

Abstract
High-affinity (Kd = 1 × 10−9 M) anti-platelet GPIIIa has been isolated from serum immune complexes of immunologic thrombocytopenic HIV-1-infected patients (HIV-1-ITP). Affinity-purified anti-platelet antibody reacted with a recombinant GPIIIa-(1–200) and -(1–66) fusion peptide and with an 18-mer GPIIIa-(49–66) peptide but not with seven other GPIIIa peptides spanning the length of GPIIIa. Most of the anti-platelet antibody (≈85%) could be adsorbed to and eluted from a GPIIIa-(49–66) affinity column. Binding of antibody to platelets could be inhibited by GPIIIa-(49–66) or an equimolar peptide-albumin conjugate (IC50 = 2 μM). Sera from 7 control subjects and 10 classic autoimmune thrombocytopenic patients gave background reactivity with GPIIIa-(49–66). HIV-1-ITP sera from 16 patients reacted with a mean OD 6-fold greater than background (range, 4- to 9-fold). Serum anti-GPIIIa-(49–66) concentration correlated inversely with platelet count, R2 = 0.51, n = 31, P < 0.0001. Because mouse platelet GPIIIa-(49–66) has 83% homology with human GPIIIa and mouse monocytes contain Fc receptors for the human IgG1-κ/λ antibody, we determined the in vivo effect of human anti-GPIIIa on mouse platelets. Affinity-purified antibody, 25–50 μg given i.p., resulted in a precipitous drop in platelet count to 30% of baseline, with nadir at 4 hr and return to normal in 36 hr. No effect was noted with control IgG. Acute thrombocytopenia could be prevented or reversed by the injection of the GPIIIa-(49–66) albumin conjugate at zero time or 2 hr after antibody, respectively, but not with a scrambled peptide-albumin conjugate. Thus HIV-1-ITP patients have high-affinity anti-platelet GPIIIa against a major antigenic determinant, GPIIIa-(49–66), which correlates inversely with platelet count and induces thrombocytopenia in mice.
Patients with HIV-1 infection develop an immunologic thrombocytopenia (ITP) that is clinically indistinguishable from classic autoimmune thrombocytopenia (ATP), seen predominantly in females (1–3). The incidence of severe thrombocytopenia (platelet count <10,000 platelets per μl) in two different HIV-1-infected cohorts is 30–52% (4, 5). However, HIV-1-ITP is different from the classic variety of ATP with respect to the predominant male incidence, markedly elevated platelet-associated IgG, IgM, and C3C4, and the presence of PEG-precipitable serum immune complexes (ICs) containing IgG, IgM, and C3C4 (2, 6). These complexes bind to the platelet surface in a saturation-dependent manner (7). We have recently reported the presence of high-affinity (Kd, 1 × 10−9 M) anti-platelet GPIIIa sequestered within the PEG-ICs of these patients by rheumatoid factor and that binding of the PEG-ICs to platelets was facilitated by anti-platelet IgG within the complex (8) as well as complement (9).
In the present report, we document the presence of a major antigenic determinant on platelet GPIIIa-(49–66) (CAPESIEFPVSEARVLED) that is pathophysiologically relevant with respect to differentiating serum of HIV-1-ITP patients from nonthrombocytopenic HIV-1-infected patients and controls. Serum anti-GPIIIa-(49–66) antibody correlates with thrombocytopenia and induces acute thrombocytopenia in mice, whose GPIIIa-(49–66) is 83% homologous with its human GPIIIa counterpart (10). The passively transferred thrombocytopenia can be prevented and reversed with an albumin conjugate of GPIIIa-(49–66).
MATERIALS AND METHODS
Population.
The population consists of 32 HIV-1-seropositive subjects without AIDS. Twenty-two were thrombocytopenic (11 homosexuals and 11 intravenous drug abusers), and 10 had normal platelet counts. Ten classic (female) autoimmune thrombocytopenic patients and 7 healthly control subjects were also studied for PEG-IC and serum binding to GPIIIa-(49–66).
ICs.
ICs were prepared from serum by PEG precipitation (6). Precipitates were dissolved in one-fifth their serum volume in 0.01 M sodium phosphate-buffered saline (pH 7.4; PBS).
Isolation of IgG from ICs.
(8). PEG-ICs were applied to a staphylococcal protein A affinity column. The bound complex was washed with PBS and eluted with 0.1 M glycine buffer (pH 2.5). The eluted material was applied to an acidified Sephadex G-200 gel filtration column preequilibrated with the same elution buffer. Effluents of the IgG peak were isolated, neutralized, dialyzed against PBS, and then applied to a rabbit anti-IgM affinity column, prepared with Affi-Gel 10 (Bio-Rad). The flow-thru material was free of contaminating IgM as determined by immunoblot and ELISA.
