer-dependent 5′-splice site activation. We conclude that splicing enhancers function similarly in activating regulated 5′- and 3′-splice sites. These results suggest that exonic splicing enhancers recruit multiple spliceosomal components required for the initial recognition of 5′- and 3′-splice sites.
Alternative pre-mRNA splicing is commonly used to regulate the expression of genes and to enrich the proteomic diversity of higher eukaryotic organisms (1-4). Current estimates suggest that ~60% of human genes are alternatively spliced (5), sometimes leading to thousands of different mRNA isoforms from a single gene. Numerous examples describe how alternative splicing regulates gene expression in humans, but the mechanisms involved are understood in only a few cases (6-17). In contrast, the use of genetics in Drosophila has aided in the identification of proteins that regulate alternative splicing. In particular, the genes involved in Drosophila sex determination have become prototypes for the study of positive and negative control of alternative splicing (18). The best characterized gene is the doublesex (dsx)1 gene, in which the use of two alternative 3′-splice sites leads to the expression of male- or female-specific gene products that initiate gender-specific sexual differentiation. Female-specific splicing of dsx requires the production of two proteins, Transformer (TRA) and Transformer2 (TRA2), and an exonic splicing enhancer (ESE) element located within the untranslated region of the fourth exon (19-21). Both TRA and TRA2 contain Arg/Ser-rich (RS) domains, protein interaction domains characteristic of the Ser/Arg-rich (SR) family of essential splicing factors. SR proteins are thought to function by binding to ESEs and by recruiting the splicing machinery to adjacent introns via protein interactions involving the RS domain (22, 23). The dsx ESE, designated dsxRE, contains six nearly identical copies of a 13-nucleotide repeat sequence (24-30) that each serve as a binding site for the cooperative assembly of a multicomponent splicing enhancer complex containing TRA, TRA2, and an SR protein (31). Most ESEs studied in mechanistic detail activate 3′-splice sites that contain suboptimal polypyrimidine tracts, the site of U2 snRNP auxiliary factor (U2AF) binding. A picture has therefore emerged suggesting that enhancer complexes assembled on ESEs recruit U2AF to a weak 3′-splice site (32-34). An alternative and equally attractive model proposes that ESEs interact with a master assembly factor, such as the splicing coactivator SRm160 (22), that assists in the assembly of the spliceosome. Thus, ESEs may recruit spliceosomal factors to the regulated 3′-splice site through direct or indirect interactions.
Although much has been learned about the mechanisms of ESE-dependent 3′-splice site activation, the mechanisms of enhancer-dependent 5′-splice site recognition are currently not well understood. It has been documented that the SR protein ASF/SF2 promotes the association of U1 snRNP to the 5′-splice site of several test RNAs (35-37). However, it has not yet been established how ASF/SF2 stabilizes U1 snRNP binding to the 5′-splice site. Several recent studies have described examples of ESE-dependent alternative 5′-splice site regulation (38-43). Although many of the current efforts are focused on the identification of the trans-acting factors that regulate each alternative 5′-splice site choice, genetic approaches have already provided this information for alternative 5′-splice site choice in the Drosophila melanogaster fruitless (fru) gene. The detailed study of fru alternative splicing therefore provides an excellent opportunity to expand our limited knowledge on the mechanisms that govern ESE-dependent 5′-splice site regulation.
fru is expressed in ~0.5% of the neurons in the central nervous system (44, 45). Cloning of fru demonstrated that sex-specific alternative splicing of the fru pre-mRNA determines the majority of male courtship behavior (38, 46, 47). Like dsx, fru is alternatively spliced in a sex-specific manner that requires TRA and TRA2 to switch from the male-specific pattern to the female-specific pattern (38, 46). However, in contrast to dsx, fru alternative splicing involves the activation of a female-specific 5′-splice site (38, 40, 46). This activation requires an ESE located immediately upstream of the female-specific 5′-splice site (40). Surprisingly, the fru enhancer, designated fruRE, contains three copies of similar 13-nucleotide repeat elements that regulate the female-specific alternative 3′-splice site in dsx (see Table I). Thus, TRA and TRA2 regulate both dsx and fru alternative splicing, presumably through the formation of similar, if not identical, enhancer complexes. In support of this suggestion, it was previously shown that the fruRE is capable of functionally replacing the dsxRE using heterologous pre-mRNAs in transfection assays (40). Interestingly, whereas the dsx enhancer complex activates an alternative 3′-splice site, the fru enhancer complex activates an alternative 5′-splice site.
Table I. Comparison of 13-nucleotide repeat elements in dsx and fru.
Repeat 1 is defined as the closest repeat to the regulated splice site (Fig. 1). NA, not applicable.
| Repeat no. | dsx | fru |
|---|---|---|
| 1 | UCUUCAAUCAACA | ACUUCAAUCAACA |
| 2 | UCUUCAAUCAACA | UCUUCAAUCAACA |
| 3 | UCUACAAUCAACA | UCAUCAAUCAACA |
| 4 | UCUUCAAUCAACA | NA |
| 5 | UCAACAAUCAACA | NA |
| 6 | UCAACGAUCAACA | NA |
Initial recognition of exon/intron junctions is achieved by several spliceosomal factors (48). U1 snRNP binds to the 5′-splice site by a combination of RNA/RNA and protein/RNA interactions (49, 50). The 3′-splice site is initially recognized by U2AF. The activation of 5′- or 3′-splice sites thus requires the recruitment of a different set of splicing factors to their respective exon/intron junction. Previous studies have demonstrated that each dsxRE interacts with only one spliceosomal target (51). However, the manner in which the alternative splicing of the fru pre-mRNA is regulated suggests that the TRA/TRA2-dependent dsx and fru ESEs may recruit distinct spliceosomal components to the adjacent intron.
This hypothesis was tested using an in vitro splicing system that allowed an in-depth investigation of the splicing enhancer that controls the recognition of the fru female-specific 5′-splice site. Mutational analysis demonstrated that one of the 13-nucleotide repeat elements present in the fruRE is sufficient to activate the regulated female-specific splice site. As was demonstrated for dsx, the fruRE can be replaced efficiently by a short element encompassing tandem 13-nucleotide repeat elements, by previously characterized heterologous ESEs, and by artificially tethering a splicing activation domain to the pre-mRNA. Finally, complementation experiments demonstrated that SR proteins facilitate ESE-dependent 5′-splice site activation. These experiments show that the fruRE and dsxRE function similarly in activating regulated 5′- and 3′-splice sites, respectively, and suggest that the fru and dsx ESE complexes are capable of recruiting targets that contain multiple spliceosomal components required for the initial recognition of 5′- and 3′-splice sites.
