

Abstract
Although histologic features of airway remodeling have been well characterized in asthma, the immunologic and inflammatory mechanisms that drive progression of asthma to remodeling are still incompletely understood. Conceptually, airway remodeling may be due to persistent inflammation and/or aberrant tissue repair mechanisms. It is likely that several immune and inflammatory cell types and mediators are involved in mediating airway remodeling. In addition, different features of airway remodeling are likely mediated by different inflammatory pathways. Several important candidate mediators of remodeling have been identified including TGF-β and Th2 cytokines (including IL-5 and IL-13), as well as VEGF, ADAM-33, and MMP-9. Mouse models of airway remodeling have provided important insight into potential mechanisms by which TGF-β activation of the Smad 2/3 signaling pathway may contribute to airway remodeling. Human studies have demonstrated that anti-IL-5 reduces levels of airway eosinophils expressing TGF-β, as well as levels of airway remodeling as assessed by bronchial biopsies. Further such studies confirming these observations, as well as alternate studies targeting additional individual cell types, cytokines, and mediators are needed in human subjects with asthma to determine the role of candidate mediators of inflammation on the development and progression of airway remodeling.
PROGRESSION OF ASTHMA TO AIRWAY REMODELING: EPIDEMIOLOGY
Asthma is a chronic inflammatory disease of the airway which affects approximately 7% of the population of the USA1. The chronic inflammatory response in the airway in asthma is characterized by the presence of increased numbers of Th2 lymphocytes, eosinophils, and activated mast cells2. In addition to the presence of inflammatory cells in the airway, the airways of patients with asthma exhibit varying levels of structural changes termed airway remodeling3–5. Characteristic structural changes of airway remodeling include epithelial cell mucus metaplasia, smooth muscle hypertrophy/hyperplasia, subepithelial fibrosis, and increased angiogenesis3–5. Studies of lung function over time have demonstrated that lung function in adult asthmatics declines at a greater rate than non-asthmatic controls6. In a study of the change in FEV1 in a general adult population of 17,506 subjects, asthmatics demonstrated a greater decline in FEV1 (38 ml per year), as compared to those without asthma (22 ml per year) over the fifteen year duration of the study (Figure 1). While such epidemiologic studies point out the significant potential for populations of asthmatics to progress with an accelerated decline in lung function over time, it is likely that both genetic and environmental factors contribute to differing rates of decline in lung function in individual asthmatic subjects (Figure 1). The potential for a subset of asthmatics to develop a more rapid disease progression to non-reversible airflow obstruction (defined as a β-agonist response <9%) was noted in 23% of 92 adult lifelong non-smoking subjects with moderate to severe asthma after 10 years7. At present there are no reliable clinical characteristics, genotypes, or biomarkers to accurately identify subsets of asthmatics that are more prone to airway remodeling or progression of their asthma (Figure 1). An improved understanding of the immune and inflammatory mechanisms which mediate the progression of asthma may provide important insight into biomarkers or genotypes to identify such patients, as well as suggest novel therapeutic interventions to prevent or reverse disease progression.
Figure 1. Asthma Progression in adults.
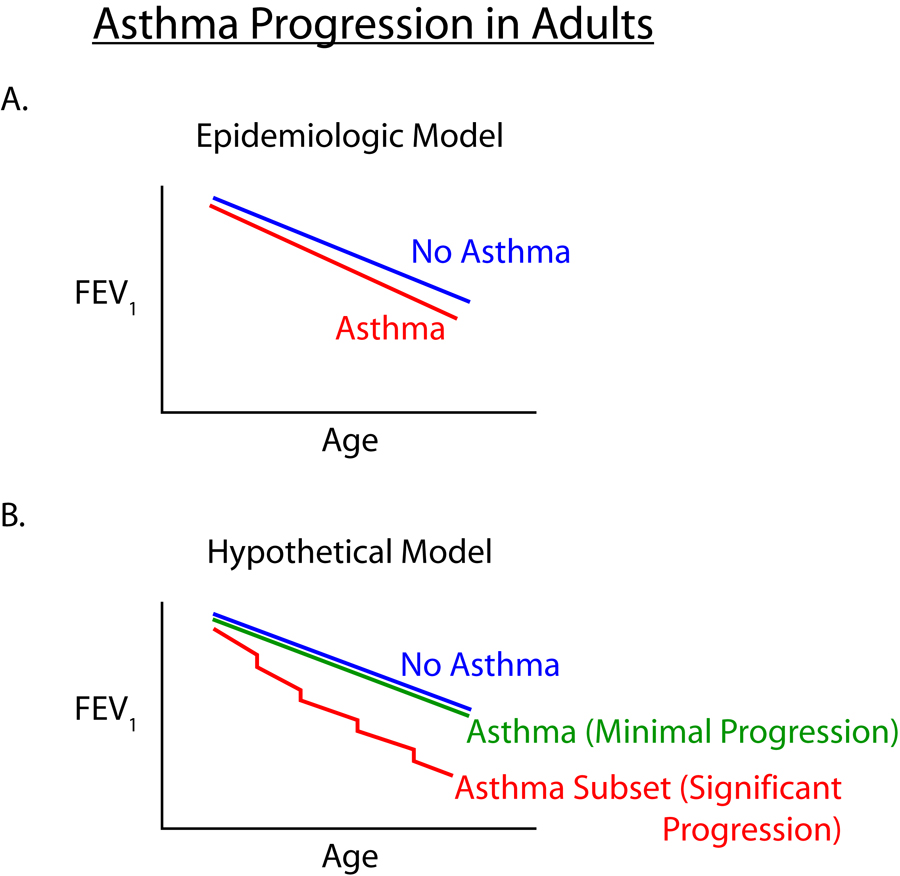
Population based studies of asthma progression in adults (e.g. reference 6)_shows an increased rate of decline in lung function compared to non-asthma controls (Panel A).Asthmatics may remodel their airways at different rates (Hypothetical Model) suggesting that certain genetic factors and/or environmental factors (exacerbations, viral infections, tobacco smoke, etc) may contribute to enhanced remodeling in a subset of asthmatics (Panel B)
IS THERE A LINK BETWEEN IMMUNE CELLS, AIRWAY INFLAMMATION, AND AIRWAY REMODELING ?
Although it is well recognized that airway inflammation is a prominent feature of asthma, the relationship between individual components of airway inflammation and the progression of inflammation to remodeling of the airways in asthma is not well understood. Evidence that immune mechanisms and inflammation are important in the pathogenesis of airway remodeling are derived either from studies in animal models of airway remodeling in asthma or from human studies of asthmatics with remodeled airways. Each of these approaches has strengths as well as limitations. For example, studies of airway remodeling in mice subjected to repetitive allergen challenge demonstrate that there is an association between sustained airway inflammation and airway remodeling8–10. Insights into which immune or inflammatory cells are important in mediating specific aspects of airway remodeling in mice can be determined from studies in mutant mice lacking either specific cell types, cytokines, or mediators8–10. The limitation of using murine models of airway remodeling is the uncertainty regarding the translation of findings in murine models to human disease. Studies in human asthmatics utilizing bronchial biopsies have the advantage of being able to characterize the presence of immune and inflammatory cells and the cytokines and mediators they express in the remodeled airway of asthmatics, as well as correlate levels of individual cell types, cytokines or mediators with levels of airway remodeling11–15. A limitation of these correlative human studies is that a cause and effect relationship between expression of a particular cell, cytokine, or mediator and the pathogenesis of airway remodeling cannot be established until specific cells have been depleted, and/or specific cytokine/mediators neutralized. In human studies of airway remodeling in asthma, only anti-IL-5 has thus far been used as a specific intervention (in this case to deplete eosinophils) and demonstrate that such a targeted intervention reduced both levels of a specific cell type (i.e. eosinophils) as well as levels of airway remodeling as assessed in bronchial biopsies16. Additional such specific intervention studies in humans with asthma targeting particular cells, cytokines, or mediators coupled with bronchial biopsies are needed to determine the relationship between individual components of the immune and inflammatory response and the pathogenesis of airway remodeling in asthma.
EVIDENCE FOR INDIVIDUAL CELLULAR COMPONENTS OF THE IMMUNE AND INFLAMMATORY RESPONSE CONTRIBUTING TO AIRWAY REMODELING
T cells
Although Th2 cells play an important role in the pathogenesis of allergic inflammation and asthma17 (Figure 2), the contribution of Th2 cells to the pathogenesis of airway remodeling requires further investigation. Insight into the role of T cell subsets in allergen induced airway remodeling has been derived from the study of transgenic mice with enhanced expression of transcription factors that direct lineage commitment of T cells to either Th1 or Th2 cells. For example, studies of airway remodeling in mice with increased numbers of Th2 cells (GATA-3 transgenic mice) demonstrate that the level of subepithelial fibrosis and airway smooth muscle hyperplasia after repeated allergen exposure were significantly enhanced18. In contrast, mice with increased numbers of Th1 cells (T-bet transgenic mice) had reduced levels of allergen-induced goblet cell hyperplasia and mucus hypersecretion18. Interestingly, human subjects with asthma and remodeled airways express increased levels of GATA-3 and reduced levels of T-bet19,20, suggesting an association between increased numbers of Th2 cells and remodeled airways of asthmatics. In addition to studies suggesting a role for Th2 cells in remodeling, there are also studies demonstrating a role for several Th2 cell derived cytokines (i.e. IL-5, IL-13) in aspects of airway remodeling8,21. Studies using either IL-5 deficient mice (reduced airway remodeling)8, or IL-13 transgenic mice (increased airway remodeling)21, support a role for Th2 cytokines in airway remodeling in vivo.