ELISA.
Antibody reactivity was measured by solid-phase ELISA (8) using serial dilutions of IgG on U-shaped polyvinyl microtiter plates (Curtin-Matheson Scientific, Houston) preincubated overnight at 4°C with 200 ng of peptide in 0.1 M sodium bicarbonate (pH 9.5) or with 1 × 107 washed platelets in PBS and blocked with 2.5% bovine serum albumin in PBS. The second antibody was a 1:500 dilution of goat F(ab′)2 anti-human IgG (γ chain specific) coupled to alkaline phosphatase (Sigma). Color was read in an automated microtiter plate reader at 405 nm. In some experiments (see Fig. 5A) antibody titer units were calculated from the reciprocal of the lowest concentration of antibody (μg per well) capable of reacting with platelets applied to a microtiter plate. In other experiments (see Fig. 5B) anti-GPIIIa-(49–66) concentration was calculated from a standard curve of affinity-purified anti-GPIIIa-(49–66) vs. the peptide.
Figure 5.

Correlation of platelet count with anti-platelet IgG and anti-GPIIIa-(49–66). (A) Anti-platelet IgG was extracted from PEG-ICs, affinity-purified with fixed platelets, and tested for its reactivity with washed platelets adsorbed to a microtiter place via a dilution determination of antibody titer, as described. A best-fit curvilinear power regression curve was selected by computer analysis (Macintosh delta graph pro 3.5) with R2 = 0.61, F(1, 25) = 40.2, and P < 0.0001. (B) Anti-GPIIIa-(49–66) concentration was measured in serum by using a standard curve of affinity-purified anti-GPIIIa-(49–66) vs. purified peptide. Ordinate represents nanograms of antibody per ml of sera, diluted 1:4, reacting with GPIIIa-(49–66) adsorbed to microtiter plates. Gray horizontal band represents mean ± 2 SD of 10 control subjects tested. A similar best-fit curvilinear power regression curve was selected with R2 = 0.51, F(1, 29) = 30.0, and P < 0.0001.
Immunoblotting with Anti-GPIIIa.
PEG-ICs (75 μg/ml) were separated by SDS/PAGE on 10% gels, transferred to a nitrocellulose membrane, and immunoblotted with mAb LK-7r (5 μg/ml) against GPIIIa (11), and bound antibody was detected by chemiluminescence (12).
Affinity Purification of Anti-Platelet IgG.
(8). Washed platelets (1 × 108 platelets) (13) were fixed with 2% paraformaldehyde for 4 hr at 4°C, washed extensively with Ringer’s solution containing 2 mM EDTA, and sedimented to remove the supernatant fluid. Affinity-purified immunoglobulin (0.4 mg) from PEG-ICs, in 1 ml of PBS, was added to the platelet pellet at room temperature for 2 hr followed by overnight gentle rocking at 4°C. The platelets were sedimented and washed three times with Ringer’s solution containing 2 mM EDTA (13), and the antibody was eluted with 0.1 M glycine buffer (pH 2.5). The eluate was neutralized with 1 M Tris buffer (pH 10.3) and dialyzed against PBS. The IgG subclass was determined by radial immunodiffusion with reagents supplied by The Binding Site (San Diego). Affinity-purified IgG was predominently IgG1 with both κ and λ light chains.
Binding and Elution of Anti-Platelet IgG from a GPIIIa-(49–66) Affinity Column.
GPIIIa-(49–66) (4 mg) was coupled to an affinity column with the heterobifunctional cross-linker sulfo-succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate, as recommended by the manufacturer (Pierce; cross-links the resin with N-terminal cysteine of the peptide) and incubated with ≈0.4 mg of affinty-purified IgG overnight at 4°C. The gel was then washed extensively with five bed volumes of PBS, five washes of PBS/0.5 M NaCl, and five washes of PBS. IgG was eluted with 0.1 M glycine (pH 2.5), monitored by absorption at 280 nm, and immediately neutralized with 1 M Tris buffer.
Conjugation of GPIIIa-(49–66) and Control Peptide with Serum Albumin.
Conjugation to bovine serum albumin (Sigma) was performed with the heterobifunctional cross-linker N-maleimidobenzoyl-N-hydroxysuccinimide ester (Pierce), which cross-links free amines on albumin with the N-terminal cysteine of the peptide. The in vivo half-life of the GPIIIa-(49–66) albumin conjugate was 6 hr as determined by solid-phase ELISA on serum aliquots removed from mice at 1, 2, 4, 6, and 24 hr, using a potent mouse anti-GPIIIa-(49–66) antibody at a dilution of >1:50,000. Unconjugated peptide had an in vivo half-life of <30 min.