EXPERIMENTAL PROCEDURES
RNAs
To generate the in vitro splicing substrate fruF-long, two fragments generated from fruM+Fwt (a gift from Bruce Baker) (40) were inserted into the same restriction sites of SP73 (Promega): a PstI fragment that contains the common 3′-exon and an EcoRI fragment containing the female-specific 5′-splice site and the upstream splicing enhancer. Substrate fruF was derived from fruF-long by deleting ~560 nucleotides of intronic sequences (XbaI and NcoI digest, followed by blunt ligation). Mutant substrates fruFmut1, fruFmut2, fruFmut3, fruFmut23, and fruFmut123 are derivatives of construct fruF. In all these constructs, the 13-nucleotide repeat sequences have been changed from NNNTCAATCAACA to NNNGGCAGCTTAC. To generate these mutations, a three-step PCR method was used. Primers spanning each repeat in forward (fwd) and reverse (rev) directions were used in combination with a reverse primer at the downstream EcoRI site (fruFrev) and an SP6 primer, respectively. The PCR fragments so generated were then annealed together and amplified. After digestion with EcoRI, mutated fragments were inserted into EcoRI-digested fruF backbone, replacing the wild-type fruF EcoRI fragment. The fru primers used were 5′-ACTGGCAGCGTACTACCCACAAA-3′ (fruFmut1fwd), 5′-GTACGCTGCCAGTATTGGTTCTT-3′ (fruFmut1rev), 5′-TCTGGCAGCTTACCTCAACCCGAACTGGGCC-3′ (fruFmut2fwd), 5′-GTAAGCTGCCAGAGCAGCTCCTCCGGGCAC-3′ (fruFmut2rev), 5′-TCAGGCAGCTTACTTCCCGTGCCCGGAGGAG-3′ (fruFmut3fwd), 5′-GTAAGCTGCCTGATATTCTCTTTCGATCTGG-3′ (fruFmut3rev), and 5′-CGAATTCGGTTGCATGTTCC-3′ (fruFrev). The cryptic splice site, aaa/gttagt (the slash indicates the exon/intron junction), was removed by PCR-based site-directed mutagenesis, resulting in the deletion of nucleotides a/gt. The primers used for site-directed mutagenesis were as followed: 5′-ATTTAGGTGACACTATA-3′ (primer 1fwd), 5′-GCCCGGCAACCTAATAGTCCTTTCATTAG-3′ (primer 2rev), 5′-CTAATGAAAGGACTATTAGGTTGCCGGGC-3′ (primer 3fwd), and 5′-GCTCGAATTCGGTTGCATGTTCC-3′ (primer 4rev). The construct fruM+F1mut contains both male- and female-specific 5′-splice sites, but lacks the cryptic 5′-splice site. To generate fruM+F1mut, an insert containing the fru male-specific exonic and 5′-splice site sequences was generated by PCR amplification using fruM+Fwt (40) as the template. The primer sequences used for amplification were 5′-GCCGCTCGAGTGCATTACGCGGCCTTGGACTT-3′ and 5′-GTAAGCTGCCTGATATTCTCTTTCGATCTGG-3′. The insert was XhoI/BbsI-digested and subcloned into fruM+F1R. A BglII/ClaI fragment containing the fru male-specific exonic and 5′-splice site sequences from fruM+F1R was then inserted 92 nucleotides upstream of a mutated 13-nucleotide repeat element to yield fruM+F1mut. The only 13-nucleotide repeat element present in this construct has been mutated as described for the fruFmut plasmids. In substrate fruF2Rs, two closely linked synthetic 13-nucleotide repeat elements replace the three dispersed 13-nucleotide repeat elements contained within the fruRE. To generate fruM+F2Rs, a BglII/ClaI fragment as described for fruM+F1mut was inserted into the EcoRV site of plasmid fruF2Rs that contains two tandem 13-nucleotide repeat elements ~10 base pairs upstream of the female-specific 5′-splice site. The fru(70)M1 pre-mRNA was generated by PCR using the fruF construct as a template and the T7 primer and the 5′-BglII fru primer (5′-CCTAAAGATCTTACCCACAAAAAAAAACAA-3′). The PCR product from this reaction was digested with BglII and XhoI and cloned into pSP72-MS2 (23) digested with XhoI and BamHI. The identities of all constructs were verified by DNA sequencing.
With the exception of fru(70)M1, all constructs were digested with XhoI to yield the SP6 RNA polymerase transcription templates. Capped, 32P-labeled RNAs were transcribed with SP6 RNA polymerase (Promega). The fru(70)M1 construct was digested with XhoI and transcribed with T7 RNA polymerase. Transcripts were gel-purified on a 7 m urea and 4% polyacrylamide gel before use.
In Vitro Splicing Assays
Conditions for in vitro splicing reactions were 30% HeLa nuclear extract (52), 1 mm ATP, 20 mm creatine phosphate, 3.2 mm MgCl2, 5 units of RNasin (Promega), 1 mm dithiothreitol, 72.5 mm KCl, 3% polyvinyl alcohol (30–70 kDa), and 12 mm HEPES (pH 7.9). Reactions for TRA/TRA2-dependent substrates were performed in the absence or presence of 500 nm TRA/TRA2, unless otherwise stated. Splicing reactions were performed for 2 h at 30 °C. Following incubation, reactions were proteinase K-digested, phenol/chloroform-extracted, and ethanol-precipitated prior to PAGE separation. Bands were visualized and quantitated using PhosphorImager analysis (Bio-Rad). Percent spliced is defined as follows: spliced products/(unspliced product + spliced products). The background was determined individually for each lane because the total number of substrate and product counts varied throughout a time course. The -fold activation of the female-specific spliced product was calculated by dividing the percent female-specific spliced product in the presence of TRA/TRA2 by the percent female-specific spliced product in the absence of TRA/TRA2. Comparison of female- and male-specific (or cryptic) splice site strength was performed by taking the ratio of percent female-specific spliced product to percent cryptic spliced product in fruF. S100 complementation experiments were performed essentially as described above, except that 40% HeLa cell cytoplasmic extract (S100) was used instead of 30% nuclear extract. Reactions were complemented with varying amounts (0–800 nm) of the baculovirus-produced SR protein 9G8. In vitro splicing reactions using the MS2-RS fusion proteins were carried out using 15% HeLa cell nuclear extract as described (53), except that the reactions were conducted for 4 h. Experiments were performed at least three times. Variations between experiments were within 2-fold.
Proteins
The MS2, MS2-RSSF2/ASF, MS2-RSp55, and MS2-RS9G8 proteins were described previously (53). The isolation of recombinant TRA and TRA2 was performed according to previously described methods (28).
RESULTS
In Vitro Splicing of fru Minigenes
As observed for dsx, the products of the sex determination genes tra and tra2 switch fru splicing from the male-specific pattern to the female-specific pattern through the activities of an ESE that contains multiple, near-identical 13-nucleotide repeat elements (38). However, whereas the dsx enhancer complex activates an alternative 3′-splice site, the fru enhancer complex activates an alternative 5′-splice site. To investigate the mechanisms of this process, an in vitro splicing system was established in which recombinant TRA/TRA2 could activate the female-specific 5′-splice site of fru. Similar to the successful approach taken to unravel the mechanisms of enhancer-dependent 3′-splice site activation in dsx, this assay relies on spliceosomal activities supplied from HeLa cell nuclear extract supplemented with the Drosophila-specific recombinant splicing factors TRA and TRA2 (28). Initially, two pre-mRNA substrates, fruF-long and fruF, were generated. Both substrates harbor the female-specific 5′-splice junction, an upstream region that contains all three 13-nucleotide repeat elements presumed to constitute the fruRE, and the common 3′-exon. The length of the intron was reduced from its natural span of 70,000 nucleotides to either 1200 or 640 nucleotides (Fig. 1A). As shown in Fig. 1B, in the absence of TRA and TRA2, the female-specific 5′-splice site of substrates fruF-long and fruF was not utilized (lanes 4, 5, 10, and 11). However, the addition of 500 nm TRA and TRA2 to the splicing reactions activated the female-specific 5′-splice site in each pre-mRNA (lanes 2, 3, 8, and 9). In addition to the TRA/TRA2-dependent female-specific splicing pathway, we routinely observed splicing to an upstream cryptic 5′-splice site located within the fruRE (Fig. 1, B and C). However, cryptic splicing was independent of the presence of TRA and TRA2. The identities of the female-specific and cryptic product bands as diagramed in Fig. 1 were established by band excision, reverse transcription-PCR, and sequencing of the amplification product (data not shown). Variation between independent experiments was observed to be within 2-fold.
Fig. 1. In vitro activation of the fru female-specific 5′-splice site.
