

Abstract
Maternal diabetes leads to an adverse in utero environment, but whether maternal diabetes impairs nephrogenesis is unknown. Diabetes was induced with streptozotocin in pregnant Hoxb7–green fluorescence protein mice at embryonic day 13, and the offspring were examined at several time points after birth. Compared with offspring of nondiabetic controls, offspring of diabetic mice had lower body weight, body size, kidney weight, and nephron number. The observed renal dysmorphogenesis may be the result of increased apoptosis, because immunohistochemical analysis revealed significantly more apoptotic podocytes as well as increased active caspase-3 immunostaining in the renal tubules compared with control mice. Regarding potential mediators of these differences, offspring of diabetic mice had increased expression of intrarenal angiotensinogen and renin mRNA, upregulation of NF-κB isoforms p50 and p65, and activation of the NF-κB pathway. In conclusion, maternal diabetes impairs nephrogenesis, possibly via enhanced intrarenal activation of the renin-angiotensin system and NF-κB signaling.
Diseases such as maternal diabetes create an adverse in utero environment that may impair the process of embryogenesis, thus predisposing infants of low birth weight (LBW) to subsequent increased risk for future disease.1–5 The developing kidney seems particularly sensitive to a high-glucose milieu, exposure to which may result in congenital renal malformations, such as renal agenesis, dysplasia, or hypoplasia.6–9
Intrauterine growth retardation, defined as birth weight below the 10th percentile for gestational age, is associated with a reduction in nephron number.10 Although the so-called “thrifty phenotype” hypothesis suggests that LBW is linked to perinatal programming,10,11 the underlying mechanisms whereby nephron number may be affected and/or nephron function altered are not yet completely delineated.
The NF-κB pathway has been reported to be a major intracellular target in hyperglycemia and oxidative stress,12,13 and its two functional pathways (canonical-classical and noncanonical) have been studied in the diabetic kidney.14–16 Five members of the NF-κB family have been identified: NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB, and c-Rel. It seems that RelA (p65) and p50, in particular, can contribute to p53-, TNF-α–, and reactive oxygen species–mediated cell apoptosis.17–21 There is a growing consensus that a high-glucose milieu and diabetes-induced activation of the NF-κB pathway have a functional impact on the course of diabetic nephropathy15,16,22,23; however, the underlying mechanisms of NF-κB signaling in impairing renal morphogenesis are not well understood.
Deficiency, mutation, or abnormal expression of genes of the intrarenal renin-angiotensin system (RAS) during organogenesis in experimental animal models often leads to abnormal kidneys,12,24–28 with a decrease in ultimate nephron numbers.29–36 Studies, including ours, have demonstrated that intrarenal RAS activation plays a key role in the development of hypertension and renal injury in diabetes.37–40 In a model of experimental diabetes in the rat, Lee et al.23 reported a functional interaction between NF-κB (in particular, the p65 subunit) and the angiotensin II (AngII) type 1 receptor. Given these data, we hypothesize that, in diabetes, enhanced intrarenal RAS activation and NF-κB signaling are two key elements intimately involved in the process of apoptosis in nascent nephrons, ultimately leading to nephron deficiency.
RESULTS
Neonatal Offspring from Control and Diabetic Hoxb7-Green Fluorescence Protein-Transgenic Mothers
The body length of neonatal offspring of streptozotocin (STZ)-induced diabetic mothers was significantly smaller as compared with those from control mothers (control versus diabetic neonate offspring in length 2.54 ± 0.27 versus 2.03 ± 0.25 cm), as shown in Figure 1.
Figure 1.