Injection of Affinity-Purified Anti-Platelet GPIIIa Into Mice.
Human anti-platelet GPIIIa or pooled human IgG (ICN; 25–50 μg) was injected i.p. into BALB/c mice (Taconic Farms) and blood was withdrawn from the orbital sinus at various times. In some experiments, albumin-conjugated GPIIIa-(49–66) or control scrambled-irrelevant GPIIIa-(49–66) (CGGGARVLEDRP) were also injected into mice at various times.
Determination of Mouse Platelet Counts.
Platelet counts were determined from 20 μl of blood drawn into Unopettes (no. 5854, Becton Dickinson), containing optimal anti-coagulant concentration and diluent for quantitating platelet counts by phase-contrast microscopy.
Proteins and Peptides.
GPIIIa-(1–66) recombinant glutathione S-transferase fusion protein was a gift of James Casella (John’s Hopkins University Medical School). GPIIIa-(1–200), -(150–400), -(350–550), -(450–700), and -(775–762) recombinant maltose-binding fusion proteins were a gift of Robert McMillan (Scripps Clinic and Research Foundation, La Jolla, CA). GPIIIa-(49–66), GPIIIa-(49–56) (CAPESIEF), GPIIIa-(57–66) preceded by cysteine (CPVSEARVLED), and CGGGARVLEDRP were synthesized by Quality Control Biochemicals (Hopkinton, MA). Irrelevant 13-mer peptide (CGYGPKKKRKVGG) and GPIIIa-(217–232) (DAPEGFDAIMCQATVC) were synthesized by the New York University Kaplan Cancer Center. GPIIIa-(1–13) (GPNICTTRGVSSQ) and GPIIIa-(204–228) (NEEVKKQSVSRNRNAPEGGFDAIMQ) were kind gifts of Barry Coller (Mt. Sinai Medical Center, NY). Ovalbumin, carbonic anhydrase, and soybean trypsin inhibition were obtained from Sigma. All peptides were verified by reverse-phase chromatography and mass spectrometry.
RESULTS
Reactivity of Affinity-Purified Anti-Platelet IgG with GPIIIa-(1–66) and GPIIIa-(49–66).
HIV-I-ITP affinity-purified anti-platelet IgG immunoprecipitates GPIIIa from 125I-surface-labeled platelets as well as several other minor proteins (8). The anti-platelet IgG is of the IgG1-κ/λ isotype (data not shown). To determine the antigenic determinant for this anti-platelet GPIIIa, we screened several GPIIIa peptides. The recombinant (r) GPIIIa-(1–66)–glutathione S-transferase fusion protein was highly reactive with the anti-platelet IgG eluted from fixed platelets. As little of the eluted protein as 38 ± 6 ng/ml gave a positive signal in wells coated with 50 ng of peptide. GPIIIa-(49–66) was selected as a likely linear immunogenic determinant because of its high concentration of basic and acidic acids, unencumbered by cysteine residues, and the two proline residues that could serve to constrain the peptide. GPIIIa-(49–66) was highly reactive. As little of the eluted Ig as 15 ± 3 ng/ml gave a positive signal. No reactivity was noted with GPIIIa-(1–13), -(204–228), or -(217–232); irrelevant scrambled GPIIIa-(49–66); rGPIIIa-(150–400), -(350–550), -(450–700), or -(715–762) fusion proteins; or an irrelevant 13-mer peptide, ovalbumin, carbonic anhydrase, or soybean trypsin inhibitor (data not shown). Reactivity was noted with rGPIIIa-(1–200).
Some reactivity was maintained when the 18-mer GPIIIa-(49–66) was fractionated into two segments, GPIIIa-(49–56) and GPIIIa-(57–66), at ≈50% of the potency of the 18-mer peptide. This was demonstrated by direct ELISA on free peptides, on albumin-conjugated peptides bound to microtiter plates (data not shown), and by peptide inhibition studies of binding of affinity-purified anti-platelet IgG to platelets bound to microtiter plates (Fig. 1). Similar inhibitory reactivity was obtained with rGPIIIa-(1–200) and GPIIIa-(49–66) with IC50 values of 4.4 and 3.5 μM, respectively, compared with ≈7 μM for GPIIIa-(49–56) and GPIIIa-(57–66).
Figure 1.