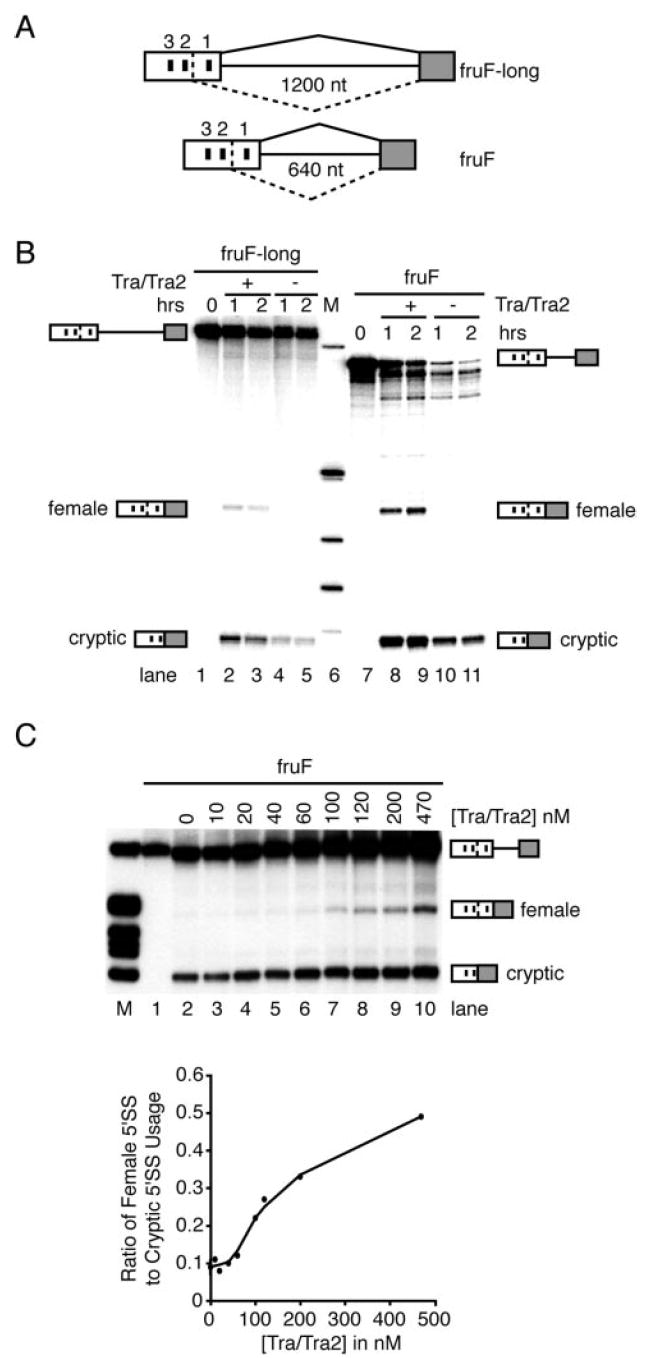
A, schematic of fru substrates used. Gray boxes, common 3′-exon; white boxes, female-specific exon containing the fruRE. Black bars within the exons represent TRA/TRA2-dependent 13-nucleotide (nt) repeat elements. The numbering of the repeats starts with the one closest to the female-specific 5′-splice site. The dashed lines between the first and second repeat elements represent a cryptic 5′-splice site. The solid lines between the boxes represents an intron. The solid lines above the pre-mRNAs illustrate the normal splicing pathway. The dashed lines below the pre-mRNAs illustrate cryptic splice site usage. B, autoradiograph of in vitro splicing reactions performed with fruF-long and fruF constructs in the presence or absence of TRA/TRA2. Lane 6 shows nucleotide length markers (M). Products of the splicing reaction are indicated on the side. C, titration of TRA/TRA2 on fruF pre-mRNA. Upper panel, TRA/TRA2 titration in 30% HeLa cell nuclear extract. Lane M shows nucleotide length markers. Lane 1 is a zero time point to show the mobility of unspliced pre-mRNA. The reactions in lanes 2–10 contained increasing concentrations of TRA/TRA2. The identities of the unspliced pre-mRNA, female-specific spliced product, and cryptic product are indicated on the right. Lower panel, the ratio of female-specific 5′-splice site (5′SS) usage to cryptic 5′-splice site usage depicted as a function of TRA/TRA2 concentration.
To evaluate the efficiency of TRA/TRA2-dependent splice site activation, a series of titration experiments were performed. As demonstrated in Fig. 1C (upper panel), high concentrations of TRA/TRA2 resulted in significant activation of the female-specific 5′-splice site. At lower concentrations of TRA/TRA2, the female-specific 5′-splice site was only marginally activated. These observations suggested that TRA and TRA2 cooperatively influence 5′-splice site choice. Indeed, quantitation of the data in Fig. 1C (lower panel) clearly illustrates the sigmoidal nature of the binding curve, characteristic of synergistic assembly of multicomponent complexes. These observations mirror the TRA/TRA2-dependent activation profile determined for dsx (51), thus providing support for the notion that the dsxRE and fruRE assemble into highly related splicing complexes. Supplementation of splicing reactions with TRA alone did not lead to any measurable activation of the female-specific splice site (data not shown). The dsx pre-mRNA was used as a control to evaluate the activity of the recombinant proteins as well as to compare the efficiencies of TRA/TRA2-dependent splice site activation in the dsx and fru systems (data not shown). PhosphorImager analysis indicated similar splicing efficiencies between the fruF and dsx pre-mRNAs (~50% spliced/unspliced at 2 h). However, the efficiency of fruF-long pre-mRNA splicing was observed to be ~25%. Together, these results demonstrate that fru female-specific 5′-splice site activation can be faithfully recapitulated in vitro using HeLa cell nuclear extracts that are complemented with the Drosophila recombinant splicing activators TRA and TRA2.
Relative Contribution of Each 13-Nucleotide Repeat Element to Female-specific 5′-Splice Site Activation
To study the relative contribution of each repeat element, a series of mutant fruF substrates were tested in the presence or absence of saturating concentrations of TRA/TRA2 (300 nm). The fruFmut1, fruFmut2, fruFmut3, fruFmut23, and fruFmut123 pre-mRNAs were derived from fruF by mutating the sequence of each 13-nucleotide repeat element (Fig. 2A) (see “Note Added in Proof ”). In the absence of TRA/TRA2, the female-specific 5′-splice site was not utilized in any of these constructs (Fig. 2B, lanes 1–6). However, in the presence of TRA/TRA2, the female-specific 5′-splice site was activated for each pre-mRNA containing at least one 13-nucleotide repeat element (lanes 7–11). Moreover, there was no significant difference in the levels of TRA/TRA2-dependent female-specific splicing, regardless of which repeat was mutated (Fig. 2, B and C). Importantly, TRA and TRA2 did not activate female-specific splicing of the fruFmut123 pre-mRNA, in which all three 13-nucleotide repeats have been mutated (Fig. 2B, lane 12), indicating that at least one repeat is necessary for TRA/TRA2-dependent splicing. By comparing the difference in the observed -fold activation of the female-specific spliced product for the tested substrates when TRA/TRA2 was added (Fig. 2C), it can be inferred that the presence of a single repeat is both necessary and sufficient for maximal activation of the female-specific 5′-splice site. We conclude that the 13-nucleotide repeat elements are the only splicing regulatory elements that significantly contribute to the activation of the fru female-specific 5′-splice site.
Fig. 2. Mutations in TRA/TRA2 enhancer elements determine the relative strength of each 13-nucleotide repeat element.
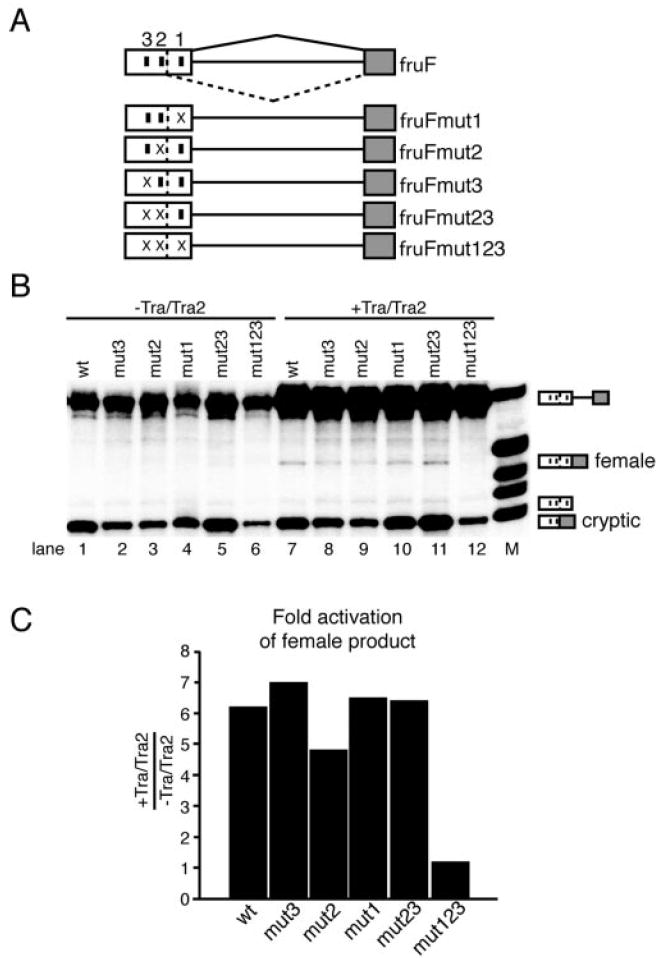
A, schematic of the substrates used. Symbols are as described in the legend to Fig. 1. Mutation of the individual 13-nucleotide repeat elements from NNNTCAATCAACA to NNNGGCAGCTTAC is indicated (×). B, patterns of fruF splicing with successive mutations in TRA/TRA2 repeat elements. The fruF minigenes were spliced in HeLa cell nuclear extract. Splicing patterns were analyzed in the absence (lanes 1–6) and presence (lanes 7–12) of TRA and TRA2. Lane M shows nucleotide length markers. The identities of the spliced products are indicated on the right. C, bar graph depicting the minimal -fold activation of female-specific 5′-splice site usage as observed in the presence of TRA/TRA2. wt, wild-type fruF.