Figure 2. Immune and inflammatory mechanisms and asthma progression.
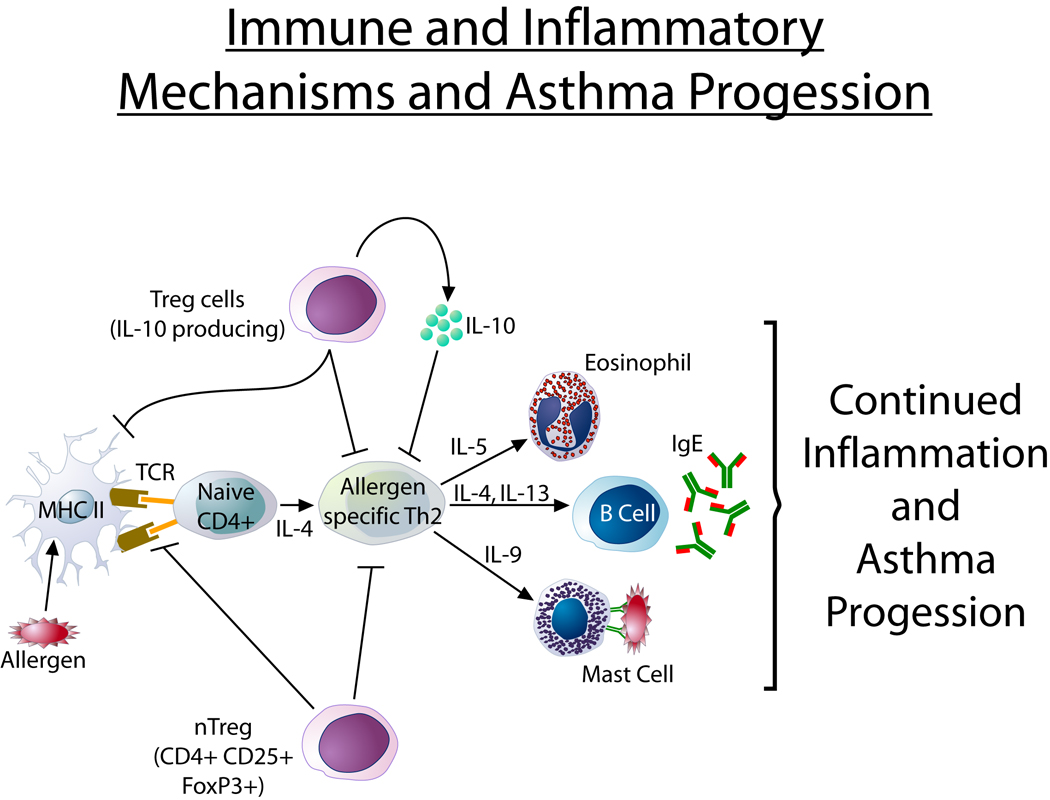
Asthma progression may be due to persistent airway inflammation and/or impaired repair mechanisms. Allergen inhalation induces activation of Th2 cells which express cytokines including IL-5 which generates TGF-β+ eosinophils that promote features of remodeling. Treg cells (natural and adaptive) have the ability to inhibit Th2 responses. Theoretically a deficiency of Treg function in asthma could promote continued Th2 mediated inflammation.
Regulatory T cells
While Th2 cells may play a role in promoting airway remodeling, regulatory T cells have the ability to down-regulate Th2 cell function and thus potentially reduce levels of airway remodeling (figure 2). A deficiency in Treg cell function in asthma could thus theoretically play an important role in asthma progression. There are two broad categories of Tregs, a) natural Tregs and b) inducible or adaptive Tregs22–26. Natural Tregs constitute 1–5% of the peripheral blood CD4+ T cell population and are defined as CD4+, CD25+, a phenotype not specific for Tregs alone. Expression of CD127 (the IL-7 receptor) is inversely correlated with Treg suppressive function and may discriminate between Tregs and activated T cells22–26. Natural Treg develop in the thymus and may expand in the periphery upon antigen exposure. Antigen specific adaptive Treg cells are induced by immunization with antigen or by natural exposure to antigen in the environment. Adaptive Tregs have many features in common with natural Tregs, but exhibit marked cytokine dependent suppressive mechanisms in vitro which are mediated through the secretion of IL-10 and TGF-β, which may suppress Th2 cells. In contrast, in vitro natural Tregs suppress through cell contact dependent mechanisms and do not secrete significant quantities of inhibitory cytokines such as IL-10 or TGF-β.
Studies have attempted to define whether Treg function is impaired in allergy and asthma. Some but not all studies of peripheral blood have reported that Treg function is impaired in some allergic patients with active disease27–29, whereas one study of pulmonary natural Tregs obtained by BAL in pediatric asthma demonstrated that these Tregs had an impaired capacity to suppress both responder T cell proliferation and Th2 cytokine synthesis30. Further studies are needed to determine whether Treg function is impaired in asthma and if so whether this impaired Treg function contributes to airway remodeling.
Eosinophils
Persistent airflow limitation has been used as a physiologic surrogate for airway remodeling. Studies of severe asthmatics have demonstrated that persistent airflow limitation (defined as either post-bronchodilator FEV1 <75% predicted, or FEV1/FVC ratio <0.75) occurred almost nine times more often in patients with sputum eosinophilia31. More recent studies suggest that eosinophil derived mediators such as TGF-β (figure 3)8,16 may play an important role in the pathogenesis of airway remodeling.
Figure 3. Eosinophil and asthma progression.
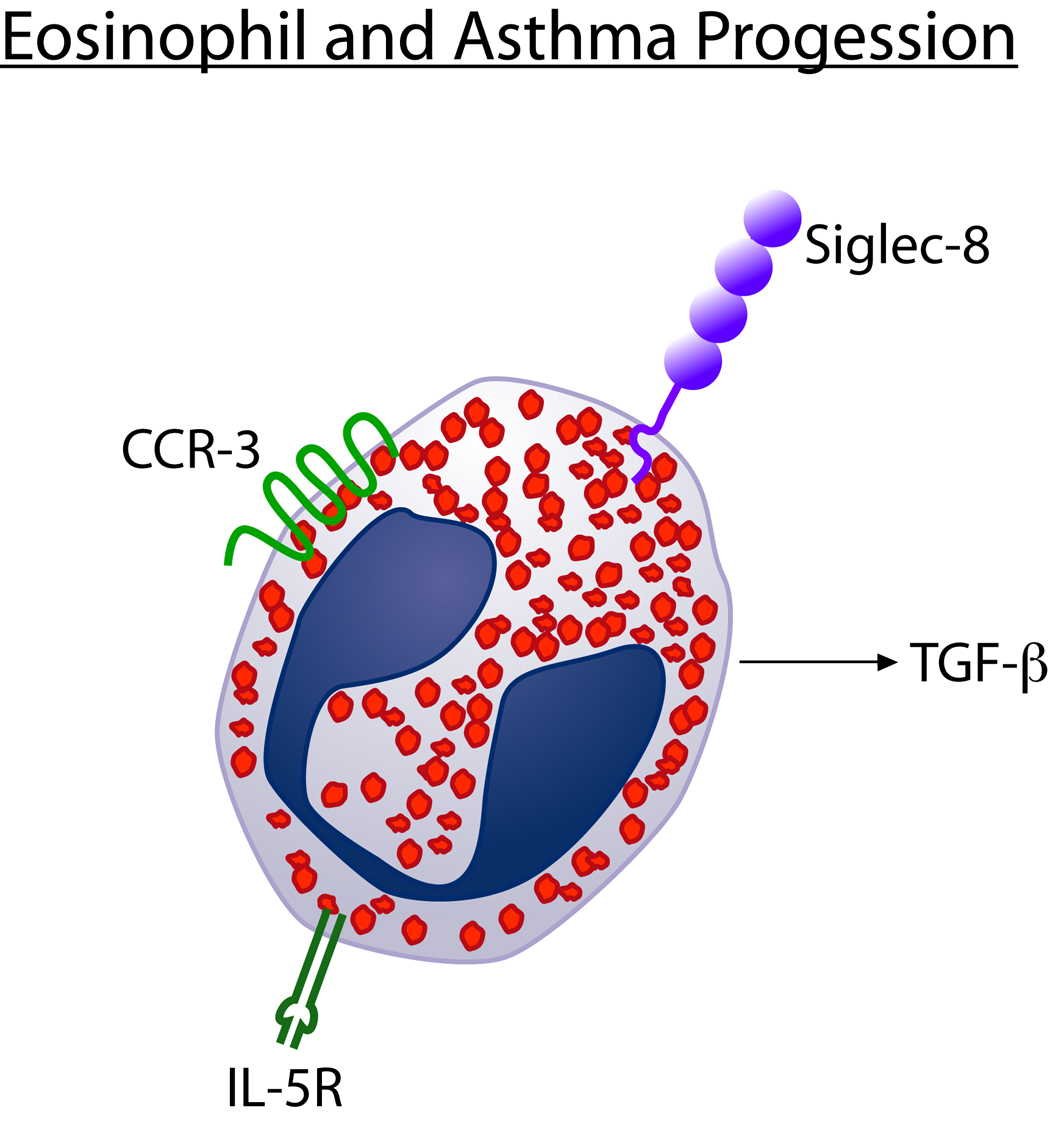
Eosinophils express receptors which induce their proliferation (IL-5R), chemo-attraction 1003 into tissues (CCR-3), and apoptosis (Siglec-8). Targeting such receptors can modulate levels of eosinophilic inflammation and the ability of eosinophils to express TGF-β an 1005 important pro-remodeling cytokine.