Inhibition of binding of anti-platelet IgG to platelets with GPIIIa-(49–66) and its smaller component peptides GPIIIa-(49–56) and GPIIIa-(57–66). Anti-platelet GPIIIa antibody was preincubated with various concentrations of the respective peptides for 2 hr at room temperature and then reacted with platelets adsorbed to a microtiter plate. Antibody reactivity was determined by ELISA. Percent inhibition of binding is calculated by subtracting the OD (405 nm) obtained with the inhibitory peptide from the OD obtained in the absence of inhibitory peptide, dividing by the OD in the absence of the peptide and multiplying by 100. Data are the mean ± SEM of results from five patients.
Adsorption of Anti-Platelet Reactivity with a GPIIIa-(49–66) Affinity Column.
Binding of anti-platelet IgG to a GPIIIa-(49–66) affinity column removed ≈85% of anti-platelet IgG reactivity (range 65–90%, n = 6; Fig. 2), as determined by flow-thru and eluted reactivity. Reapplication of flow-thru material to a fresh column resulted in 100% of the nonadsorbed reactivity remaining unbound. Reactivity of anti-platelet IgG with an irrelevant-scrambled GPIIIa peptide affinity column (CGGGARVLEDRP) revealed no adsorption (data not shown).
Figure 2.

Binding and elution of anti-platelet IgG with a GPIIIa-(49–66) affinity column. Affinity-purified anti-platelet IgG of a representative patient was applied to a GPIIIa-(49–66) affinity column overnight at 4°C, washed, and eluted with 0.1 M glycine (pH 2.5). The original material, eluate, flow thru, and second eluate from the flow-thru reapplied to the column were then reacted against platelets. Antibody binding at doubling dilutions was determined by ELISA as in Fig. 1. Protein was assayed prior to ELISA determination by the Bio-Rad method. Nonadsorbed (flow-thru) anti-platelet reactivity was 32%. Five additional patients gave similar results.
Effect of Centrifugation on Serum Anti-GPIIIa-(49–66) Reactivity.
Because serum and PEG-ICs from HIV-1-ITP patients bind to platelets and PEG-ICs bind in part via anti-platelet IgG within the complex (6–8), we determined whether patient serum also bound to GPIIIa-(49–66). Fig. 3 demonstrates the effect of centrifugation at 100,000 × g for 1 hr on serum reactivity with GPIIIa-(49–66). Approximately 75% of serum reactivity was removed by centrifugation. The ability of serum ICs to bind to GPIIIa-(49–66) was confirmed with PEG-ICs, which also bound to GPIIIa-(49–66), not to GPIIIa-(1–13) or GPIIIa-(204–228) (data not shown).
Figure 3.

Serum anti-GPIIIa-(49–66) reactivity before and after centrifugation at 100,000 × g for 1 hr. Antibody reactivity was determined by ELISA, against GPIIIa-(49–66)-coated wells. Data are the mean results from eight patients.
A similar loss in anti-platelet IgG reactivity after centrifugation at 100,000 × g was noted when platelets were used as antigen (data not shown).
Differentiation of HIV-1-ITP from Nonthrombocytopenic HIV-1-Infected Patients, Chronic Classic ATP Patients, and Healthy Controls by Reactivity of Serum with GPIIIa-(49–66).
Fig. 4 demonstrates a clear cut distinction between HIV-1-ITP patients and the other groups tested. The HIV-1-ITP group reacted with a mean ELISA OD value that was 6-fold greater than background (range, 4- to 9-fold), whereas nonthrombocytopenic HIV-1-infected patients reacted with a mean OD value that was 3-fold background (range, 3- to 4-fold). Centrifugation reduced the reactivity of HIV-1 serum with GPIIIa-(49–66). No such change was noted with the background reactivity of classic ATP patients or control subjects.
Figure 4.

Scattergram of serum anti-GPIIIa-(49–66) reactivity in four cohorts of subjects and patients: 7 control subjects, 10 classic ATP patients, 16 HIV-1-ITP patients, and 10 HIV-1-infected patients. Samples were analyzed before (B) and after (A) centrifugation at 100,000 × g. Reactivity represents the ELISA OD (405 nm) obtained at a 1:4 dilution of serum.
Correlation of Anti-Platelet and Anti-GPIIIa-(49–66) Reactivity with Platelet Count.