Regulation of the fru Female-specific 5′-Splice Site Requires Competing Splice Sites
Because TRA is expressed only in female flies (54), a possible mechanism of regulation is that the female-specific 5′-splice site is weak and requires ESEs for activation (40). Thus, in male flies, in which TRA is not expressed, the default splicing pathway favors the stronger male-specific splice site. In support of this hypothesis, the sequences for the male-specific, cryptic, and female-specific 5′-splice site are TAG/gtaagc, AAA/gttagt, and GCG/gtaagt, respectively. Compared with the 5′-splice site consensus sequence (YAG/gtragt), we predicted that the male-specific splice site would be the strongest, followed by the female-specific splice site and finally the cryptic splice site. However, in vitro splicing reactions of fruF indicated that the cryptic 5′-splice site 150 nucleotides upstream of the female-specific splice site, positioned in between the first and second repeat elements (Fig. 3A), was efficiently utilized (Figs. 1B, 2B, and 3B). Therefore, in the case of fru, splice site usage does not correlate well with splice site strength based on a consensus sequence match (55). Quantitation of the female-specific spliced product over the cryptic spliced product revealed that the cryptic splicing pathway was dominant and independent of TRA/TRA2 (Fig. 3B, lanes 1–3). Upon addition of TRA/TRA2, the ratio of female-specific splice site choice to cryptic splice site choice increased by ~65-fold (Fig. 3C). To investigate TRA/TRA2-dependent regulation of the female-specific 5′-splice site without interference from constitutive splice sites, the cryptic 5′-splice site was deleted by site-directed mutagenesis (Fig. 3A). Surprisingly, removal of the competing splice site resulted in loss of regulation by TRA/TRA2 (Fig. 3B, lanes 4 –6). Quantitation showed equal efficiency of female-specific splice site activation in the presence and absence of TRA/TRA2 (Fig. 3, B and D), even at decreased levels of nuclear extract (data not shown). These results indicate that activation of the fru female-specific 5′-splice site requires the presence of competing 5′-splice sites. Although the fru female-specific 5′-splice site presumably provides a suboptimal binding site for spliceosomal components, recognition of the female-specific 5′-splice site by the spliceosome occurs efficiently in the absence of competing 5′-splice sites. Therefore, alternative 5′-splice site choice in the fru pre-mRNA is potentiated in part by the relative activities of competing splice sites.
Fig. 3. fru female-specific 5′-splice site strength.
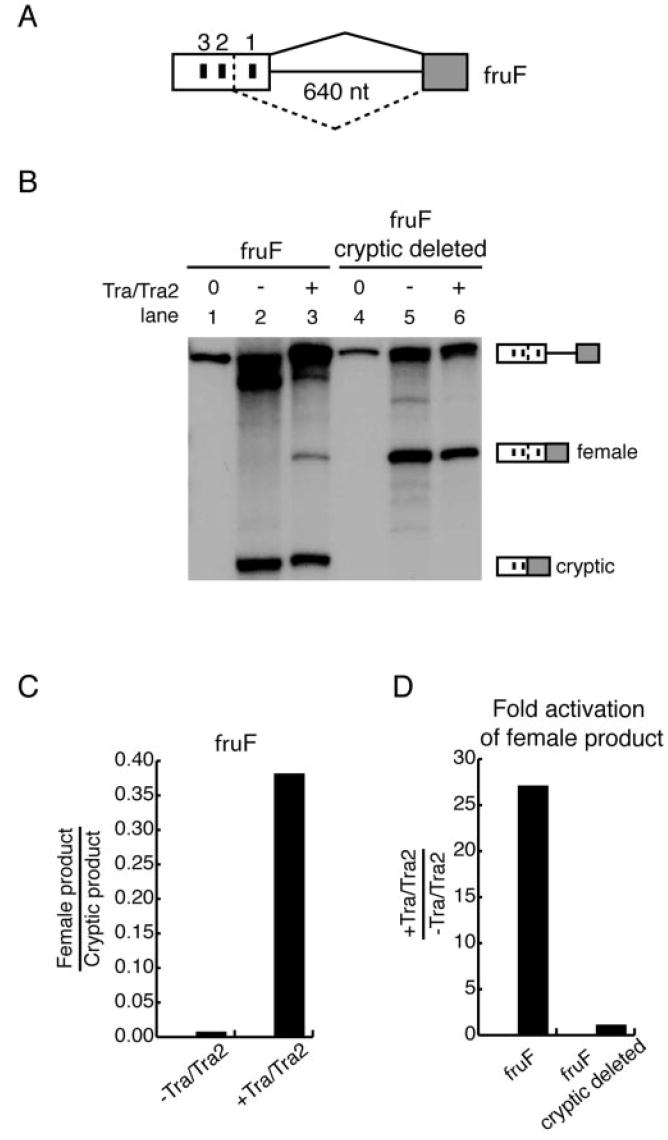
A, schematic of the fruF pre-mRNA construct as described in the legend to Fig. 1. B, autoradiograph of in vitro splicing reactions performed with fruF constructs. Lanes 1–3, fruF; lanes 4–6, fruF with the cryptic splice site deleted. The reactions in lanes 1 and 4 contained unspliced pre-mRNAs. The reactions in lanes 2 and 5 contained no TRA/TRA2. The reactions in lanes 3 and 6 were complemented with 500 nm TRA/TRA2. The identities of unspliced and spliced RNAs are indicated on the right. C, bar graph comparing female-specific and cryptic splice site activation in the fruF construct. D, bar graph depicting the -fold activation of the female-specific spliced product upon addition of TRA/TRA2 in fruF and in fruF with the cryptic splice site deleted. nt, nucleotides.
Other Well Characterized ESEs Can Replace the fruRE
Two tandem 13-nucleotide repeat elements have been shown to be sufficient to activate the TRA/TRA2-dependent 3′-splice site in dsx (51, 56) or to trigger constitutive activation when moved closer to the regulated 3′-splice site (30, 57). In addition, many other ESEs have been shown to activate the weak female-specific 3′-splice site of dsx (58) and other pre-mRNAs. To test if other ESEs can substitute for the fruRE, we prepared two different pre-mRNAs that contained the male- and female-specific 5′-splice sites. As outlined in Fig. 4A, the fruRE was replaced in the context of competing male- and female-specific 5′-splice sites either with two tandem 13-nucleotide repeat elements, fruM+F2Rs, or with an ESE containing both the well characterized dsx purine-rich element (PRE) and a pyrimidine-rich enhancer element isolated from in vitro selection experiments (designated 624), fruM+F624/PRE (59). As a control, we tested fruM+F1mut, which contains a single mutated repeat element. These ESEs were placed 10 nucleotides upstream of the regulated 5′-splice site to promote constitutive activation by SR proteins present in HeLa cell nuclear extracts. As expected, male-specific 5′-splice site usage was by far the predominant splicing pathway of fruM+F1mut in the absence of TRA and TRA2 (Fig. 4B, lane 2). Replacement of the fruRE either with tandem 13-nucleotide repeats (fruM+F2Rs) or with a purine- and pyrimidine-rich ESE (fruM+F624/PRE) significantly activated the female-specific 5′-splice site (lanes 4 and 6). Quantitative analysis revealed that the ratio of female- to male-specific splicing changed up to 150- and 30-fold for fruM+F2Rs and fruM+F624/PRE, respectively, compared with the fruM+F1mut pre-mRNA. Thus, the 2Rs ESE in fruM+F2Rs is clearly more potent in directing the splicing machinery to the female-specific splice site compared with the 624/PRE ESE in fruM+F624/PRE. To test if activation of the female-specific splice site is SR protein-dependent, we tested fruM+F2Rs splicing in SR protein-deficient cytoplasmic extracts (S100). As illustrated in Fig. 4C, the addition of the recombinant SR protein 9G8, which interacts with the 13-nucleotide repeat element,2 complemented S100 experiments in a concentration-dependent manner (lanes 5–7). We conclude that heterologous ESEs can qualitatively substitute for the fruRE. As demonstrated for ESE-dependent 3′-splice sites, the activation of the female-specific 5′-splice site in fru can be mediated by at least one member of the SR protein family.