IL-5
The potential importance of eosinophils to airway remodeling has been demonstrated in mouse models of allergen induced airway remodeling8 as well as in human studies16. Studies have examined levels of airway remodeling in IL-5 deficient mice which lack the ability to generate eosinophils. These studies demonstrated that IL-5 deficient mice chronically challenged with allergen had significantly lower levels of peribronchial fibrosis as well as significantly reduced thickness of the peribronchial smooth muscle layer8. The mechanism by which eosinophils contribute to airway remodeling in mice is likely due to eosinophil expression of TGF-β as levels of TGF-β were significantly reduced in the remodeled airways of IL-5 deficient mice. The importance of IL-5 and eosinophils to airway remodeling in humans with asthma is suggested from studies in which anti-IL-5 was administered to asthmatics in a double blind placebo controlled study16. Asthmatics enrolled in the study had a baseline bronchial biopsy prior to receiving a single dose of anti-IL-5 or placebo and had a repeat bronchial biopsy three months later. Anti-IL-5 significantly reduced levels of BAL eosinophils and levels of remodeling as assessed by reduced deposition of the extracellular matrix associated remodeling proteins pro-collagen and tenascin. The reduced levels of total TGF-β as well as the reduced levels of TGF-β+ eosinophils in anti-IL-5 treated subjects suggested that eosinophil expression of TGF-β contributed to airway remodeling in asthma.
The potential importance of eosinophils to tissue remodeling in humans has also been demonstrated in patients with eosinophilic esophagitis (EE)32. EE is characterized by extensive eosinophilia of the esophagus. A subset of EE subjects develop esophageal strictures32. Recent studies have demonstrated evidence of remodeling in the esophagus in subjects with EE. The remodeling changes include increased levels of collagen deposition and increased numbers of angiogenic blood vessels32. The increased number of eosinophils in EE are associated with increased levels of TGF-β as well as increased activation of the phosphorylated-Smad 2/3 signaling pathway which is activated by TGF-β32. Thus there is evidence in at least two human diseases associated with eosinophilic inflammation (asthma and EE) that eosinophils and TGF-β may play an important role in tissue remodeling.
CCR-3
Although targeting IL-5 significantly reduces levels of eosinophils in mice, in human studies airway eosinophils were only reduced approximately 50–60% by anti-IL-533 suggesting that other strategies were needed to more completely deplete tissue eosinophils. An alternative strategy to targeting IL-5 to reduce levels of eosinophils, is to inhibit recruitment of eosinophils into the airway by targeting chemokine receptors expressed by eosinophils. One such important chemokine receptor expressed by eosinophils is CCR3 which mediates eosinophil chemotaxis in response to chemokines including eotaxin and RANTES both of which are expressed in the airway in asthma. Evidence that targeting CCR3 reduces eosinophilic inflammation are derived from studies of CCR3 deficient mice, or eotaxin deficient mice, which have significantly decreased levels of airway eosinophilia and associated decreased mucus production10. Studies with low molecular weight antagonists of CCR3 in a mouse model of airway remodeling have demonstrated reduced subepithelial fibrosis and goblet cell hyperplasia34. Similarly, administration of a bi-specific antibody recognizing CCR3 and the inhibitory receptor CD300a expressed on eosinophils and mast cells, reversed allergen induced airway inflammation and significantly inhibited goblet cell hyperplasia, mucus production, deposition of collagen, and smooth muscle thickness in a murine model35. Thus, targeting CCR-3 may provide a complementary approach to targeting IL-5 to reduce levels of eosinophilic inflammation and associated airway remodeling.
Siglec-8
While much is known about the pathways which induce eosinophilic airway inflammation (i.e. IL-5, CC chemokines), there is more limited information regarding the pathways which mediate resolution of eosinophilic inflammation. Knowledge of the pathways that mediate resolution of eosinophilic tissue inflammation could theoretically be harnessed to treat eosinophil diseases associated with tissue remodeling. One such candidate molecule to mediate resolution of eosinophilic inflammation is Siglec-8 which is highly expressed on human eosinophils36,37 (figure 3). In vitro studies have demonstrated that cross-linking Siglec-8 receptors on eosinophils induces an apoptotic signal37. Interestingly, neither IL-5 nor GM-CSF (eosinophil survival-promoting cytokines) are able to counteract the ability of Siglec-8 cross-linking to induce eosinophil apoptosis37.
Siglecs (sialic-acid-binding immunoglobulin-like lectins) are a family of thirteen human and nine mouse molecules involved in innate immunity38. Siglecs are transmembrane receptors characterized by an extracellular N terminal V-set domain important in ligand binding to sialic acid containing ligands, a variable number of extracellular immunoglobulin like C2-set domains, and a cytosolic tyrosine based signaling motif (ITIM/ITAM) which is present in several Siglecs. Because mouse Siglec-F shares many properties with human Siglec-8, including predominant expression on eosinophils, and shared unique ligand specificity39,40, studies of mouse Siglec-F have provided insight into the potential role of Siglec-8 in human allergic disease. Studies using Siglec-F deficient mice have demonstrated an important role for Siglec-F in mediating the resolution of eosinophilic inflammation in the airway following allergen challenge41. Siglec-F deficient mice challenged with inhaled allergen have significantly enhanced levels of eosinophilic airway inflammation as well as delayed resolution of eosinophilic airway inflammation suggesting that Siglec-F normally functions to down-regulate eosinophilic inflammation41. At present there are no studies demonstrating whether targeting Siglecs on eosinophils reduces levels of airway remodeling in asthma.
Mast cells
In studies of asthmatics, mast cells have been noted to infiltrate airway smooth muscle in particular suggesting a potential mast cell smooth muscle cell remodeling interaction42. Several mast cell derived mediators including tryptase, histamine, and cytokines such as TNF have been postulated to contribute to airway remodeling43. In addition to effects on smooth muscle, mast cells may also play a role in other features of remodeling including angiogenesis. Endobronchial biopsy specimens from asthmatic patients have demonstrated increased numbers of chymase positive mast cells associated with both increased numbers of blood vessels, and increased numbers of cells expressing the pro-angiogenic cytokine VEGF compared to healthy controls subjects44. Co-localization immunohistochemistry studies demonstrated that mast cells were a cellular source of VEGF. Recent studies of airway remodeling in mast cell deficient mice chronically challenged with allergen have also supported a role for mast cells in mediating chronic airway inflammation (including infiltration by eosinophils and lymphocytes), as well as features of remodeling including increased airway mucus expression, and increased pulmonary collagen levels45. Thus, mast cells may potentially modulate levels of airway remodeling either directly through effects on structural cells, or indirectly through effects on levels of eosinophilic and T cell mediated inflammation.
Epithelium
Epithelial cells are an example of a structural cell that can undergo both remodeling changes as well as contribute to inflammation by producing pro-inflammatory molecules. Epithelial cells are a significant source of pro-inflammatory molecules which modulate the recruitment and function of immune and inflammatory cells46,47. Thus, epithelial cell signaling molecules and mediators have the potential to play a significant role in airway remodeling.
NF-kb
Studies utilizing cre/lox molecular techniques have examined whether inhibiting NF-κB expression only in airway epithelial cells would reduce levels of airway remodeling in a mouse model48. Transcription factors such as NF-κB are important regulators of the expression of several genes (e.g. cytokines, chemokines, and adhesion molecules) important to the pathogenesis of airway inflammation in asthma48. Mice with selective airway epithelial ablation of the inhibitor of kappa B kinase β (Ikkβ), which is required for activation of NF-κB, had significantly decreased peribronchial fibrosis, levels of BAL TGF-β, and numbers of peribronchial TGF-β1 positive cells48. Levels of airway mucus, airway eosinophils, and peribronchial CD4+ cell were also reduced significantly upon airway epithelial Ikk-β ablation. The diminished airway inflammatory response was associated with reduced expression of NF-κB-regulated chemokines, including eotaxin-1 and thymus- and activation-regulated chemokine, which attract eosinophils and Th2 cells, respectively, into the airway. These results support the potential important role of the bronchial epithelium, and specifically the epithelial NF-κB pathway, in the process of remodeling.