Fig. 5 demonstrates an inverse curvilinear correlation between platelet count and antibody reactivity for intact platelets (Fig. 5A) and GPIIIa-(49–66) (Fig. 5B). Figue 5A depicts a correlation between the antibody titer of affinity-purified anti-platelet IgG against intact platelets with platelet count of the patient. A clear distinction is observed between antibody titer of thrombocytopenic vs. nonthrombocytopenic patients. Whereas 12 of 15 thrombocytopenic patients (<150 × 103 platelets per μl) had antibody titers >10 reciprocal units, none of 12 nonthrombocytopenic patients had antibody titers >10. Fig. 5B demonstrates a similar clear distinction between serum anti-GPIIIa-(49–66) antibody concentrations of thrombocytopenic vs. nonthrombocytopenic patients. Whereas 19 of 21 thrombocytopenic patients had serum anti-GPIIIa-(49–66) concentrations >100 ng/ml, only 1 of 10 nonthrombocytopenic patients had such a value. These correlations support the observation that GPIIIa-(49–66) is a major antigenic determinant for anti-platelet antibody in HIV-1-ITP patients.
Inhibition of Binding of Anti-Platelet IgG to Platelets with GPIIIa-(49–66) and Its Albumin Conjugate.
Fig. 6 demonstrates potent in vitro inhibition of binding of affinity-purified anti-platelet IgG to platelets with free peptide, as well as its molar equivalent of albumin conjugate with an IC50 value of 3.5 nm/ml.
Figure 6.
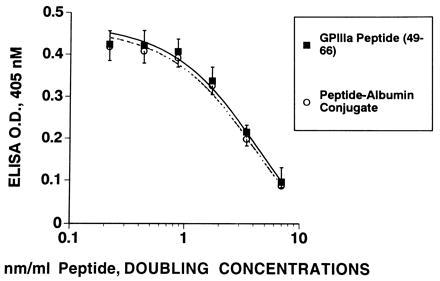
Inhibition of binding of anti-platelet IgG to platelets with GPIIIa-(49–66) and its albumin conjugate. Anti-platelet IgG (0.043 nM) was preincubated with GPIIIa-(49–66) or its equimolar GPIIIa-(49–66)-albumin conjugate for 2 hr at room temperature prior to its addition to washed platelets adsorbed to a microtiter plate. Data are the mean of results from four patients.
Induction of Thrombocytopenia in BALB/c Mice with Affinity-Purified Anti-Platelet IgG: Inhibition and Reversal with GPIIIa-(49–66) Albumin Conjugate.
Because mouse GPIIIa-(49–66) has 83% homology with human GPIIIa-(49–66) (10) and mice have Fc receptors for human IgG1 (14), an experiment was designed to test the pathophysiologic relevance of the affinity-purified human anti-GPIIIa. Fig. 7A demonstrates a precipitous drop in platelet count to 30% of control values, following the i.p. injection of a single dose of 30 μg of anti-platelet IgG into mice. A nadir was reached at 4–5 hr with recovery to normal at 36 hr. No effect was noted with control IgG. Fig. 7B demonstrates inhibition of the induction of thrombocytopenia induced by 25 μg of anti-platelet IgG, with the simultanenous injection of 226 nmol of GPIIIa-(49–66) albumin conjugate in the contralateral i.p. site. Control animals were simultaneously injected with an albumin conjugate of the scrambled irrelevant peptide CGGGARVLEDRP. Fig. 7C demonstrates reversal of the thrombocytopenia induced by 50 μg of anti-platelet IgG in animals treated at 2 hr with GPIIIa-(49–66) albumin conjugate. The unaffected control group was treated with the scrambled irrelevant peptide albumin conjugate.
Figure 7.

In vivo induction of thrombocytopenia in mice injected with affinity-purified anti-platelet IgG of HIV-1-ITP patients and its reversal with GPIIIa-(49–66). (A) Thirty micrograms of anti-platelet IgG of patients 6 and 11 or purified control human IgG was injected i.p. into eight experimental and six control mice, respectively. Platelet counts were obtained from 20 μl of blood withdrawn from the orbital sinus at the times indicated. (B) Twelve mice were injected i.p. with 25 μg of anti-platelet IgG of patient 11, as above. Seven of these mice were simultaneously injected in the opposite flank with 226 nmol of GPIIIa-(49–66)-albumin conjugate, whereas five were simultaneously injected with the irrelevant scrambled GPIIIa-albumin conjugate (CGGGARVLEDRP) at the same concentration. (C) Ten mice were injected i.p. with 50 μg of anti-platelet IgG of patients 10 and 11, as above. At 2 hr, six of these mice were injected with GPIIIa-(49–66)-albumin conjugate, whereas four were injected with the irrelevant-scrambled GPIIIa-albumin conjugate.