Fig. 4. Heterologous splicing enhancers activate the fru female-specific 5′-splice site.
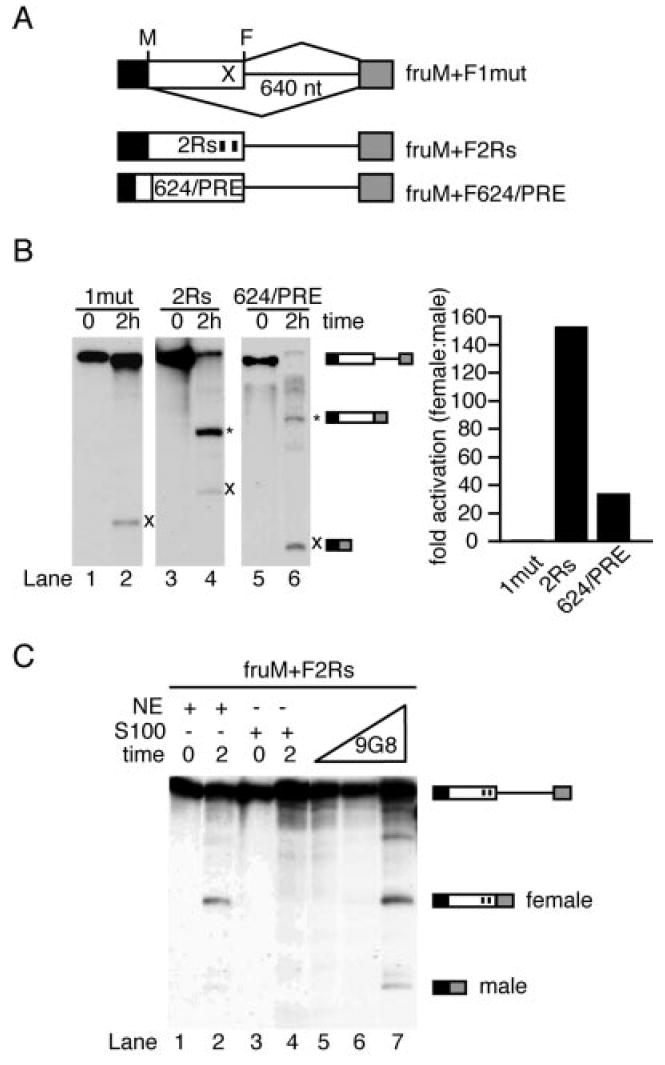
A, RNA substrates used. Symbols are as described in the legends to Figs. 1 and 2. The black boxes represent the upstream male-specific exon. M and F indicate the positions of the male- and female-specific 5′-splice sites, respectively. 2Rs indicates two 13-nucleotide (nt) repeat elements placed 10 nucleotides upstream of the fru female-specific 5′-splice site. The box 624/PRE indicates replacement of the fruRE with a pyrimidine (624)/purine (PRE)-rich enhancer. B, left panel, autoradiograph of in vitro splicing reactions. The identities of the spliced products are indicated on the right. The female-specific (*) and male-specific (×) products are indicated. Right panel, bar graph showing the -fold activation of female- to male-specific 5′-splice site usage. Each value was normalized to the ratio observed for fruM+F1mut. C, autoradiograph of S100 complementation reactions performed with fruM+F2Rs. The recombinant SR protein 9G8 was added to S100 splicing reactions to final concentrations of 200, 400, and 800 nm. The identities of the spliced products are indicated on the right. NE, HeLa cell nuclear extract.
Different RS Domains Vary in Their Ability to Activate the fru 5′-Splice Site
Similar to dsx, TRA and TRA2 complement the fruRE, and heterologous ESEs recognized by SR proteins can efficiently activate the fru female-specific 5′-splice site in vitro. It has previously been shown that hybrid proteins containing the MS2 bacteriophage coat protein fused to a human RS domain can activate the dsx female-specific 3′-splice site when artificially tethered to the pre-mRNA (53). Thus, the MS2-RS fusion proteins can functionally substitute for the dsx ESE complex. To test whether the same is true for the fru ESE, we generated fru(70)M1, in which the fru enhancer has been replaced by a single MS2-binding site (Fig. 5A). This pre-mRNA was incubated in HeLa cell nuclear extract in the presence or absence of saturating concentrations of a variety of MS2 fusion proteins (Fig. 5B, left panel). The fru(70)M1 pre-mRNA was poorly spliced in the absence of fusion protein (lane 1) and in the presence of the MS2 protein lacking an RS domain (lane 2). In contrast, the addition of any of three different fusion proteins containing a natural RS domain (MS2-RSSF2/ASF, MS2-RSp55, or MS2-RS9G8) activated splicing of the fru(70)M1 pre-mRNA (lanes 3–5). Similar results were obtained with the same proteins on the dsx(70)M1 pre-mRNA (Fig. 5B, right panel, lanes 1–5). The relative potency of each fusion protein was calculated in comparison with the most potent activator of each pre-mRNA, MS2-RS9G8, which was arbitrarily set at a value of 1.0. This analysis revealed that the relative potency of each activator was similar on each of the two pre-mRNAs (Fig. 5C). This suggests that the RS domains within enhancer complexes may be involved in similar types of protein/protein interactions to activate 5′- or 3′-splice sites.
Fig. 5. Tethering RS domains to the pre-mRNA can substitute for the fruRE.
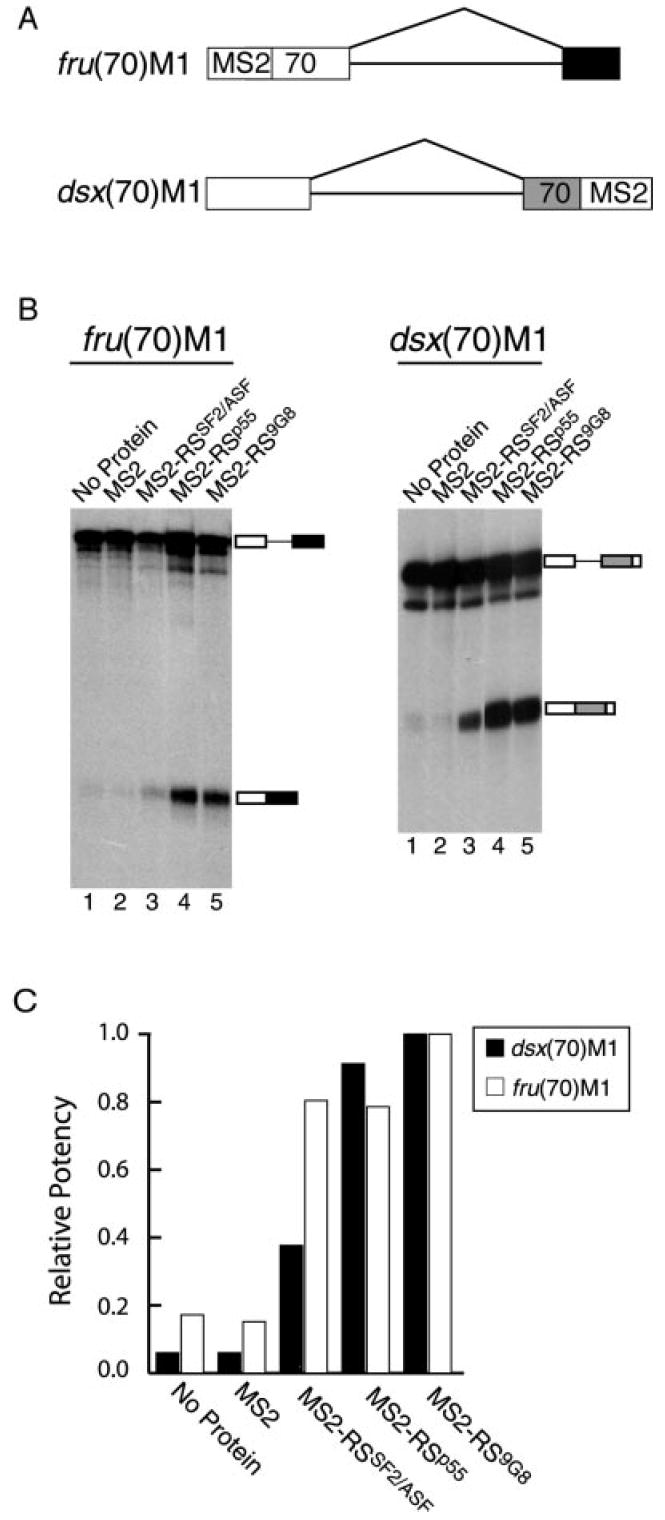
A, the fru(70)M1 pre-mRNA contains a single MS2-binding site located 70 nucleotides upstream of the female-specific 5′-splice site. The dsx(70)M1 pre-mRNA was described previously (53). B, shown is an autoradiograph of in vitro splicing reactions. The fru(70)M1 (left panel) and dsx(70)M1 (right panel) pre-mRNAs were incubated in 15% HeLa cell nuclear extract under splicing conditions for 4 h at 30 °C in the presence of saturating concentrations of the indicated recombinant proteins. C, quantitation of the data in B. The efficiency of splicing in each reaction was quantitated and normalized to the reactions containing the most potent activator, MS2-RS9G8, which was arbitrarily set to a value of 1.0.