Thymic stromal lymphopoieten (TSLP)
Transgenic mice which overexpress TSLP develop increased levels of allergic inflammation49–51. Lung epithelial cells are an important source of TSLP49,52 and increased levels of TSLP have been detected in asthmatic airway50. One mechanism by which TSLP increases allergic inflammation is through its role in enhancing the capacity of dendritic cells to induce the development of Th2 cells51. Stimuli which are known to induce epithelial secretion of TSLP include IL-4, dsRNA, and rhinovirus.
IL-25 (IL-17E)
Following initial studies demonstrating an important role for IL-25, a member of the IL-17 cytokine family, in Th2 cell-mediated immunity to parasitic infection53, subsequent studies have suggested an important role for IL-25 in the initiation of asthma54. IL-25 is expressed by lung epithelial cells as a result of innate immune responses to allergens54 Transgenic overexpression of IL-25 by these cells leads to mucus production and airway infiltration of macrophages and eosinophils, whereas blockade of IL-25 conversely reduces the airway inflammation and Th2 cytokine production in an allergen-induced asthma model54.
At present there are no studies demonstrating whether either TSLP or IL-25 are important in the progression of asthma as opposed to the initiation of asthma.
SELECTED CANDIDATE MEDIATORS OF AIRWAY REMODELING
Although multiple mediators are likely to participate in the pathogenesis of airway remodeling, several mediators (TGF-β, MMP-9, ADAM33, VEGF) have been the focus of considerable investigation.
TGF-β
Evidence to support the contribution of TGF-β to airway remodeling in asthma is derived from animal model studies with either mice deficient in Smad-3 (Smad3 mediates signaling in response to TGF-β)55, or studies with wild type mice administered an anti-TGF-β Ab56. Neutralizing either TGF-β, or TGF-β signaling through Smad-3, significantly reduces peribronchial fibrosis, airway smooth muscle proliferation, and mucus production without an associated significant change in levels of airway inflammation55,56. Interestingly, Smad-3 deficient mice chronically challenged with allergen have reduced numbers of peribronchial myofibroblasts suggesting an important role for TGF-β in vivo in the differentiation of fibroblasts to myofibroblasts55.
TGF-β, which is expressed in the airway in asthma57, stimulates fibroblasts to produce extracellular matrix proteins (collagen, fibronectin)58. In addition, TGF-β decreases the production of enzymes that degrade the extracellular matrix (collagenase), and increases the production of proteins that inhibit enzymes that degrade the extracellular matrix (Tissue inhibitor of metalloprotease, or TIMP)58 The net effect of TGF-β acting on fibroblasts is to increase the production of extracellular matrix proteins. There is evidence that the subepithelial fibrosis component of airway remodeling in asthma is mediated through induction of TGF-β expression with consequent activation of myofibroblasts to produce extracellular matrix proteins such as collagen. In addition to effects on fibrosis, TGF-β also plays an important role in airway smooth muscle proliferation through paracrine as well as potential autocrine pathways of TGF-β acting on smooth muscle59.
An important role for eosinophil expressed TGF-β in airway remodeling in humans with asthma is also suggested from studies in which TGF-β levels in the lung of asthmatics were reduced by depleting eosinophils which express TGF-β16. In these studies utilizing anti-IL-5 (which depletes eosinophils), there was a parallel decrease in levels of airway TGF-β, eosinophil TGF-β expression, and associated airway remodeling16. These studies suggest that eosinophil expression of TGF-β is an important contributor to airway remodeling in human asthma. In subjects with asthma increased levels of TGF-β have been reported in BAL60 and biopsy specimens57. TGF-β expression correlates with the degree of subepithelial fibrosis, and levels of TGF-β are significantly increased in patients with severe asthma who have prominent airway eosinophilic inflammation61.
MMP-9
Matrix metalloproteinase-9 (MMP-9) belongs to a family of extra-cellular proteases that are responsible for the degradation of the extracellular matrix during tissue remodeling62. Levels of MMP-9 (gelatinase B) are significantly increased in bronchoalveolar lavage (BAL) fluid, blood, and sputum from allergic asthmatic patients63,64,65. In addition, allergen challenge in asthmatics induces expression of MMP-9 in the airway63. Studies in MMP-9 deficient mice demonstrate that when challenged with allergen they have slightly less peribronchial fibrosis and total lung collagen compared wild type mice66. However, MMP-9 deficient mice do not have reductions in mucus expression, smooth muscle thickness or airway responsiveness66. Thus, therapies targeting MMP-9 to reduce airway remodeling in asthma may result in modest reductions in the levels of peribronchial fibrosis and may not influence levels of mucus expression, thickness of the smooth muscle layer, or airway responsiveness.
ADAM33
Like MMP-9, ADAM33 (A Disintegrin And Metaloproteinase 33) is a metalloprotease that is expressed in asthma and has been investigated for its potential role in airway remodeling67. A genetic polymorphism in ADAM33 has been associated with an accelerated decline in lung function over time in asthma, suggesting a role for ADAM33 in chronic airway injury and repair68. ADAM33 mRNA expression is significantly higher in both moderate and severe asthma compared with mild asthma and controls69. Immunostaining for ADAM33 is increased in the epithelium, submucosal cells, and smooth muscle in severe asthma compared with mild disease and controls69. However, in a mouse model of allergic asthma, ADAM33 deficient mice showed normal allergen-induced airway hyperreactivity, immunoglobulin E production, mucus metaplasia, and airway inflammation70. Studies of peribronchial fibrosis and smooth muscle remodeling were not evaluated in ADAM33 deficient mice in this acute allergen challenge protocol70.
VEGF
Increased angiogenesis, as well as increased levels of the pro-angiogenic cytokine VEGF and its receptors have been noted in asthmatic airway71,72 and animal models of asthma73–75. VEGF is elevated in induced sputum, BALF and bronchial biopsies of asthmatic patients71,72.Lung-targeted VEGF165 transgenic mice have an asthma-like phenotype with not only vascular remodeling, but also inflammation, edema, mucus metaplasia, myocyte hyperplasia and airway hyperresponsiveness73,74. Studies with VEGF lung transgenic mice suggest that VEGF is a mediator of both vascular remodeling as well as extravascular remodeling and inflammation. Interestingly, studies with inhibitors of nitric oxide (NO) suggest that NO is an important mediator of the extravascular VEGF remodeling effects74.
ASTHMA GENES AND AIRWAY REMODELING
While over 100 genes have been linked to asthma76, these studies have generally focused on identifying linkage of genes to airway responsiveness, IgE, atopy, and asthma, rather than linkage of genes to airway remodeling. The identification that a genetic polymorphism in ADAM33 is associated with an accelerated decline in lung function over time in asthma68, suggests that there may be a subset of genes associated with airway remodeling. Further studies are needed to confirm this observation and determine which genes may have linkage to airway remodeling.
ASTHMA EXACERBATIONS, IMMUNE AND INFLAMMATORY RESPONSE, AND RATE OF AIRWAY REMODELING
Acute exacerbations of asthma and airway remodeling
Although patients with asthma exhibit a greater decline in lung function compared to non-asthma controls, analysis of the rate of decline in lung function in asthmatics as a group may obscure a greater decline in a subset of asthmatic subjects. Thus, it is possible that asthmatics who have inadequate airway repair mechanisms and/or inadequately treated episodes of inflammation may have prolonged episodes of airway inflammation which lead to significantly remodeled airways in a subset of asthmatics. In this model, airway remodeling would not be a linear decrease in adult lung function over time but rather a step wise decrease in lung function following acute exacerbations in a subset of genetically predisposed individuals. Studies have started to examine whether acute exacerbations of asthma associated with increased levels of airway inflammation may account for increased levels of airway remodeling and an accelerated decline in lung function. A study examining the rate of decline in lung function in a cohort of 93 non-smoking asthmatics with moderate to severe asthma followed for at least five years demonstrated that those asthmatics with the highest frequency of acute exacerbations had a significantly greater rate of decline in lung function compared to asthmatics with the lowest frequency of acute exacerbations77 One severe asthma exacerbation/year was associated with an excess decline in FEV1 of approximately 30 ml/year77. No airway biopsies were obtained in this study so it is not possible to determine the relationship between the increased decline in lung function in patients with frequent exacerbations and changes in levels of airway remodeling.
Studies of endobronchial biopsies derived from mild asthmatics challenged with an inhaled allergen have also provided insight into the effect of such an “allergen induced exacerbation” on inflammatory responses and airway remodeling. Allergen challenge was associated with increased airway inflammation as well as increased expression of remodeling genes (including procollagen, tenascin, α-smooth muscle actin)78. Interestingly, while the increase in airway inflammation after allergen challenge resolved by seven days, the increases in markers of remodeling persisted78. Thus, acute increases in airway inflammation may contribute to more prolonged periods of airway remodeling.