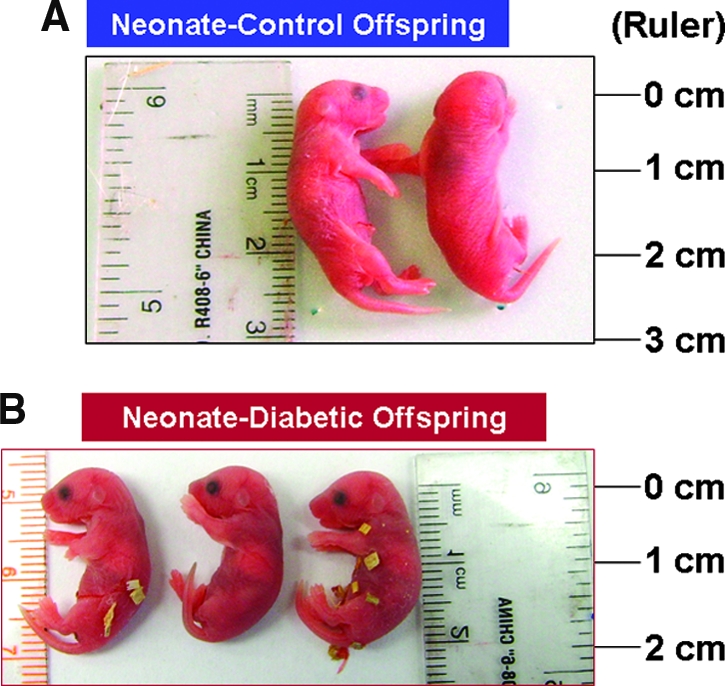
Image of representative newborn Hoxb7-GFP mice from nondiabetic and diabetic mothers. The offspring of diabetic mothers are significantly smaller than those of nondiabetic mothers (control).
Biologic Parameters
Offspring from diabetic mice remained significantly smaller and lighter (average 20% less) than control offspring during the entire suckling period. Figure 2A shows body weight (BW) for control versus STZ offspring (1.414 ± 0.110 versus 1.030 ± 0.070 g [P ≤ 0.01] in neonates; 3.744 ± 0.900 versus 3.00 ± 1.010 g [P ≤ 0.05] in 1-wk-old mice; 6.93 ± 1.32 versus 5.71 ± 1.11 [P ≤ 0.01] in 2-wk-old mice; 12.12 ± 1.16 versus 9.79 ± 2.07 [P ≤ 0.01] in 3-wk-old mice), and Figure 2B indicates the ratio of kidney weight to body weight, suggesting that kidneys of diabetic offspring are relatively large for their BW.
Figure 2.

Physical parameters in offspring of Hoxb7-GFP-Tg mice. (A) Offspring from diabetic mother had significantly lower body weight (BW; 20% less on average) than control offspring during the entire suckling period (neonate to 3 wk of age). (B) The ratios of kidney weight (KW) to BW in offspring from diabetic mother were also significantly higher than those of control offspring. □, control offspring; ▪, offspring from diabetic mother. *P < 0.05; **P < 0.01.
Renal Morphology, Nephron Number, and Glomerular Volume in Neonatal Kidneys
Hematoxylin- and eosin-stained sections of whole-mount neonatal kidneys indicated that offspring from diabetic pregnant mice had smaller kidneys with smaller glomeruli size as compared with the kidneys of control offspring (Figure 3A). We found that glomerular volume (VG) of young offspring of diabetic mothers (from neonate to 2 wk old) was persistently less than that of control animals (Figure 3B). By carefully counting the number of nephrons, we observed that neonatal nephron number in offspring of diabetic dams was significantly lower than in control animals (average 40% less; control versus diabetic neonate offspring in number 3038.00 ± 175.52 [n = 6] versus 1862.00 ± 128.74 [n = 5; P ≤ 0.001]; Figure 3C).
Figure 3.

Renal morphology and VG measurement. (A) hematoxylin and eosin (HE) staining indicates that kidney and glomerular size of neonatal offspring from diabetic mother (right) are smaller as compared with control offspring (left). (B) Quantification of VG value in control and diabetic offspring from neonate to 3 wk of age. The y axis shows the percentage of VG value compared with control animal (100%). Blue bar, control offspring (neonate: n = 9; 1 wk old: n = 12; 2 wk old: n = 9; 3 wk old: n = 8); red bar, diabetic offspring (neonate: n = 8; 1 wk old: n = 8; 2 wk old: n = 7; 3 wk old: n = 8). **P ≤ 0.01. (C) Quantification of neonatal nephron number. The y axis shows the percentage of nephron number compared with control animal (100%). Blue bar, control offspring (neonate: n = 6); red bar, diabetic offspring (neonate: n = 5). ***P ≤ 0.001.