DISCUSSION
The TRA/TRA2-dependent activation of the dsx 3′-splice site has served as an excellent model system to study the mechanisms of enhancer-dependent 3′-splice site activation. Similarly, our analysis of the TRA/TRA2-dependent regulation of fru alternative splicing should provide an analogous mechanistic framework for the regulation of enhancer-dependent alternative 5′-splice site choice. Overall, the structural and functional resemblance of the fruRE and dsxRE is remarkable. Both ESEs consist of multiple, highly conserved 13-nucleotide repeat elements that are dispersed throughout a stretch of ~300 untranslated nucleotides. However, the distance between the first repeat element and the regulated splice site is noticeably different: only 38 nucleotides separate the enhancer and the splice site in fru compared with 300 nucleotides in dsx. As observed for dsx, heterologous ESEs that function in the absence of TRA and TRA2 can substitute for the fruRE. In addition, previous work demonstrated that the fruRE could activate heterologous 5′- and 3′-splice sites (40).
Thus far, the only discernible difference between the activities of the dsxRE and fruRE appears to be the manner in which multiple repeat elements affect splice site activation. Our data indicate that only one repeat is both necessary and sufficient to maximally activate female-specific splicing of the fru pre-mRNA (Fig. 2). This differs from the situation in dsx, where splicing efficiency is directly proportional to the number of repeat elements present (51). These observations do not necessarily indicate that multiple repeat elements are not able to further augment spliceosomal recognition of the female-specific 5′-splice site. However, they demonstrate that multiple repeats do not further increase the measured rate-limiting step that is reached by a single repeat element in vitro. Thus, the recruitment of the splicing machinery to the female-specific 5′-splice site in fru may require less assistance from splicing enhancers. One repeat element could then be sufficient to lower the activation barrier. However, the use of a slightly different pre-mRNA substrate design in the splicing assays might provide an alternative explanation for this difference. Unlike the dsx substrates used, all fru pre-mRNAs tested in Fig. 2 contain a competing cryptic splice site. As has been argued recently, it is very likely that the nature of the splice site competition amplifies relatively small changes in ESE-induced splice site strength (60).
Mutation of all three repeat elements led to the loss of TRA/TRA2-dependent 5′-splice site activation. These results are in agreement with earlier cell transfection studies (40) and demonstrate that the 13-nucleotide repeat elements are the only cis-acting sequences that significantly contribute to female-specific 5′-splice site activation in fru. In addition, the conservation of the repeat elements (Table I), the thermodynamics of complex formation, and the efficiencies of splice site activation are strikingly similar between fru and dsx. Assembly of the TRA/TRA2-dependent enhancer complexes in fru and dsx is cooperative in nature, with comparable apparent dissociation constants: ~60 nm for fru (Fig. 1C) and ~20 nm for dsx (51). These observations provide compelling, although indirect evidence suggesting that the enhancer complex assembled on each of the fruREs is identical in composition to the enhancer complexes formed on each of the dsxREs. Thus, it is very likely that the presence of TRA and TRA2 allows the formation of three heterotrimeric enhancer complexes, each containing TRA, TRA2, and an additional SR protein, on the fruRE, identical to those that have been described in detail for the dsxRE (31).
SR Proteins Activate the Female-specific 5′-Splice Site
Our experiments demonstrate that several heterologous ESEs can functionally replace the fruRE. These ESEs were active in female-specific splice site activation not only in the context of a competing male-specific 5′-splice site (Fig. 4), but also when assayed in isolation under more stringent splicing conditions (Fig. 5). As expected from the location and nature of the replacement ESEs, the exchange of the fruRE caused constitutive splice site activation and, consequently, loss of TRA/TRA2-dependent regulation. In addition, the SR protein 9G8, which is known to interact with the 13-nucleotide repeat element, was sufficient to activate the female-specific 5′-splice site. To address whether different SR proteins function similarly during 5′-splice site activation, we used an approach that artificially tethers an RS domain to the pre-mRNA upstream of the fru female-specific 5′-splice site using the MS2 bacteriophage coat protein. As was previously shown for the dsx pre-mRNA (53), the MS2-RS proteins could functionally substitute for the entire fru splicing enhancer complex. The observation that the relative potency of three different RS domains was similar for the dsx and fru pre-mRNAs suggests that the RS domains within splicing enhancer complexes involved in activating 5′- or 3′-splice sites may be involved in similar types of protein interactions. These enhancer complexes may target the same protein. Alternatively, these proteins may target different splicing factors that have similar protein interaction surfaces.
Mechanism of ESE-dependent Splice Site Activation
Given the extensive analogies to enhancer-dependent activation of 3′-splice sites, it is reasonable to suggest that the fru splicing enhancer complex increases the local concentration of the splicing machinery at the regulated fru female-specific 5′-splice site. It is well established that 3′- and 5′-splice sites are initially recognized by different components of the splicing machinery (61). How then is it possible that the seemingly identical TRA and TRA2 enhancer complexes assembled on the dsxRE and fruRE recruit different targets and with different directionality? One potential explanation stems from the domain structure of TRA2, one of the protein components of the enhancer complex (31). TRA2 contains two separate RS domains, one on the N terminus and the other on the C terminus (62). It is conceivable that each of the bipartite RS domains is responsible for the recruitment of specific factors to either the 5′- or 3′-splice site (Fig. 6A). In three-dimensional space, these RS domains could be located on opposing ends of TRA2, therefore providing directionality. However, the demonstration that the fruRE complexes can be replaced by other SR proteins or by MS2-RS fusion proteins that do not have bipartite RS domain structures does not lend strong support for this explanation. An alternative model proposes the presence of cell-specific splicing factors that mediate interactions between the enhancer complex that activates the 5′-splice site and particular components of the splicing machinery (Fig. 6B). Although we cannot rule out this model, it is not very likely to occur because all heterologous ESEs tested activated the female-specific 5′-splice, suggesting that the presumed additional factor interacts with any RS domain indiscriminately. In addition, all MS2-RS fusion proteins tested activated both 5′- and 3′-splice sites to similar extents.
Fig. 6. Models for TRA/TRA2-dependent activation of the female-specific 5′-splice site in fru.