Although these studies suggest that acute exacerbations of asthma may be important in mediating airway remodeling there are other studies which dissociate the number of acute exacerbations from the decline in lung function. For example, in the START trial (Steroid Treatment As Regular Therapy), low-dose inhaled corticosteroid therapy in patients with mild persistent asthma (of less than two years duration) reduced asthma exacerbations but did not reduce the decline in lung function (post-bronchodilator FEV1 percent predicted)79. Differences in results between studies77,79 may be due to differences in severity of asthma in the different study populations. However, more studies are needed to address whether acute exacerbations drive asthma progression in a subset of asthmatics.
Viruses and airway remodeling
As viruses are the most common cause of acute exacerbations of asthma80, if acute exacerbations of asthma were an important trigger of remodeling then viruses could play an important role in airway remodeling. Studies in mouse models have demonstrated that a single paramyxovirus infection causes both acute airway inflammation/hyperreactivity and chronic airway remodeling/hyperreactivity phenotypes (the latter by a hit-and-run strategy, since viral effects persist after viral clearance)81. Further human studies are needed to determine the role of viruses in airway remodeling and whether particular viruses are more likely to play a role in inducing airway remodeling.
Environmental tobacco smoke and airway remodeling
Epidemiologic studies in asthma demonstrate that asthmatics who smoke have a greater decline in forced expiration volume in 1 s when followed over a 15-year period compared with asthmatics who do not smoke6, suggesting that tobacco smoke may contribute to the decline in lung function and airway remodeling. Studies using a mouse model of allergen-induced airway remodeling have demonstrated that chronic co-exposure of mice to the combination of ETS and allergen induces significantly increased levels of airway remodeling compared with levels of airway remodeling noted in mice from chronic exposure to either stimulus alone82. In particular, the combination of chronic ETS and chronic allergen co-exposure significantly increased the thickness of the peribronchial smooth muscle layer and this was associated with a significant increase in AHR to Mch. Constituents of ETS in combination with allergen stimulate airway remodeling by inducing increased expression of eosinophil chemoattractants such as eotaxin-1 by airway epithelial cells, and increased Th2 cytokines82, which recruit larger numbers of TGF-β1+ eosinophils to the airway than that induced by allergen alone. The increased levels of peribronchial eosinophils and cells expressing TGF-β1 are likely to contribute to airway remodeling.
IS AIRWAY REMODELING REVERSIBLE ? ENDOBRONCHIAL BIOPSY STUDIES
Corticosteroids are the most effective anti-inflammatory therapy in asthma. Several studies using bronchial biopsies have also demonstrated that corticosteroids reduce airway inflammation in asthma83. Although the ability of corticosteroids to improve features of airway inflammation is well established, the ability of corticosteroids to reduce airway remodeling based on airway biopsy studies is not as well established. There are airway biopsy studies that support as well as refute a role for corticosteroids in reducing airway remodeling84–92. Although the majority of studies which have taken biopsies of the airway demonstrate that corticosteroids reduce features of airway remodeling in asthma86,88,89,90,91,92, several other studies do not support a role for corticosteroids in reducing levels of airway remodeling 84,85,87. The differences in the results may be due to differences in the dose, duration, and compliance with corticosteroid therapy, as well as the severity of asthma and remodeling end points studied.
Studies in corticosteroid resistant asthmatics have also suggested a potential role for corticosteroids in airway remodeling93. For example, corticosteroids are unable to enhance TIMP-1 production in corticosteroid resistant asthmatics and this may promote proteolytic activity in the airways of such patients with corticosteroid resistant asthma contributing to chronic airway remodeling93.
The role of leukotriene inhibitors in airway remodeling has also been investigated. Studies in mouse models demonstrate that a CysLT Receptor 1 inhibitor reverses features of airway remodeling94. Subsequent studies in human asthmatics have demonstrated that treatment of asthmatics with a CysLT receptor 1 antagonist inhibits the number of myofibroblasts in the airway following allergen challenge95. Further human studies are needed to determine whether CysLT receptor 1 antagonists inhibit cardinal features of remodeling (fibrosis, smooth muscle changes, angiogenesis, etc) in subjects with persistent asthma not undergoing allergen challenge.
LIMITATIONS OF CURRENT APPROACHES TO QUANTITATING AIRWAY REMODELING IN ASTHMA
In general, studies of airway remodeling in asthma utilize either non-invasive or invasive methods to quantitate levels of airway remodeling. Each of these approaches has strengths and weaknesses which need to be taken into account when interpreting the results of such studies.
Non-invasive methods used to assess airway remodeling
The decline in the post-bronchodilator FEV1 and FEV1/FVC ratio is frequently used in studies as a surrogate end-point for structural changes of airway remodeling in asthma. Although intuitively appealing, no studies have directly demonstrated that a decline in lung function with time is synonymous with histologic changes in the degree of airway remodeling in airway biopsies. Thus, further studies are needed to determine whether particular histologic changes in levels of airway remodeling correlate with a decline in lung function. Although imaging techniques such as CT scans have been used to quantitate airway wall thickness96,97 as a surrogate for airway remodeling, further improvements in imaging techniques especially of the small airways are needed for this to be of practical utility in the clinic.
Invasive methods used to assess airway remodeling
Although bronchial biopsy studies have an advantage over non-invasive studies in directly being able to quantify different aspects of airway remodeling there are also limitations with this approach. Bronchial biopsies are frequently taken from large proximal airways in asthma that might not reflect the site of disease activity and airway remodeling in small airways in asthma98. For example, postmortem studies of central cartilaginous airways and peripheral membranous small airways in the same asthmatic subject suggest that reticular basement membrane thickness measured in a bronchial biopsy from the central airways reflects airway remodeling in central, but not peripheral, airways98. Transbronchial biopsies have been utilized in a small number of studies99–103 to sample the distal lung, including the distal airway wall and the alveolar tissues. The primary reason for limited studies is the concern about major complications, such as bleeding and pneumothorax103. Interestingly, studies of fibroblasts obtained from the proximal and distal airway of asthmatics demonstrate phenotypic differences104 which may contribute to the variable fibroblast response to injury and repair between proximal airways and distal lung/parenchyma.
Relationship between airway inflammation, airway remodeling, and airway responsiveness
Although there is evidence to support the relationship between airway inflammation and airway remodeling, as well as between airway inflammation and airway hyperresponsiveness, the relationship between airway remodeling and airway responsiveness may be more complex than we initially appreciated. For example, airway remodeling with increased smooth muscle mass is likely to lead to increased airway responsiveness105. However, studies in human asthmatics have also demonstrated that airway wall thickening in asthma is associated with reduced rather than increased airway reactivity to MCh106. In these human asthma studies, noninvasive high-resolution computerized tomography scanning methods were used to assess airway wall thickening and, surprisingly, demonstrated that airway wall thickening in asthma is associated with reduced rather than increased airway reactivity to MCh106. One potential explanation suggested by Pare107 for the discrepancy in results between the mathematical modeling studies in asthma and the computerized tomography scan studies in asthma evaluating airway wall thickness and airway responsiveness is that the mathematical modeling studies were primarily based on altered airway geometry and did not fully take into account the potential effect of airway wall thickening on the mechanical properties of the airway, e.g., stiffness of the airway107.
KEY CONCEPTS AND THERAPEUTIC IMPLICATIONS
Airway remodeling may be due to persistent inflammation and/or aberrant tissue repair mechanisms. Different features of airway remodeling are likely mediated by different inflammatory pathways.
Several important candidate mediators of remodeling have been identified including TGF-β and Th2 cytokines (including IL-5 and IL-13), as well as VEGF, ADAM-33, and MMP-9.
Although decline in lung function is used as a very important surrogate for airway remodeling in asthma, longitudinal studies measuring decline in lung function and features of airway remodeling on airway biopsies have not yet been performed.
Asthmatics remodel their airways at different rates suggesting that certain genetic factors and/or environmental factors (exacerbations, viral infections, tobacco smoke, etc) may contribute to enhanced remodeling in a subset of asthmatics.
Human studies have demonstrated that anti-IL-5 reduces levels of airway eosinophils expressing TGF-β, as well as levels of airway remodeling as assessed by bronchial biopsies. Further such studies confirming these observations, as well as alternate studies targeting additional individual cell types, cytokines, and mediators are needed in human subjects with asthma to determine the role of candidate mediators of inflammation on the development and progression of airway remodeling.
Acknowledgments
Source of funding: NIH
Glossary
- ANTI-IL-5
Biologic agent blocking IL-5 induced eosinophilopoesis and tissue trafficking to lungs, GI tract, and skin. Therapeutic trials using anti-IL-5 include asthma, hypereosinophilic syndrome, and eosinophilic esophagitis treatment.