DISCUSSION
High-affinity F(ab′)2 anti-platelet IgG (Kd, 1.8 × 10−9 M) has recently been isolated from the PEG-ICs of HIV-1-ITP patients and contribute to the binding of these ICs to platelets. Its major reactivity against platelet GPIIIa was documented by immunoprecipitation of GPIIIa from 125I-surface-labeled platelets, which revealed a predominant GPIIIa band as well as several minor bands and its number of high-affinity binding sites on platelets (44,000–54,000 sites) equivalent to that expected for platelet GPIIIa (8).
A major antigenic determinant, GPIIIa-(49–66), has now been recognized for anti-platelet GPIIIa of HIV-1-ITP in 19 of 21 patients studied. This is documented by (i) the adsorption and elution of ≈85% of the purified anti-platelet IgG with a GPIIIa-(49–66) affinity column; (ii) the ability of GPIIIa-(49–66) antigen to differentiate HIV-1-ITP from nonthrombocytopenic HIV-1, as well as control or classic ATP sera; (iii) an inverse correlation between serum anti-GPIIIa-(49–66) reactivity and platelet count; (iv) the ability of affinity-purified anti-platelet IgG to induce thrombocytopenia in vivo in mice injected i.p. with this antibody—and prevention or reversal of the thrombocytopenia with a GPIIIa-(49–66) peptide-albumin conjugate. Mouse GPIIIa-(49–66) has 83% homology with its human counterpart (10).
The ability to separate serum-reactive anti-GPIIIa-(49–66) into a sedimentable and soluble form by centrifugation at 100,000 × g is of interest, indicating that a major fraction of anti-platelet GPIIIa is sequestered within ICs. It is possible that further centrifugation for longer periods of time would sediment all of the anti-GPIIIa-(49–66). However, this was not evident after a second centrifugation at 100,000 × g for 1 hr, which reduced the serum reactivity by an additional 5–10%. This does not rule out the possibility that anti-GPIIIa-(49–66) could be present as a small nonsedimentable IC. As recently reported (8), it is likely that anti-platelet GPIIIa-(49–66) is sequestered within ICs by rheumatoid factor (8), anti-F(ab′)2 antibodies (15, 16), or anti-idiotype antibodies (unpublished data). If these sequestering agents form complexes that are responsible for sedimentable anti-platelet IgG, as proposed, then most, if not all, anti-platelet GPIIIa-(49–66) should be on the platelet surface or within these circulating ICs. It is therefore likely that high-affinity anti-platelet GPIIIa-(49–66) first binds to the platelet surface and then as a consequence of alterations in conformation reacts with either or all of the above-mentioned anti-IgG agents. The sedimentable and/or PEG-ICs could then be derived from shed ICs on the platelet surface. An alternative possibility is that soluble ICs form within the circulation and then deposit on platelets via sequestered anti-GPIIIa-(49–66) and/or complement receptors (9). We have been unable to demonstrate Fc receptor binding (9).
Nonthrombocytopenic HIV-1-infected patients also had anti-GPIIIa-(49–66) antibody but at a considerably lower concentration than HIV-1-ITP patients. Absence of thrombocytopenia in these patients could be explained by their lower density of platelet antibody, possible impaired phagocytic function (17), and/or relatively less inhibition of impaired platelet production (18).
A wide variety of anti-platelet antibodies have been described in patients with classic ATP. Anti-GPIIb–GPIIIa complex (19–22), GPIIIa (21, 23–27), GPIIb (28, 29), and GPIb (19, 30, 31) antibodies have been reported in 75% of classic ATP patients (20). Linear segments of the intracytoplasmic domain of GPIIIa (21, 26, 27) and a cysteine-rich extracellular 36-kDa fragment of GPIIIa (25) have been shown to react with anti-platelet antibodies in classic ATP by using a monoclonal antibody immobilization test and immunoblotting. Other reports of platelet-associated and serum anti-platelet IgG suggest the requirement for a conformational epitope in classic ATP, since anti-platelet GPIIb-IIIa reactivity could not be inhibited with EDTA-dissociated IIb–IIIa complexes or purified IIIa (22) and recombinant GPIIIa linear segments encompassing the extracellular domain of GPIIIa were unreactive (27). However, the pathophysiologic relevance of these antibodies has not been demonstrated. Our finding of a predominant GPIIIa antigenic determinant in HIV-1-ITP with in vivo relevance is different from in vitro reports in classic ATP. Although not all of the anti-platelet IgG of HIV-1-ITP patients could be adsorbed to a GPIIIa-(49–66) affinity column, most of the activity reacted with this defined peptide. It is therefore likely that the pathophysiology of HIV-1-ITP is different from classic ATP in the demonstration of a major linear GPIIIa-(49–66) antigen, which is still reactive when further fractionated into its 8-mer and 10-mer components. Differences in pathophysiology are also supported by the markedly elevated platelet-associated IgG, IgM, and C3C4 in HIV-1-ITP (compared with classic ATP patients) and the presence of PEG-ICs in HIV-1-ITP (2, 6, 7) (but absence in classic ATP).