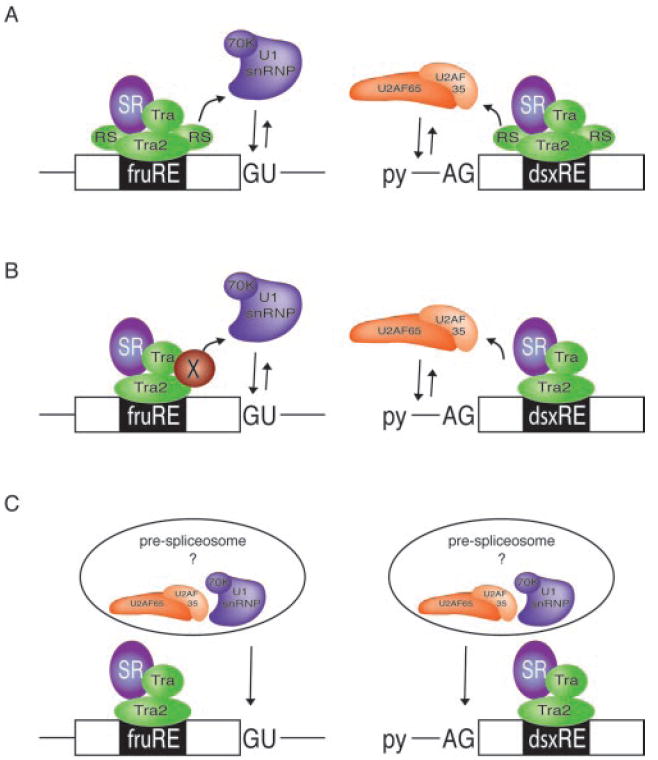
TRA and TRA2 activate a female-specific 5′-splice site in fru and a female-specific 3′-splice site in dsx through the actions of the fruRE and dsxRE, respectively. A, splice site-specific recruitment of spliceosomal factors that initially recognize the 5′- or 3′-splice sites of the regulated exons (U1 snRNP and U2AF) may be achieved by the directional activities of the separated RS domains within TRA2, an integral component of either enhancer complex. B, alternatively, splice site-specific recruitment of U1 snRNP to the regulated 5′-splice sites of fru may be mediated by the activity of a tissue-specific splicing coactivator, designated X. C, both enhancer complexes recruit the same target to the regulated exon. As illustrated, this target may be a prespliceosomal complex that contains at least U1 snRNP and U2AF or other splicing factors that provide a platform for the efficient recruitment of either U1 snRNP or U2AF or both. py, polypyrimidine tract.
Comparison of the fundamental principles that govern dsx and fru alternative splicing demonstrated striking parallels between the action of ESEs on regulated 5′- and 3′-splice sites. These observations could imply that splicing enhancers employ the same mechanisms to increase 3′- or 5′-splice site recognition. Therefore, an attractive interpretation of the data presented here suggests a model in which the dsxRE and fruRE recruit identical components not specifically to splice sites, but to the regulated exon (Fig. 6C). Consistent with the proposal that ESEs simultaneously assist 3′- and 5′-splice site recognition (63), these components might be the metazoan version of a pre-assembled pentaspliceosome similar to the one recently described in yeast (64, 65). The prespliceosome must contain, at minimum, the components required for initial recognition of the 3′- and 5′-splice sites. Alternatively, the enhancer complex might interact with a master assembly factor, such as the splicing coactivator SRm160 (22), that may provide a more stable platform for the assembly of the spliceosome. No doubt, further work is necessary to evaluate ESE-dependent occupancy at the regulated 5′-splice site and to determine the direct interactions by which SR protein-dependent splicing enhancer complexes activate regulated splice sites.
Acknowledgments
We thank Kristen Lynch and members of the Hertel laboratory for helpful comments on the manuscript. We also thank Bruce Baker for sharing fru minigene plasmid constructs.
Footnotes
This work was supported by National Institutes of Health Grants GM62287 (to K. J. H.) and GM62516 (to B. R. G.).
The abbreviations used are: dsx, doublesex; TRA, Transformer; TRA2, Transformer2; ESE, exonic splicing enhancer; RS, Arg/Ser-rich; SR, Ser/Arg-rich; dsxRE, dsx splicing enhancer repeat element; snRNP, small nuclear ribonucleoprotein; U2AF, U2 auxiliary factor; ASF, alternative splicing factor; SF2, splicing factor-2; fru, fruitless; fruRE, fru splicing enhancer repeat element; PRE, purine-rich element.
K. J. Hertel, unpublished data.
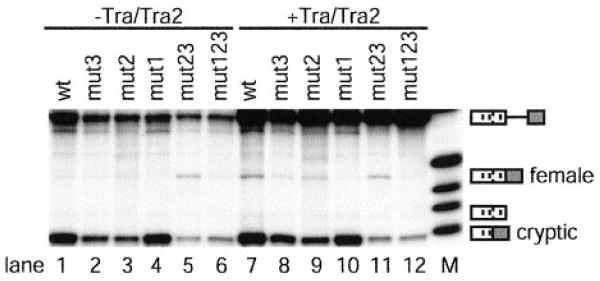
Note Added in Proof—Since acceptance and on-line publication of this report we have discovered in a follow-up experiment of the mutational analysis described in Fig. 2 that the substrates fruFmut23 and fruFmut1 did not contain the anticipated mutations within the repeats. To rectify this discrepancy, fruFmut23 and fruFmut1 were remade. After sequence verification, the in vitro splicing experiments described in Fig. 2 were carried out three times. The results obtained with the corrected mutant substrates were significantly different, suggesting an alternate interpretation of the mutational analysis of the fruRE. Unlike what was observed in Fig. 2, activation of the female-specific 5′-splice site was detected at maximal levels for fruFmut23 in the absence of TRA and TRA2 (lane 5), while all other substrates tested behaved as illustrated in Fig. 2 (lanes 1–4 and 6). The presence of TRA and TRA2 increased female-specific 5′-splice site usage only slightly for fruFmut23 (1.3-fold) but significantly for wild type (6.5-fold), fruFmut3 (6-fold), and fruFmut2 (4-fold) (lanes 7–12). Similar to the triple mutant fruFmut123, fruFmut1 was not able to support female-specific splice site activation even in the presence of TRA and TRA2 (lanes 5, 6, 11, and 12). Efficient cryptic splice site usage depended on the presence of at least one upstream 13-nucleotide repeat element. Thus, cryptic splice site choice was severely decreased for fruFmut23 and fruFmut123 (lanes 5, 6, 11, and 12).
The repeated experiments agree with the conclusion of our study that one repeat element is sufficient to promote female-specific 5′-splice site usage. However, the new data do not support the notion that each repeat element is equally efficient in this activity. Rather the data suggest that repeats 2 and 3 do not directly participate in female-specific splice site activation as their enhancement appears to be solely concentrated on activating the cryptic 5′-splice site. These observations are consistent with the following model. The 13-nucleotide splicing enhancer elements 3 and 2 almost exclusively activate the cryptic splice site, while repeat 1 is responsible for the activation of the female-specific 5′-splice site. This observation is significant as it further demonstrates that the location of splicing enhancer elements relative to potential splice sites dictates the nature of exon definition. The new data agree with the conclusion that the 13-nucleotide repeat elements are the only regulatory elements that promote activation of the female-specific splice site, however, with the added complexity that repeats 2 and 3 establish the preference of a competing splicing pathway. The fact that fruFmut23 activates female-specific splicing in the absence of TRA and TRA2 is also consistent with the observations from dsx studies that 13-nucleotide repeat elements have constitutive enhancing potential as long as they are within close proximity to the regulated splice site and as long as no alternate splicing pathways of preference exist.
While the repeated experiments influence the interpretation of the mutational analysis described in Fig. 2, they do not alter the major conclusions of the study as outlined in the abstract.