- CD25
The α chain of the IL-2 receptor, can be expressed on IL-10 producing T regulatory cells
- CD300a
Also known as IRp60, expressed on eosinophils and mast cells, suppresses IL-5, GM-CSF, and eotaxin effects
- CHYMASE
Mast cell subsets can be characterized by protease production, e.g. as tryptase, or chymase/tryptase; chymase can play a role in airway remodeling by activating MMP-9 and inducing VEGF.
- EOTAXIN-1
Eotaxins are a family of CC chemokines that bind to CCR3 and induce eosinophil trafficking. Eotaxins-1, -2 target eosinophils to the lung and lower gastrointestinal tract while eotaxin-3 functions as a chemoattractant for esophageal eosinophils.
- GATA-3
Member of the GATA family of transcription factors, increases transcription of the IL-5, -13, and -4 genes. The Gata3 gene is activated by IL-4, STAT-6, and GATA-3.
- ITIM, ITAM (Immunoreceptor Tyrosine Activation/Inhibitory Motifs)
Immunoglobulin receptors have activating and inhibiting motifs that interact with cellular kinases and phosphatases to regulate cell signaling.
- MMP (Matrix Metalloproteinase)
Activity of MMPs is balanced by Tissue Inhibitors of Metalloproteinases (TIMPs); while MMPs increase breakdown of extracellular matrix and promote tissue remodeling, TIMPs inhibit their function. In asthmatic airway remodeling, the balance is tipped in favor of MMP function.
- NFkB
A family of proteins that contain a Rel DNA binding domain, that form homo- and hetero-dimers and are inactivated when bound to IkB proteins. Phosphorylation of IkB proteins by IkB kinases (IKK) allows release of NFkB from the cytoplasm
- REPEATED AIRWAY CHALLENGE
Murine asthma models involve a sensitization period (often intraperitoneal on days 0, 12) followed by allergen challenge. Short allergen challenge (often 3 challenges over 1 week) results in an “acute” model of Th2 and eosinophilic airway inflammation; longer allergen challenges cause chronic inflammation and airway remodeling.
- Smad3
One of a family of Smad proteins, forms heteromeric protein complexes that regulate gene transcription; TGFβ, activins, and bone morphogenetic proteins utilize the phosphorylated Smad signaling pathway; Smad3 complexes with Smad2 and Smad4 while Smad7 inhibits signaling.
- T regulatory cells
Regulatory CD4+ or CD8+ cells, can express the transcription factor FoxP3 and/or TGFβ, CD25, and IL-10. Function to dampen the immune response; therapies that successfully treat autoimmunity or allergy can be associated with elevated numbers of Treg cells.
- T-bet
Transcription factor essential for Th1 cell induction and IFNγ expression; expression of T-bet can cause shift of Th2 to Th1 cells.
- TGFβ (Transforming growth factor β)
A pro-fibrotic agent, inactive form bound to LAP, can be activated by mast cell proteases, e.g. chymase, tryptase. TGFβ mediated fibrosis is implicated in many diseases including pulmonary and renal fibrosis; TGFβ gene SNPs can be associated with asthma.
- TSLP
(Thymic stromal lymphopoeitin): Expressed in activated epithelium, e.g. bronchial epithelial cells in asthmatic patients, promotes antigen presentation by dendritic cells by inducing expression of second signal molecules such as OX40, CD40, and CD80
- VEGF
(Vascular Endothelial Growth Factor): Required for angiogenesis, VEGF deficient mice have an embryonic lethal phenotype; the balance between pro- and anti-angiogenic factors (e.g. endostatin) is tipped toward increased vascularity in asthma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 2.Cohn L, Elias JA, Chupp GL. Asthma: Mechanisms of Disease Persistence and Progression. Annu. Rev. Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 3.Mauad T, Bel EH, Sterk PJ. Asthma therapy and airway remodeling. Journal of Allergy and Clinical Immunology. 2007;120:997–1009. doi: 10.1016/j.jaci.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 4.Boulet LP, Sterk PJ. Airway remodeling: the future. Eur Resp J. 2007;30:831–834. doi: 10.1183/09031936.00110107. [DOI] [PubMed] [Google Scholar]
- 5.Pascual RM, Peters SP. Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. J Allergy Clin Immunol. 2005;116:477–486. doi: 10.1016/j.jaci.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med. 1998;339:1194–1200. doi: 10.1056/NEJM199810223391703. [DOI] [PubMed] [Google Scholar]
- 7.Ulrik CS, Backer V. Nonreversible airflow obstruction in life-long nonsmokers with moderate to severe asthma. Eur Respir J. 1999;14:892–896. doi: 10.1034/j.1399-3003.1999.14d27.x. [DOI] [PubMed] [Google Scholar]
- 8.Cho JY, Miller M, Baek KJ, Han JW, Nayar J, Lee SY, et al. Inhibition of airway remodeling in IL-5 deficient mice. J Clin Invest. 2004;133:551–560. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 10.Fulkerson PC, Fischetti CA, McBride ML, Hassman LM, Hogan SP, Rothenberg ME. A central regulatory role for eosinophils and the eotaxin/CCR3 axis in chronic experimental allergic airway inflammation. Proc Natl Acad Sci. 2006;103:16418–16423. doi: 10.1073/pnas.0607863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vignola AM, Kips J, Bousquet J. Tissue remodeling as a feature of persistent asthma. J Allergy Clin Immunol. 2000;105:1041–1053. doi: 10.1067/mai.2000.107195. [DOI] [PubMed] [Google Scholar]
- 12.Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol. 2003;111:215–225. doi: 10.1067/mai.2003.128. [DOI] [PubMed] [Google Scholar]
- 13.Pepe C, Foley S, Shannon J, Lemiere C, Olivenstein R, Ernst P, et al. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;166:544–549. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Chu HW, Halliday JL, Martin RJ, Leung DY, Szefler SJ, Wenzel SE. Collagen deposition in large airways may not differentiate severe asthma from milder forms of the disease. Am J Respir Crit Care Med. 1998;158:1936–1944. doi: 10.1164/ajrccm.158.6.9712073. [DOI] [PubMed] [Google Scholar]
- 15.Djukanovic R, Roche WR, Wilson JW, Beasley CRW, Twentyman OP, Howarth PH, et al. Mucosal inflammation in asthma. Am Rev Respir Dis. 1990;142:434–457. doi: 10.1164/ajrccm/142.2.434. [DOI] [PubMed] [Google Scholar]
- 16.Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, Ludwig MS, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 18.Kiwamoto T, Ishii Y, Morishima Y, Yoh K, Maeda A, Ishizaki K, et al. Transcription factors T-bet and GATA-3 regulate development of airway remodeling. Am J Resp Crit Care Med. 2006;174:142–151. doi: 10.1164/rccm.200601-079OC. [DOI] [PubMed] [Google Scholar]
- 19.Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura Y, Ghaffar O, Olivenstein R, Taha RA, Soussi-Gounni A, Zhang DH, et al. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J Allergy Clin Immunol. 1999;103:215–222. doi: 10.1016/s0091-6749(99)70493-8. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Oosterhout AJM, Bloksma N. Regulatory T-lymphocytes in asthma. Eur Resp J. 2005;26:918–932. doi: 10.1183/09031936.05.00011205. [DOI] [PubMed] [Google Scholar]
- 23.Larche M. Regulatory T Cells in Allergy and Asthma. Chest. 2007;132:1007–1014. doi: 10.1378/chest.06-2434. [DOI] [PubMed] [Google Scholar]
- 24.Umetsu DT, DeKruyff RH. Immune dysregulation in asthma. Current Opin Immunol. 2006;18:727–732. doi: 10.1016/j.coi.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Tournoy KG, Van Hove C, Grooten J, Moerloose K, Brusselle GG, Joos GF. Animal models of allergen-induced tolerance in asthma: are T-regulatory-1 cells (Tr-1) the solution for T-helper-2 cells (Th-2) in asthma? Clin Exp Allergy. 2006;36:8–20. doi: 10.1111/j.1365-2222.2005.02385.x. [DOI] [PubMed] [Google Scholar]
- 26.Seroogy CM, Gern JE. The role of T regulatory cells in asthma. J Allergy Clin Immunol. 2005;116:996–999. doi: 10.1016/j.jaci.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 27.Ling EM, Smith T, Nguyen XD, Pridgeon C, Dallman M, Arbery J, et al. Relation of CD4+CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. Lancet. 2004;363:608–615. doi: 10.1016/S0140-6736(04)15592-X. [DOI] [PubMed] [Google Scholar]
- 28.Grindebacke H, Wing K, Andersson AC, Suri-Payer E, Rak S, Rudin A. Defective suppression of Th2 cytokines by CD4CD25 regulatory T cells in birch allergics during birch pollen season. Clin Exp Allergy. 2004;34:1364–1372. doi: 10.1111/j.1365-2222.2004.02067.x. [DOI] [PubMed] [Google Scholar]