STZ Toxicity Effect
STZ is an unstable product with a biologic half-life in cell culture medium of approximately 19 min (http://www.sigmaaldrich.com). Because STZ administration does not induce diabetes 100% of the time, we had the opportunity to examine nephrogenesis in fetuses of STZ-exposed mice with or without diabetes. We performed additional experiments to determine whether STZ could affect nephrogenesis in Nephrin-Cyan Fluorescence Protein-Transgenic (Nephrin-CFP-Tg) mice in vivo. We observed that renal damage (kidney size and number of glomeruli forming) seemed to depend on the level of maternal hyperglycemia (Figure 4A) but be independent of STZ administration (Figure 4B) or the length of exposure to STZ (from 0 to 5 d; Figure 4C). Thus, we believe that it is unlikely that a small amount of STZ (if it crosses the placenta) exerts toxicity in the fetus in utero in our model.
Figure 4.

STZ toxicity studies in Nephrin-CFP-Tg mice. (A) E16 kidney isolated from pregnant mice with three different maternal hyperglycemic levels after 3 d of STZ administration (150 mg/kg, intraperitoneally, at E13): Normal (6.0 mmol), mild (14.3 mmol), and severe (24.9 mmol). (B) E16 kidneys isolated from pregnant mice in normal maternal glucose range with or without administration of STZ at E13. (C) E16 and E18 kidneys isolated from pregnant mice with normal maternal glucose level with STZ administration at E13.
Apoptosis in Kidneys of Offspring of Diabetic Mothers
Terminal transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay revealed that apoptotic cells seem to be increased in the collapsed nephron region in neonates and 1-wk-old offspring of diabetic mothers as compared with control offspring at the same ages (Figure 5A). Double immunostaining with anti–Wilms’ tumor-1 (WT-1) and anti-active cleaved caspase-3 antibody indicated that glomerular podocytes undergo apoptosis, which ultimately results in nephron collapse (Figure 5B). Similarly, we observed augmented cleaved caspase-3 immunostaining in renal tubule of offspring of diabetic mothers from neonate to 3 wk of age as shown in Figure 6. Taken together, these data suggest that high-glucose milieu creates an adverse in utero environment that dynamically triggers nascent nephron apoptosis during nephrogenesis, consequently resulting in dysmorphogenesis with small kidneys.
Figure 5.

Apoptotic assay (TUNEL; A) in kidneys of offspring from nondiabetic and diabetic mothers: Neonate (C0 and D0), offspring at 1 wk of age (C1 and D1), offspring at 2 wk of age (C2 and D2), and offspring at 3 wk of age (C3 and D3). (B) Double immunostaining of WT-1 (a) and active caspase-3 expression (b) as well as a merged image (c) in neonatal kidneys of offspring from nondiabetic and diabetic mothers. Magnification, ×60. Pink arrows indicate the apoptotic cells.
Figure 6.

Active caspase-3 expression in kidneys of offspring from nondiabetic (control) and diabetic mothers: Neonate (C0 and D0), offspring at 1-wk old (C1 and D1), offspring at 2-wk old (C2 and D2), and offspring at 3 wk-old (C3 and D3). Magnification, ×60.
Activation of the Intrarenal RAS and NF-κB Pathways in Offspring of Diabetic Mothers
Real-time quantitative PCR (RT-qPCR) assays revealed that high-glucose milieu in utero is capable of affecting intrarenal RAS gene expression in kidneys of offspring of diabetic dams; in particular, angiotensinogen and renin mRNA expression was persistently upregulated from the neonatal period to 3 wk of age (Figure 7). Immunohistochemical examination of renal sections confirmed that augmented angiotensinogen protein expression was generally localized to the proximal tubule region in kidneys of offspring of diabetic mother (Figure 8), whereas cells positive for renin appeared in a glomerular or tubular position in offspring of diabetic mothers rather than in the juxtaglomerular apparatus as in normal offspring (Figure 9). Moreover, we observed that the p50 and p65 subunits of NF-κB were upregulated and translocated from the cytosol to the nucleus in the proximal tubules of offspring from diabetic mothers (Figure 10A). In gel mobility shift assays (GMSA), we found that NF-κB activation was more elevated in the neonatal kidneys of offspring from diabetic mothers compared with the offspring of control dams (Figure 10B). Taken together, our data indicate that a hyperglycemic environment in utero reduces kidney size and triggers apoptosis of nascent nephrons, possibly via the activation of the intrarenal RAS and NF-κB pathways.