What could be responsible for the presence of such a major antigen determinant? One should consider the possibility that the GPIIIa-(49–66) antigen molecularly mimics a conformational epitope of HIV-1 gp120, which has similar or greater affinity for a conformational epitope on GPIIIa. Indeed such molecular mimicry has been recently reported (32). However, our anti-GPIIIa antibodies neither react with HIV-1 gp120 (8) nor any of multiple HIV-1 antigens displayed on immunoblot with positive anti-HIV-1 sera (unpublished data). It is possible that other infectious agents could induce molecular mimicry antibody, as recently reported for varicella zoster (33).
Regardless of the apparent differences of antibody specificity with classic ATP, as well as the lack of a molecular mimicry explanation for our findings, it is clear that this anti-GPIIIa antibody is pathophysiologically relevant. The induction of thrombocytopenia in mice with anti-platelet GPIIIa-(49–66) is of particular interest and of clinical relevance. Acute thrombocytopenia, with nadir at 4 hr could be induced in mice treated i.p. with as little as 25–50 μg of affinity-purified anti-platelet IgG and prevented or reversed with GPIIIa-(49–66) coupled to albumin, but not by a scrambled-irrelevant peptide. Such a peptide-conjugate with a half-life of 6 hr in the mouse could be useful in the treatment of severe HIV-1-ITP, rather than high dose i.v. gamma-globulin, particularly if the half-life can be prolonged by coupling the peptide to other agents known to increase the half-life of drugs (34).
Acknowledgments
This work was supported by National Institute of Health Grants HL-13336-25 and DA-04315-09 and by the Boulanger-Glazier Immunology Research Fund and Dorothy and Seymour Weinstein Platelet Research Fund.
ABBREVIATIONS
- HIV-1-ITP
immunologic thrombocytopenic HIV-1-infected patient
- ATP
autoimmune thrombocytopenia
- IC
immune complex
- r
recombinant
References
- 1.Morris L, Distenfeld A, Amorosi E, Karpatkin S. Ann Int Med. 1982;96:714–717. doi: 10.7326/0003-4819-96-6-714. [DOI] [PubMed] [Google Scholar]
- 2.Savona S, Nardi M A, Karpatkin S. Ann Int Med. 1985;102:737–741. doi: 10.7326/0003-4819-102-6-737. [DOI] [PubMed] [Google Scholar]
- 3.Ratnoff O D, Menitove J E, Aster R N, Lederman M M. N Engl J Med. 1983;308:439–442. doi: 10.1056/NEJM198302243080808. [DOI] [PubMed] [Google Scholar]
- 4.Murphy M F, Metcalfe P, Waters A H. Br J Haematol. 1987;66:337–340. doi: 10.1111/j.1365-2141.1987.tb06920.x. [DOI] [PubMed] [Google Scholar]
- 5.Jost J, Tauber M G, Luthy R, Siegenthaler W. Schweiz Med Wschr. 1988;118:206–212. [PubMed] [Google Scholar]
- 6.Walsh C M, Nardi M A, Karpatkin S. N Engl J Med. 1984;311:635–639. doi: 10.1056/NEJM198409063111004. [DOI] [PubMed] [Google Scholar]
- 7.Karpatkin S, Nardi M A. J Clin Invest. 1992;89:356–364. doi: 10.1172/JCI115593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karpatkin S, Nardi M A, Hymes K B. Proc Natl Acad Sci USA. 1995;92:2263–2267. doi: 10.1073/pnas.92.6.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hymes K B, Nardi M A, Leaf A, Karpatkin S. Blood. 1993;81:2375–2380. [PubMed] [Google Scholar]
- 10.Cientat A M, Rosa J P, Letourner F, Poncz M, Rifat S. Biochem Biophys Res Commun. 1993;193:771–778. doi: 10.1006/bbrc.1993.1692. [DOI] [PubMed] [Google Scholar]