- 29.Bellinghausen I, Klostermann B, Knop J, Saloga J. Human CD4+CD25+ T cells derived from the majority of atopic donors are able to suppress TH1 and TH2 cytokine production. J Allergy Clin Immunol. 2003;111:862–868. doi: 10.1067/mai.2003.1412. [DOI] [PubMed] [Google Scholar]
- 30.Hartl D, Koller B, Mehlhorn AT, Reinhardt D, Nicolai T, Schendel DJ, et al. Quantitative and functional impairment of pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. J Allergy Clin Immunol. 2007;119:1258–1266. doi: 10.1016/j.jaci.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 31.ten Brinke A, Zwinderman AH, Sterk PJ, Rabe KF, Bel EH. Factors associated with persistent airflow limitation in severe asthma. Am J Respir Crit Care Med. 2001;164:744–748. doi: 10.1164/ajrccm.164.5.2011026. [DOI] [PubMed] [Google Scholar]
- 32.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2007;119:206–212. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 33.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil's role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 34.Wegmann M, Goggel R, Sel S, Erb KJ, Kalkbrenner F, Renz H, et al. Effects of a low-molecular-weight CCR-3 antagonist on chronic experimental asthma. Am J Respir Cell Mol Biol. 2007;36:61–67. doi: 10.1165/rcmb.2006-0188OC. [DOI] [PubMed] [Google Scholar]
- 35.Munitz A, Bachelet I, Levi-Schaffer F. Reversal of airway inflammation and remodeling in asthma by a bispecific antibody fragment linking CCR3 to CD300a. J Allergy Clin Immunol. 2006;118:1082–1089. doi: 10.1016/j.jaci.2006.07.041. [DOI] [PubMed] [Google Scholar]
- 36.Floyd H, Ni J, Cornish AL, Zeng Z, Liu D, Carter KC, et al. Siglec-8. A novel eosinophil-specific member of the immunoglobulin superfamily. J Biol Chem. 2000;275:861–866. doi: 10.1074/jbc.275.2.861. [DOI] [PubMed] [Google Scholar]
- 37.Nutku E, Aizawa H, Hudson SA, Bochner BS. Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood. 2003;101:5014–5020. doi: 10.1182/blood-2002-10-3058. [DOI] [PubMed] [Google Scholar]
- 38.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 39.Tateno H, Crocker PR, Paulson JC. Mouse Siglec-F and human Siglec-8 are functionally convergent paralogs that are selectively expressed on eosinophils and recognize 6'-sulfo-sialyl Lewis X as a preferred glycan ligand. Glycobiology. 2005;15:1125–1135. doi: 10.1093/glycob/cwi097. [DOI] [PubMed] [Google Scholar]
- 40.Bochner BS, Alvarez RA, Mehta P, Bovin NV, Blixt O, White JR, et al. Glycan array screening reveals a candidate ligand for Siglec-8. J Biol Chem. 2005;280:4307–4312. doi: 10.1074/jbc.M412378200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A. Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood. 2007;109:4280–4287. doi: 10.1182/blood-2006-08-039255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346:1699–1705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 43.Okayama Y, Ra C, Saito H. Role of mast cells in airway remodeling. Curr Opin Immunol. 2007 doi: 10.1016/j.coi.2007.07.018. Epub, Aug 28. [DOI] [PubMed] [Google Scholar]
- 44.Zanini A, Chetta A, Saetta M, Baraldo S, D'Ippolito R, Castagnaro A, et al. Chymase-positive mast cells play a role in the vascular component of airway remodeling in asthma. J Allergy Clin Immunol. 2007;120:329–333. doi: 10.1016/j.jaci.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 45.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest. 2006;116:1633–1641. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc Am Thorac Soc. 2004;1:93–98. doi: 10.1513/pats.2306034. [DOI] [PubMed] [Google Scholar]
- 47.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: At the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–1284. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broide DH, Lawrence T, Doherty T, Cho JY, Miller M, McElwain K, et al. Allergen-induced peribronchial fibrosis and mucus production mediated by IkappaB kinase beta-dependent genes in airway epithelium. Proc Natl Acad Sci U S A. 2005;102:17723–17728. doi: 10.1073/pnas.0509235102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu YJ. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J Allergy Clin Immunol. 2007;120:238–244. doi: 10.1016/j.jaci.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 51.Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 52.Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, Chartier S, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le AV, Cho JY, Miller M, McElwain S, Golgotiu K, Broide DH. Inhibition of allergen-induced airway remodeling in smad 3-deficient mice. J Immunol. 2007;178:7310–7316. doi: 10.4049/jimmunol.178.11.7310. [DOI] [PubMed] [Google Scholar]
- 56.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect of the Smad signaling pathway. J Immunol. 2005;174:5774–5780. doi: 10.4049/jimmunol.174.9.5774. [DOI] [PubMed] [Google Scholar]
- 57.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 58.Wynn A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie S, Sukkar MB, Issa R, Khorasani NM, Chung KF. Mechanisms of induction of airway smooth muscle hyperplasia by transforming growth factor-{beta} Am J Physiol Lung Cell Mol Physiol. 2007;293:L245–L253. doi: 10.1152/ajplung.00068.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642–647. doi: 10.1164/ajrccm.156.2.9605065. [DOI] [PubMed] [Google Scholar]
- 61.Ohno I, Nitta Y, Yamauchi K, Hoshi H, Honma M, Woolley K, et al. Transforming growth factor beta 1 (TGF beta 1) gene expression by eosinophils in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1996;15:404–409. doi: 10.1165/ajrcmb.15.3.8810646. [DOI] [PubMed] [Google Scholar]
- 62.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28:12–24. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 63.Kelly EA, Busse WW, Jarjour NN. Increased matrix metalloproteinase-9 in the airway following allergen challenge. Am J Respir Crit Care Med. 2000;162:1157–1161. doi: 10.1164/ajrccm.162.3.9908016. [DOI] [PubMed] [Google Scholar]
- 64.Lee YC, Lee HB, Rhee YK, Song CH. The involvement of matrix metalloproteinase-9 in airway inflammation of patients with acute asthma. Clin Exp Allergy. 2001;31:1623–1630. doi: 10.1046/j.1365-2222.2001.01211.x. [DOI] [PubMed] [Google Scholar]
- 65.Mautino G, Oliver N, Chanez P, Bousquet J, Capony F. Increased release of matrix metalloproteinase-9 in bronchoalveolar lavage fluid and by alveolar macrophages of asthmatics. Am J Respir Cell Mol Biol. 1997;17:583–591. doi: 10.1165/ajrcmb.17.5.2562. [DOI] [PubMed] [Google Scholar]
- 66.Lim DH, Cho JY, Miller M, McElwain K, McElwain S, Broide DH. Reduced peribronchial fibrosis in allergen-challenged MMP-9-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:L265–L271. doi: 10.1152/ajplung.00305.2005. [DOI] [PubMed] [Google Scholar]
- 67.Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 68.Jongepier H, Boezen HM, Dijkstra A, Howards TD, Vonk JM, Koppelman GH, et al. Polymorphisms of the ADAM33 gene are associated with accelerated lung function decline in asthma. Clin Exp Allergy. 2004;34:757–760. doi: 10.1111/j.1365-2222.2004.1938.x. [DOI] [PubMed] [Google Scholar]
- 69.Foley SC, Mogas AK, Olivenstein R, Fiset PO, Chakir J, Bourbeau J, et al. Increased expression of ADAM33 and ADAM8 with disease progression in asthma. J Allergy Clin Immunol. 2007;119:863–871. doi: 10.1016/j.jaci.2006.12.665. [DOI] [PubMed] [Google Scholar]
- 70.Chen C, Huang X, Sheppard D. ADAM33 is not essential for growth and development and does not modulate allergic asthma in mice. Mol Cell Biol. 2006;26:6950–6956. doi: 10.1128/MCB.00646-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Asai K, Kanazawa H, Otani K, Shiraishi S, Hirata K, Yoshikawa J. Imbalance between vascular endothelial growth factor and endostatin levels in induced sputum from asthmatic subjects. J Allergy Clin Immunol. 2002;110:571–575. doi: 10.1067/mai.2002.127797. [DOI] [PubMed] [Google Scholar]
- 72.Hoshino M, Nakamura Y, Hamid QA. Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. J Allergy Clin Immunol. 2001;107:1034–1038. doi: 10.1067/mai.2001.115626. [DOI] [PubMed] [Google Scholar]