Figure 7.

Mouse angiotensinogen (mANG) and renin mRNA expression assayed by RT-qPCR. (A and B) Expression levels of ANG (A) and renin (B) mRNA in kidneys of offspring from nondiabetic and diabetic mothers from neonate to 3 wk of age. **P ≤ 0.01; ***P ≤ 0.001.
Figure 8.

ANG protein expression in kidneys of offspring from nondiabetic (control) and diabetic mothers: Neonate (C0 and D0), offspring at 1 wk of age (C1 and D1), offspring at 2 wk of age (C2 and D2), and offspring at 3 wk of age (C3 and D3). Magnification, ×60.
Figure 9.

Renin protein expression in kidneys of offspring from nondiabetic (control) and diabetic mothers: Neonate (C0 and D0), offspring at 1 wk of age (C1 and D1), offspring at 2 wk of age (C2 and D2), and offspring at 3 wk of age (C3 and D3). Magnification, ×60.
Figure 10.

NF-κB expression and localization as well as activation in neonate kidneys from control and diabetic mothers. (A) Expression and localization of two isoforms of NF-κB, p50 and p65, were displayed by immunostaining. (B) GMSA assay. The labeled DNA probe (0.1 pmol) was incubated without (lane 1) or with BSA (10 μg; lane 2) or renal nuclear protein(s) (N.P.; 10 μg each) of neonatal kidney (lanes 3 through 8) in the presence of 0.3 U of poly dI-dC. Renal N.P. from neonatal control (lanes 3, 5, and 7) and diabetic offspring (lanes 4, 6, and 8) is incubated with consensus NF-κB DNA cold probe (lanes 3 and 4), consensus NF-κB DNA probe (lanes 5 and 6), and mutant NF-κB DNA probe (lanes 7 and 8). Magnification ×60.
DISCUSSION
In this work, we aimed to delineate the functional role of maternal diabetes in modulating renal morphogenesis in their offspring and to study their underlying mechanisms. Our data indicate that a hyperglycemic environment in utero reduces kidney size and triggers nascent nephron apoptosis via intrarenal RAS activation and NF-κB signaling.
Maternal diabetes presents an environmental challenge in utero and may fundamentally and dynamically impair the process of embryogenesis, thus predisposing to LBW.1–5 By comparing the global phenotypes displayed in young offspring of control and diabetic mothers, we observed that LBW pups with small kidneys were frequent in the offspring of diabetic dams. In the kidneys, we observed that glomeruli were smaller and that there were a relatively low number of nephrons; there is evidence of nephron collapse in these kidneys. These findings may constitute the genesis of low glomerular endowment.
In the 1980s, Brenner and associates41–44 hypothesized that “low glomerular endowment” or “fewer numbers of nephrons” are a risk factor for hypertension and ESRD in adulthood. In principle, decreased nephron number leads to renal hyperfiltration (higher filtration pressure and an increased GFR per glomerulus). Consequently, later in life, pressure natriuresis curves shift, leading to increase in BP, thereby enhancing the risk for injury as a result of hypertension and ESRD. Although outcomes such as LBW, small kidneys, and fewer nephron numbers resulting from an adverse intrauterine environment that might predispose to future hypertension are known, the mechanisms by which this occurs remain incompletely delineated.
Increased apoptosis in the blastocyst and, later, in embryonic kidneys has been reported in rodent embryos developing in diabetic dams.45–50 We suggest that apoptosis, in particular differential apoptosis of specific renal lineages during nephrogenesis, induced by a high-glucose milieu, is the major mechanism by which renal function is ultimately affected in diabetic offspring over time. The activation of intrarenal RAS and NF-κB pathways are two key mechanisms that seem critical in the apoptosis induced by an intrauterine high-glucose milieu.