References
- 1.Smith CW, Valcarcel J. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 2.Black DL. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 3.Graveley BR. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 4.Maniatis T, Tasic B. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- 5.Modrek B, Lee C. Nat Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 6.Rio DC. Gene Expr. 1992;2:1–5. [PMC free article] [PubMed] [Google Scholar]
- 7.Horowitz DS, Krainer AR. Trends Genet. 1994;10:100–106. doi: 10.1016/0168-9525(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 8.Black DL. RNA (N Y) 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 9.Adams MD, Rudner DZ, Rio DC. Curr Opin Cell Biol. 1996;8:331–339. doi: 10.1016/s0955-0674(96)80006-8. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Manley JL. Curr Opin Genet Dev. 1997;7:205–211. doi: 10.1016/s0959-437x(97)80130-x. [DOI] [PubMed] [Google Scholar]
- 11.Lopez AJ. Annu Rev Genet. 1998;32:279–305. doi: 10.1146/annurev.genet.32.1.279. [DOI] [PubMed] [Google Scholar]
- 12.Stojdl DF, Bell JC. Biochem Cell Biol. 1999;77:293–298. [PubMed] [Google Scholar]
- 13.Chou MY, Rooke N, Turck CW, Black DL. Mol Cell Biol. 1999;19:69–77. doi: 10.1128/mcb.19.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modafferi EF, Black DL. RNA (N Y) 1999;5:687–706. doi: 10.1017/s1355838299990155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Shambaugh ME, Rottman FM, Bokar JA. RNA (N Y) 2000;6:1847–1858. doi: 10.1017/s1355838200000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, Mayeda A, Krainer AR. Mol Cell. 2001;8:1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 17.Expert-Bezancon A, Le Caer JP, Marie J. J Biol Chem. 2002;277:16614–16623. doi: 10.1074/jbc.M201083200. [DOI] [PubMed] [Google Scholar]
- 18.Baker BS. Nature. 1989;340:521–524. doi: 10.1038/340521a0. [DOI] [PubMed] [Google Scholar]
- 19.McKeown M, Belote JM, Boggs RT. Cell. 1988;53:887–895. doi: 10.1016/s0092-8674(88)90369-8. [DOI] [PubMed] [Google Scholar]
- 20.Nagoshi RN, McKeown M, Burtis KC, Belote JM, Baker BS. Cell. 1988;53:229–236. doi: 10.1016/0092-8674(88)90384-4. [DOI] [PubMed] [Google Scholar]
- 21.Nagoshi RN, Baker BS. Genes Dev. 1990;4:89–97. doi: 10.1101/gad.4.1.89. [DOI] [PubMed] [Google Scholar]
- 22.Blencowe BJ. Trends Biochem Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- 23.Graveley BR. RNA (N Y) 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burtis KC, Baker BS. Cell. 1989;56:997–1010. doi: 10.1016/0092-8674(89)90633-8. [DOI] [PubMed] [Google Scholar]
- 25.Hedley ML, Maniatis T. Cell. 1991;65:579–586. doi: 10.1016/0092-8674(91)90090-l. [DOI] [PubMed] [Google Scholar]
- 26.Ryner LC, Baker BS. Genes Dev. 1991;5:2071–2085. doi: 10.1101/gad.5.11.2071. [DOI] [PubMed] [Google Scholar]
- 27.Hoshijima K, Inoue K, Higuchi I, Sakamoto H, Shimura Y. Science. 1991;252:833–836. doi: 10.1126/science.1902987. [DOI] [PubMed] [Google Scholar]
- 28.Tian M, Maniatis T. Science. 1992;256:237–240. doi: 10.1126/science.1566072. [DOI] [PubMed] [Google Scholar]
- 29.Tian M, Maniatis T. Cell. 1993;74:105–114. doi: 10.1016/0092-8674(93)90298-5. [DOI] [PubMed] [Google Scholar]
- 30.Tian M, Maniatis T. Genes Dev. 1994;8:1703–1712. doi: 10.1101/gad.8.14.1703. [DOI] [PubMed] [Google Scholar]
- 31.Lynch KW, Maniatis T. Genes Dev. 1996;10:2089–2101. doi: 10.1101/gad.10.16.2089. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Hoffmann HM, Grabowski PJ. RNA (N Y) 1995;1:21–35. [PMC free article] [PubMed] [Google Scholar]
- 33.Zuo P, Maniatis T. Genes Dev. 1996;10:1356–1368. doi: 10.1101/gad.10.11.1356. [DOI] [PubMed] [Google Scholar]
- 34.Graveley BR, Hertel KJ, Maniatis T. RNA (N Y) 2001;7:806–818. doi: 10.1017/s1355838201010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohtz JD, Jamison SF, Will CL, Zuo P, Lührmann R, Garcia-Blanco MA, Manley JL. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 36.Jamison SF, Pasman Z, Wang J, Will C, Luhrmann R, Manley JL, Garcia-Blanco MA. Nucleic Acids Res. 1995;23:3260–3267. doi: 10.1093/nar/23.16.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuo P, Manley JL. Proc Natl Acad Sci U S A. 1994;91:3363–3367. doi: 10.1073/pnas.91.8.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryner LC, Goodwin SF, Castrillon DH, Anand A, Villella A, Baker BS, Hall JC, Taylor BJ, Wasserman SA. Cell. 1996;87:1079–1089. doi: 10.1016/s0092-8674(00)81802-4. [DOI] [PubMed] [Google Scholar]
- 39.Elrick LL, Humphrey MB, Cooper TA, Berget SM. Mol Cell Biol. 1998;18:343–352. doi: 10.1128/mcb.18.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinrichs V, Ryner LC, Baker BS. Mol Cell Biol. 1998;18:450–458. doi: 10.1128/mcb.18.1.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bourgeois CF, Popielarz M, Hildwein G, Stevenin J. Mol Cell Biol. 1999;19:7347–7356. doi: 10.1128/mcb.19.11.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cote J, Simard MJ, Chabot B. Nucleic Acids Res. 1999;27:2529–2537. doi: 10.1093/nar/27.12.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Souza I, Schellenberg GD. J Biol Chem. 2000;275:17700–17709. doi: 10.1074/jbc.M909470199. [DOI] [PubMed] [Google Scholar]
- 44.Hall JC. Science. 1994;264:1702–1714. doi: 10.1126/science.8209251. [DOI] [PubMed] [Google Scholar]
- 45.Taylor BJ, Villella A, Ryner LC, Baker BS, Hall JC. Dev Genet. 1994;15:275–296. doi: 10.1002/dvg.1020150309. [DOI] [PubMed] [Google Scholar]
- 46.Ito H, Fujitani K, Usui K, Shimizu-Nishikawa K, Tanaka S, Yamamoto D. Proc Natl Acad Sci U S A. 1996;93:9687–9692. doi: 10.1073/pnas.93.18.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujii S, Amrein H. EMBO J. 2002;21:5353–5363. doi: 10.1093/emboj/cdf556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reed R. Curr Opin Gen Dev. 1996;6:215–220. doi: 10.1016/s0959-437x(96)80053-0. [DOI] [PubMed] [Google Scholar]
- 49.Du H, Rosbash M. Nature. 2002;419:86–90. doi: 10.1038/nature00947. [DOI] [PubMed] [Google Scholar]
- 50.Lund M, Kjems J. RNA (N Y) 2002;8:166–179. doi: 10.1017/s1355838202010786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hertel KJ, Maniatis T. Mol Cell. 1998;1:449–455. doi: 10.1016/s1097-2765(00)80045-3. [DOI] [PubMed] [Google Scholar]
- 52.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graveley BR, Maniatis T. Mol Cell. 1998;1:765–771. doi: 10.1016/s1097-2765(00)80076-3. [DOI] [PubMed] [Google Scholar]
- 54.Boggs RT, Gregor P, Idriss S, Belote JM, McKeown M. Cell. 1987;50:739–747. doi: 10.1016/0092-8674(87)90332-1. [DOI] [PubMed] [Google Scholar]
- 55.Burge CB, Tuschl T, Sharp PA. In: The RNA World. 2. Gesteland RF, Cech TR, Atkins JF, editors. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1999. pp. 525–560. [Google Scholar]
- 56.Inoue K, Hoshijima K, Higuchi I, Sakamoto H, Shimura Y. Proc Natl Acad Sci U S A. 1992;89:8092–8096. doi: 10.1073/pnas.89.17.8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lynch KW, Maniatis T. Genes Dev. 1995;9:284–293. doi: 10.1101/gad.9.3.284. [DOI] [PubMed] [Google Scholar]
- 58.Lorson CL, Androphy EJ. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 59.Schaal TD, Maniatis T. Mol Cell Biol. 1999;19:1705–1719. doi: 10.1128/mcb.19.3.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim SR, Hertel KJ. J Biol Chem. 2001;276:45476–45483. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- 61.Reed R, Palandjian L. In: Eukaryotic mRNA Processing. Krainer AR, editor. IRL Press; Oxford: 1997. pp. 103–129. [Google Scholar]
- 62.Amrein H, Hedley ML, Maniatis T. Cell. 1994;76:735–746. doi: 10.1016/0092-8674(94)90512-6. [DOI] [PubMed] [Google Scholar]
- 63.Lam BJ, Hertel KJ. RNA (N Y) 2002;8:1233–1241. doi: 10.1017/s1355838202028030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stevens SW, Ryan DE, Ge HY, Moore RE, Young MK, Lee TD, Abelson J. Mol Cell. 2002;9:31–44. doi: 10.1016/s1097-2765(02)00436-7. [DOI] [PubMed] [Google Scholar]
- 65.Nilsen TW. Mol Cell. 2002;9:8–9. doi: 10.1016/s1097-2765(02)00430-6. [DOI] [PubMed] [Google Scholar]