- 73.Lee GL, Lin H, Baluk P, Homer RJ, Chapoval S, Bhandari V, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med. 2004;10:1095–1103. doi: 10.1038/nm1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhandari V, Choo-Wing R, Chapoval S, Lee CG, Tang C, Kim YK, et al. Essential role of nitric oxide in VEGF-induced asthma-like angiogenic, inflammatory, mucus, and physiologic responses in the lung. Proc Natl Acad Sci. 2006;103:11021–11026. doi: 10.1073/pnas.0601057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee SY, Cho JY, Miller M, McElwain K, McElwain S, Sriramarao P, et al. Immunostimulatory DNA inhibits allergen-induced peribronchial angiogenesis in mice. J Allergy Clin Immunol. 2006;117:597–603. doi: 10.1016/j.jaci.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 76.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immunity. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 77.Bai TR, Vonk JM, Postman DS, Boezen Severe exacerbations predict excess lung function decline in asthma. Eur Resp J. 2007;30:452–456. doi: 10.1183/09031936.00165106. [DOI] [PubMed] [Google Scholar]
- 78.Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am J Resp Crit Care Med. 2007;175:896–904. doi: 10.1164/rccm.200609-1260OC. [DOI] [PubMed] [Google Scholar]
- 79.Pauwels RA, Pedersen S, Busse WW, Tan WC, Chen Y, Ohlsson SV, et al. Early intervention with budesonide in mild persistent asthma: a randomized, double-blind trial. Lancet. 2003;361:1071–1076. doi: 10.1016/S0140-6736(03)12891-7. [DOI] [PubMed] [Google Scholar]
- 80.Gern JE, Busse WW. Relationship of viral infections to wheezing illnesses and asthma. Nature Rev Immunol. 2002;2:132–138. doi: 10.1038/nri725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holtzman MJ, Tyner JW, Kim EY, Lo MS, Patel AC, Shornick LP, et al. Acute and chronic airway responses to viral infection: implications for asthma and chronic airway responses to viral infection: implications for asthma and chronic obstructive pulmonary disease. Proc Am Thor Soc. 2005;2:132–140. doi: 10.1513/pats.200502-015AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Min MG, Song DJ, Miller M, Cho JY, McElwain S, Ferguson P, et al. Coexposure to environmental tobacco smoke increases levels of allergen-induced airway remodeling in mice. J Immunol. 2007;178:5321–5328. doi: 10.4049/jimmunol.178.8.5321. [DOI] [PubMed] [Google Scholar]
- 83.Djukanovic R, Wilson JW, Britten KM, Wilson SJ, Walls AF, Roche WR, et al. Effect of an inhaled corticosteroid on airway inflammation and symptoms in asthma. Am Rev Respir Dis. 1992;145:669–674. doi: 10.1164/ajrccm/145.3.669. [DOI] [PubMed] [Google Scholar]
- 84.Boulet LP, Turcotte H, Laviolette M, Naud F, Bernier MC, Martel S, et al. Airway hyperresponsiveness, inflammation, and subepithelial collagen deposition in recently diagnosed versus long-standing mild asthma. Influence of inhaled corticosteroids. Am J Respir Crit Care Med. 2000;162:1308–1313. doi: 10.1164/ajrccm.162.4.9910051. [DOI] [PubMed] [Google Scholar]
- 85.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 86.Hoshino M, Takahash M, Takai Y, Sim J. Inhaled corticosteroids decrease subepithelial collagen deposition by modulation of the balance between matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 expression in asthma. J Allergy Clin Immunol. 1999;104:356–363. doi: 10.1016/s0091-6749(99)70379-9. [DOI] [PubMed] [Google Scholar]
- 87.Jeffery PK, Godfrey RW, Adelroth E, Nelson F, Rogers A, Johansson SA. Effects of treatment on airway inflammation and thickening of basement membrane reticular collagen in asthma. Am Rev Respir Dis. 1992;145:890–899. doi: 10.1164/ajrccm/145.4_Pt_1.890. [DOI] [PubMed] [Google Scholar]
- 88.Laitinen A, Altraja A, Kampe M, Linden M, Virtanen I, Laitinen LA. Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am J Respir Crit Care Med. 1997;156:951–958. doi: 10.1164/ajrccm.156.3.9610084. [DOI] [PubMed] [Google Scholar]
- 89.Olivieri D, Chetta A, Del Donno M, Bertorelli G, Casalini A, Pesci A, et al. Effect of short-term treatment with low-dose inhaled fluticasone propionate on airway inflammation and remodeling in mild asthma: a placebo-controlled study. Am J Respir Crit Care Med. 1997;155:1864–1871. doi: 10.1164/ajrccm.155.6.9196087. [DOI] [PubMed] [Google Scholar]
- 90.Sont JK, Willems LN, Bel BH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med. 1999;159:1043–1051. doi: 10.1164/ajrccm.159.4.9806052. [DOI] [PubMed] [Google Scholar]
- 91.Trigg CJ, Manolitsas ND, Wang J, Calderon MA, McAulay A, Jordan SE, et al. Placebo-controlled immunopathologic study of four months of inhaled corticosteroids in asthma. Am J Respir Crit Care Med. 1994;150:17–22. doi: 10.1164/ajrccm.150.1.8025745. [DOI] [PubMed] [Google Scholar]
- 92.Ward C, Pais M, Bish R, Reid D, Feltis B, Johns D, et al. Airway inflammation, basement membrane thickening and bronchial hyperresponsiveness in asthma. Thorax. 2002;57:309–316. doi: 10.1136/thorax.57.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goleva E, Hauk PJ, Boguniewicz J, Martin RJ, Leung DY. Airway remodeling and lack of bronchodilator response in steroid-resistant asthma. J Allergy Clin Immunol. 2007;120:1065–1072. doi: 10.1016/j.jaci.2007.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Henderson WR, Jr, Chiang GK, Tien YT, Chi EY. Reversal of allergen-induced airway remodeling by CysLT1 receptor blockade. Am J Respir Crit Care Med. 2006;173:718–728. doi: 10.1164/rccm.200501-088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kelly MM, Chakir J, Vethanayagam D, Boulet LP, Laviolette M, Gauldie J, O'Byrne PM. Montelukast treatment attenuates the increase in myofibroblasts following low-dose allergen challenge. Chest. 2006;130:741–753. doi: 10.1378/chest.130.3.741. [DOI] [PubMed] [Google Scholar]
- 96.De Jong PA, Muller NL, Pare PD, Coxson HO. Computed tomographic imaging of the airways: relationship to structure and function. Eur Respir J. 2005;26:140–152. doi: 10.1183/09031936.05.00007105. [DOI] [PubMed] [Google Scholar]
- 97.Niimi A, Matsumoto H, Takemura M, Ueda T, Mishima M. Relationship of airway wall thickness to airway sensitivity and airway reactivity in asthma. Am J Respir Crit Care Med. 2003;168:983–988. doi: 10.1164/rccm.200211-1268OC. [DOI] [PubMed] [Google Scholar]
- 98.James AL, Maxwell PS, Pearce-Pinto G, Elliot JG, Carroll NG. The relationship of reticular basement membrane thickness to airway wall remodeling in asthma. Am J Respir Crit Care Med. 2002;166:1590–1595. doi: 10.1164/rccm.2108069. [DOI] [PubMed] [Google Scholar]
- 99.Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase-positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med. 2005;171:431–439. doi: 10.1164/rccm.200407-949OC. [DOI] [PubMed] [Google Scholar]
- 100.Hauber HP, Gotfried M, Newman K, et al. Effect of HFA-flunisolide on peripheral lung inflammation in asthma. J Allergy Clin Immunol. 2003;112:58–63. doi: 10.1067/mai.2003.1612. [DOI] [PubMed] [Google Scholar]
- 101.Kraft M, Djukanovic R, Wilson S, Holgate ST, Martin RJ. Alveolar tissue inflammation in asthma. Am J Respir Crit Care Med. 1996;154:1505–1510. doi: 10.1164/ajrccm.154.5.8912772. [DOI] [PubMed] [Google Scholar]
- 102.Jeffery P, Holgate S, Wenzel S. Methods for the assessment of endobronchial biopsies in clinical research: application to studies of pathogenesis and the effects of treatment. Am J Respir Crit Care Med. 2003;168:S1–S17. doi: 10.1164/rccm.200202-150WS. [DOI] [PubMed] [Google Scholar]
- 103.Bergeron C, Tullic MK, Hamid Q. Tools used to measure airway remodelling in research. Eur Respir J. 2007;29:596–604. doi: 10.1183/09031936.00019906. [DOI] [PubMed] [Google Scholar]
- 104.Kotaru C, Schoonover KJ, Trudeau JB, Huynh ML, Zhou X, Hu H, Wenzel SE. Regional fibroblast heterogeneity in the lung: implications for remodeling. Am J Respir Crit Med. 2006;173:1208–1215. doi: 10.1164/rccm.200508-1218OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.An SS, Bai TR, Bates JHT, Black JL, Brown RH, Brusasco V, et al. Airway smooth muscle dynamics: a common pathway of airway obstruction in asthma. Eur Resp J. 2007;29:834–860. doi: 10.1183/09031936.00112606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Niimi A, Matsumoto H, Takemura M, Ueda T, Chin K, Mishima M. Relationship of airway wall thickness to airway sensitivity and airway reactivity in asthma. Am. J. Respir. Crit. Care Med. 2003;168:983–988. doi: 10.1164/rccm.200211-1268OC. [DOI] [PubMed] [Google Scholar]
- 107.Pare PD. Airway hyperresponsiveness in asthma geometry is not everything! Am. J. Respir. Crit. Care Med. 2003;168:913–914. doi: 10.1164/rccm.2307005. [DOI] [PubMed] [Google Scholar]