In normal nephrogenesis, apoptotic events occur normally throughout renal organogenesis until the formation of the final kidney is complete. For example, the undifferentiated stromal mesenchyme either becomes interstitial cells or is destined to undergo apoptosis to make space for the expanding loops of Henle; in contrast, the differentiated metanephric mesenchyme (MM) normally undergoes epithelialization as a result of mesenchymal-to-epithelial transformation and becomes the proximal portion of the nephron.51,52 Under certain circumstances, however, for example, in maternal diabetes, if the resultant high-glucose milieu triggers apoptotic events in cells that do not normally undergo apoptosis (e.g., differentiated mesenchymal mesenchyme), then nephron formation may be altered and result in nephron collapse. Indeed, our data suggest that a high-glucose milieu in utero retards renal morphogenesis by inducing a significantly higher number of apoptotic podocytes in the developing glomeruli and inducing a high level of caspase-3 activity in the renal tubule, perhaps via activation of NF-κB pathways and the intrarenal RAS.
On the basis of our observations and those of others, we propose that high-glucose–induced cell apoptosis resulting in nephron collapse in diabetic offspring may be due to several factors. First, although the NF-κB pathway has been reported as a major intracellular target in hyperglycemia and oxidative stress,12,13,53 the expression pattern of NF-κB in the kidneys of diabetic adults is still controversial.14–16 Regarding the offspring of diabetic mothers, the functional impact of NF-κB pathways on apoptosis is unknown. We observed that NF-κB pathway was upregulated in kidneys of diabetic offspring. Furthermore, p50 and p65 subunits of NF-κB were markedly upregulated, and these subunits were translocated from the cytosol to the nucleus in proximal tubular cells of diabetic offspring. Linking this observation to apoptosis, a first possibility might be that NF-κB activation evokes several pro- and antiapoptotic genes, including Fas (CD95); TRAIL receptors (DR4, 5, and 6); the death-inducing ligands FasL, TNF-α, and TRAIL; tumor suppressor p53; Bcl-xL; and Bcl-xS,17,19 which could lead to apoptosis of proximal tubular cells, consequently resulting in nephron collapse and, ultimately, in nephron deficiency. Second, the intrarenal RAS, which has been extensively linked to diabetes-induced apoptosis in both human54 and experimental animal models,55 may play a role. Angiotensinogen and renin are major contributors to the production of AngII, the most physiologically active peptide of the RAS. We observed that the expressions of angiotensinogen and renin were dramatically activated and correlated with apoptotic events in kidneys of offspring of diabetic mothers. In addition, the cells that express renin, mainly in the renal juxtaglomerular apparatus56–58 in the kidneys of the normal offspring, are found in the glomerular or tubular region in kidneys of offspring from diabetic mothers. This shift in renin expression together with increased mouse angiotensinogen expression might be capable of stimulating increased local AngII formation, which could contribute to the observed increase in glomerular or tubular apoptosis. Finally, the cross-talk between NF-κB pathways and the intrarenal RAS23,59 may be fundamentally associated with nephron deficiency.
In conclusion, our results demonstrate that maternal diabetes impairs renal development and induces nascent nephron cell apoptosis via enhanced intrarenal RAS activation and NF-κB signaling.
CONCISE METHODS
Animals
For these in vivo studies, we used two fertile Tg mouse lines that have normal phenotype: Hoxb7-Green Fluorescence Protein-Tg (Hoxb7-GFP-Tg) and Nephrin-CFP-Tg. Hoxb7-GFP-Tg mice were provided by Dr. Frank Costantini (Columbia University Medical Center, New York, NY),60,61 which have been used to study ureteral bud branching in nephrogenesis.53 Nephrin-CFP-Tg mice, which have CFP expression driven by the podocyte-specific nephrin promoter in glomeruli, were obtained from Dr. Susan Quaggin (University of Toronto, Toronto, ON, Canada)62; these mice permit us to follow glomerular development during nephrogenesis.
Animal care in this set of studies met the standards set forth by the Canadian Council on Animal Care, and the procedures used were approved by the Institutional Animal Care Committee of the Centre Hospitalier de l’Université de Montréal. Mice were housed under standard humidity and lighting conditions (12-h light-dark cycles) and were allowed free access to standard mouse food and water ad libitum. Timed-pregnant Hoxb7-GFP mice aged 8 to 10 wk were used in all experiments. Vaginal wet mounts were made to determine the estrous cycles of the mice. On the evening before estrus, female mice were housed overnight with male mice; the presence of spermatozoa in a vaginal smear the next morning was defined as day 1 of pregnancy.