- 11.Liu L-X, Nardi M A, Nierodzik M L, Karpatkin S. Br J Haematol. 1995;91:976–982. doi: 10.1111/j.1365-2141.1995.tb05422.x. [DOI] [PubMed] [Google Scholar]
- 12.Thorpe G H G, Kricka L J, Mosely S B, Whitehead T P. Clin Chem. 1985;31:1335–1341. [PubMed] [Google Scholar]
- 13.Lyman B, Rosenberg L, Karpatkin S. J Clin Invest. 1971;50:1854–1863. doi: 10.1172/JCI106677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hulett M D, Hogarth P M. Adv Immunol. 1994;57:1–127. doi: 10.1016/s0065-2776(08)60671-9. [DOI] [PubMed] [Google Scholar]
- 15.Yu J-R, Lennette E T, Karpatkin S. J Clin Invest. 1986;77:1756–1761. doi: 10.1172/JCI112498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Susal C, Oberg H H, Daniel V, Dorr C, Terness P, Huth-Kuhne A, Zimmerman R, Opelz G. Vox Sang. 1994;63:37–45. doi: 10.1111/j.1423-0410.1994.tb00274.x. [DOI] [PubMed] [Google Scholar]
- 17.Frank M M, Hamburger M I, Lawley T J, Kimberly R P, Plotz P H. N Engl J Med. 1979;300:518–523. doi: 10.1056/NEJM197903083001002. [DOI] [PubMed] [Google Scholar]
- 18.Ballem P, Belzberg A, Devine D, Lyster D, Spruston B, Chambers H, Doubroff P, Mikulash K. N Engl J Med. 1992;327:1779–1784. doi: 10.1056/NEJM199212173272503. [DOI] [PubMed] [Google Scholar]
- 19.VanLeeuwen E F, VanderVen J T H M, Egelfriet C P, von dem Bourne A E G T. Blood. 1982;59:23–26. [Google Scholar]
- 20.McMillan R, Tani P, Millard F, Berchtold P, Renshaw L, Woods V L J. Blood. 1987;70:1040–1045. [PubMed] [Google Scholar]
- 21.Fujisawa K, O’Toole T E, Tani P, Loftus J C, Plow E F, Ginsberg M H, McMillan R. Blood. 1991;77:2207–2213. [PubMed] [Google Scholar]
- 22.Fujisawa K, Tani P, McMillan R. Blood. 1993;81:1284–1289. [PubMed] [Google Scholar]
- 23.Beardsley D J S, Spiegel J E, Jacobs M M, Handin R I, Lux S E I. J Clin Invest. 1984;74:1701–1707. doi: 10.1172/JCI111587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mason D, McMillan R. Br J Haematol. 1984;56:529–534. doi: 10.1111/j.1365-2141.1984.tb02177.x. [DOI] [PubMed] [Google Scholar]
- 25.Kekomaki R, Dawson B, McFarland J, Kunicki T J. J Clin Invest. 1991;88:847–854. doi: 10.1172/JCI115386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujisawa K, Tani P, O’Toole T E, Ginsberg M H, McMillan R. Blood. 1992;79:1441–1446. [PubMed] [Google Scholar]
- 27.Bowditch R D, Tani P, McMillan R. Br J Haematol. 1995;91:178–184. doi: 10.1111/j.1365-2141.1995.tb05266.x. [DOI] [PubMed] [Google Scholar]
- 28.Varon D, Karpatkin S. Proc Natl Acad Sci USA. 1983;80:6992–6995. doi: 10.1073/pnas.80.22.6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomiyama Y, Take H, Honda S, Furubayashi T, Mizutani H, Tsubakio T, Kurata Y, Yonezawa T, Tarui S. Br J Haematol. 1989;71:77–83. doi: 10.1111/j.1365-2141.1989.tb06278.x. [DOI] [PubMed] [Google Scholar]
- 30.Woods V L J, Kurata Y, Montgomery R R, Tani P, Mason D, Oh E H, McMillan R. Blood. 1984;64:156–160. [PubMed] [Google Scholar]
- 31.Szatkowski N S, Kunicki T J, Aster R H. Blood. 1986;67:310–315. [PubMed] [Google Scholar]
- 32.Bettaieb A, Fromont P, Louache F, Olsenhendler E, Vainchenker W, Duédari N, Bierling P. Blood. 1992;80:162–169. [PubMed] [Google Scholar]
- 33.Wright J F, Blanchette V S, Wang H, Naveen A, Petric M, Semple J W, Chia W-K, Freedman J. Br J Haematol. 1996;95:145–152. doi: 10.1046/j.1365-2141.1996.d01-1872.x. [DOI] [PubMed] [Google Scholar]
- 34.Delgado C, Francis G E, Fisher D. Crit Rev Ther Drug Carrier Systems. 1992;9:249–304. [PubMed] [Google Scholar]