Animal Model and Experimental Design
On the basis of our previous report63 as well as those of others,64–70 maternal diabetes was induced by a single intraperitoneal injection of STZ (Sigma, St. Louis, MO) at a dosage of 150 mg/kg body weight at embryonic day 13 (E13) in Hoxb7-GFP mice. Meanwhile, we performed pilot studies regarding STZ potential toxicity effect on nephron formation in Nephrin-CFP mice.
Offspring from diabetic Hoxb7-GFP pregnant mice were killed at four time periods after birth (n = 24 at each time point): Neonate, 1 wk, 2 wk, and 3 wk. Offspring from nondiabetic pregnant mice at same time point were used as controls.
Isolation of Metanephroi
Post-STZ embryos were microdissected aseptically from timed-pregnant Nephrin-CFP mice (E16 and E18), and the metanephroi were isolated under sterile conditions as previously report.53,63 Glomerular images and quantification in Nephrin-CFP-Tg mice were analyzed by fluorescence microscopy (Nikon Eclipse TE 2000-S Microscope; Nikon, Montreal, QC, Canada).
Biologic Parameters, Renal Morphology Review, and Renal Endowment Measurement
Biologic parameters such as kidney weight, body weight, and body length were carefully monitored during the entire suckling period. Hematoxylin and eosin staining was used to review renal morphology.63 We measured glomerular size using an estimate of mean VG and also quantified nephron number. VG was determined by the method of Weibel71 with the aid of an image analysis software system (Motics Images Plus 2.0; Motic, Richmond, BC, Canada). The VG was estimated by the mean glomerular tuft area (AT) derived from the light microscopic measurement of 30 random sectional profiles of glomeruli from each group (n = 6 animals per group) using the formula VG = β/k × AT1.5, where β = 1.382 (shape coefficient for spheres) and k = 1.1 (size distribution coefficient). Quantification of nephron number was adapted from Bertram's72 method using serial sections.
RT-qPCR
RT-qPCR (iQ SYBR Green Supermix Kit and MiniOpticon Real-Time PCR machine; Bio-Rad Laboratories, Mississauga, ON, Canada) was performed as reported previously.36,53 The forward and reverse primers corresponding to mouse angiotensinogen, mouse renin, and β-actin cDNA36,53 in RT-qPCR assays were as follows: Mouse angiotensinogen forward primer 5′-CCA CGC TCT CTG GAT TTA TC-3′ and reverse primer 5′-ACA GAC ACC GAG ATG CTG TT-3′ (NM_007428), mouse renin forward primer 5′-CTG GCC AAG TTT GAC GGT GTT-3′ and reverse primer 5′-GTG TCC ACC ACT ACC GCA CAG-3′ (BC061053), and β-actin forward primer 5′-CGT GCG TGA CAT CAA AGA GAA-3′) and reverse primer 5′-GCT CGT TGC CAA TAG TGA TGA-3′ (NM_007393).
TUNEL Assay
Paraffin-embedded kidney sections (5 μm) fixed in 4% paraformaldehyde were deparaffinized in xylene and rehydrated. Apoptosis was quantified with a TUNEL kit (La Roche Biochemicals, Laval, QC, Canada) according to the supplier's instructions.
Immunohistochemistry and Immunofluorescence Staining
Paraffin-embedded kidney sections (5 μm) fixed in 4% paraformaldehyde were deparaffinized in xylene and rehydrated. Immunohistochemical examination for angiotensinogen, renin, caspase-3, and NF-κB pathway (p50 and p65) was performed by the standard avidin-biotin-peroxidase complex method (ABC Staining System; Santa Cruz Biotechnologies, Santa Cruz, CA).39,73 The primary antibodies used included a polyclonal anti-angiotensinogen antibody39,73 (gift from Dr. John S.D. Chan, CHUM-Hôtel Dieu Hospital, Montreal, QC, Canada) in a 1:100 dilution, anti–WT-1 (clone 6F-H2; Dako Cytomation, Carpinteria, CA) in 1:100 dilution, anti-cleaved caspase-3 polyclonal antibody (Cell Signaling Technology, Inc., Danvers, MA) in a 1:100 dilution, a polyclonal NF-κB pathway antibody (p50/p65; Santa Cruz Biotechnologies) in 1:100 dilution, and a polyclonal anti-renin antibody (cat. no. RDI-rtreninabm; Research Diagnostics, Concord, MA) in a 1:500 dilution.
GMSA
Nuclear protein extracts were prepared from neonatal kidneys of control and diabetic offspring. GMSA were performed as described previously,74,75 using 32P-labeled NF-κB probes (NF-κB consensus oligonucleotide: C1 [5′-AGT TGA GGG GAC TTT CCC AGG C-3′] and C2 [5′-GCC TGG GAA AGT CCC CTC AAC T-3′]; NF-κB mutant oligonucleotide: M1 [5′-AGT TGA GGC GAC TTT CCC AGG C-3′] and M2 [5′-GCC TGG GAA AGT CGC CTC AAC T-3′]; cat. no. SC-2505 and SC-2511, Santa Cruz Biotechnologies).
Statistical Analyses
Statistical significance between experimental groups was analyzed initially by t test or by one-way ANOVA followed by the Bonferroni test as appropriate. Three to four separate experiments were performed for each protocol. Data are expressed as means ± SD. P ≤ 0.05 was considered statistically significant.
DISCLOSURES
None.
Acknowledgments
This research was supported by Kidney Research Scientist Core Education and National Training Program (KRESCENT)-Canadian Institute of Health Research (CIHR) and Kidney Foundation of Canada and a New Investigator Award (KRESCENT-CIHR scholarship) to S.-L.Z.
We acknowledge the kind gifts of Hoxb7-GFP mice from Dr. Frank Costantini (Columbia University, New York, NY). We also thank Dr. Indra R. Gupta (Montreal Children's Hospital, Montreal, QC, Canada), who taught us to count the number of nephrons using J.F. Bertram's method in serial sections, and Dr. Maxime Bouchard (McGill Cancer Center, Montreal, QC, Canada), who provided us with anti–WT-1 antibody. Special thanks to Dr. John S.D. Chan (CHUM-Hôtel-Dieu, Montreal, QC, Canada) and Dr. Julie R. Ingelfinger (Massachusetts General Hospital, Boston, MA) for unconditional support and discussion of this project.
Published online ahead of print. Publication date available at www.jasn.org.
S.T. and Y.-W.C. contributed equally in this work.
See related editorial, “Perinatal Nephron Programming is not So Sweet in Maternal Diabetes,” on pages 837–839.
REFERENCES
- 1.Holemans K, Aerts L, Van Assche FA: Fetal growth and long-term consequences in animal models of growth retardation. Eur J Obstet Gynecol Reprod Biol 81: 149–156, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Holemans K, Gerber RT, Meurrens K, De Clerck F, Poston L, Van Assche FA: Streptozotocin diabetes in the pregnant rat induces cardiovascular dysfunction in adult offspring. Diabetologia 42: 81–89, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Holemans K, Caluwaerts S, Van Assche FA: Unravelling the fetal origins hypothesis. Lancet 360: 2073–2075, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Holemans K, Aerts L, Van Assche FA: Fetal growth restriction and consequences for the offspring in animal models. J Soc Gynecol Investig 10: 392–399, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Holemans K, Aerts L, Van Assche FA: Lifetime consequences of abnormal fetal pancreatic development. J Physiol 547: 11–20, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitzmiller JL, Gavin LA, Gin GD, Jovanovic-Peterson L, Main EK, Zigrang WD: Preconception care of diabetes: Glycemic control prevents congenital anomalies. JAMA 265: 731–736, 1991 [PubMed] [Google Scholar]
- 7.Lynch SA, Wright C: Sirenomelia, limb reduction defects, cardiovascular malformation, renal agenesis in an infant born to a diabetic mother. Clin Dysmorphol 6: 75–80, 1997 [PubMed] [Google Scholar]
- 8.Soler NG, Walsh CH, Malins JM: Congenital malformations in infants of diabetic mothers. Q J Med 45: 303–313, 1976 [PubMed] [Google Scholar]
- 9.Woolf AS: Multiple causes of human kidney malformations. Arch Intern Med 77: 471–473, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zandi-Nejad K, Luyckx VA, Brenner BM: Adult hypertension and kidney disease: The role of fetal programming. Hypertension 47: 502–508, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Manning J, Vehaskari VM: Low birth weight-associated adult hypertension in the rat. Pediatr Nephrol 16: 417–422, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Mercurio F, Manning AM: NF-kappaB as a primary regulator of the stress response. Oncogene 18: 6163–6171, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP: The role of oxidative stress and NF-kappaB activation in late diabetic complications. Biofactors 10: 157–167, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Flyvbjerg A, Denner L, Schrijvers BF, Tilton RG, Mogensen TH, Paludan SR, Rasch R: Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes 53: 166–172, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Morcos M, Sayed AA, Bierhaus A, Yard B, Waldherr R, Merz W, Kloeting I, Schleicher E, Mentz S, Abd el Baki RF, Tritschler H, Kasper M, Schwenger V, Hamann A, Dugi KA, Schmidt AM, Stern D, Ziegler R, Haering HU, Andrassy M, van der Woude F, Nawroth PP: Activation of tubular epithelial cells in diabetic nephropathy. Diabetes 51: 3532–3544, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Starkey JM, Haidacher SJ, LeJeune WS, Zhang X, Tieu BC, Choudhary S, Brasier AR, Denner LA, Tilton RG: Diabetes-induced activation of canonical and noncanonical nuclear factor-kappaB pathways in renal cortex. Diabetes 55: 1252–1259, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Perkins ND, Gilmore TD: Good cop, bad cop: The different faces of NF-kappaB. Cell Death Differ 13: 759–772, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Janssen I, Katzmarzyk PT, Boyce WF, King MA, Pickett W: Overweight and obesity in Canadian adolescents and their associations with dietary habits and physical activity patterns. J Adolesc Health 35: 360–367, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C: Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene 25: 6800–6816, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K: Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ 13: 730–737, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H: Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem 280: 33917–33925, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP: Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50: 2792–2808, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Lee FT, Cao Z, Long DM, Panagiotopoulos S, Jerums G, Cooper ME, Forbes JM: Interactions between angiotensin II and NF-kappaB-dependent pathways in modulating macrophage infiltration in experimental diabetic nephropathy. J Am Soc Nephrol 15: 2139–2151, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Li W, Yoshida H, Tsuchida S, Nishimura H, Takemoto F, Okubo S, Fogo A, Matsusaka T, Ichikawa I: Targeting deletion of angiotensin type 1B receptor gene in the mouse. Am J Physiol 272: F299–F304, 1997 [DOI] [PubMed] [Google Scholar]
- 25.Okubo S, Niimura F, Matsusaka T, Fogo A, Hogan BL, Ichikawa I: Angiotensinogen gene null-mutant mice lack homeostatic regulation of glomerular filtration and tubular reabsorption. Kidney Int 53: 617–625, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Matsusaka T, Kon V, Takaya J, Katori H, Chen X, Miyazaki J, Homma T, Fogo A, Ichikawa I: Dual renin gene targeting by Cre-mediated interchromosomal recombination. Genomics 64: 127–131, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Tsuchida S, Matsusaka T, Chen X, Okubo S, Niimura F, Nishimura H, Fogo A, Utsunomiya H, Inagami T, Ichikawa I: Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J Clin Invest 101: 755–760, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimura H, Yerkes E, Hohenfellner K, Miyazaki Y, Ma J, Hunley TE, Yoshida H, Ichiki T, Threadgill D, Phillips JA III, Hogan BM, Fogo A, Brock JW III, Inagami T, Ichikawa I: Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol Cell 3: 1–10, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Lasaitiene D, Friberg P: The renin-angiotensin system in kidney development. Acta Physiol Scand 181: 529–535, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Ingelfinger JR, Woods LL: Perinatal programming, renal development, and adult renal function. Am J Hypertens 15: 46S–49S, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Iosipiv IV, Schroeder M: A role for angiotensin II AT1 receptors in ureteric bud cell branching. Am J Physiol Renal Physiol 285: F199–F207, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Moritz KM, Dodic M, Wintour EM: Kidney development and the fetal programming of adult disease. Bioessays 25: 212–220, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Pan L, Gross KW: Transcriptional regulation of renin: An update. Hypertension 45: 3–8, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Solhaug MJ, Bolger PM, Jose PA: The developing kidney and environmental toxins. Pediatrics 113: 1084–1091, 2004 [PubMed] [Google Scholar]
- 35.Wolf G: Angiotensin II and tubular development. Nephrol Dial Transplant 17[Suppl 9]: 48–51, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Zhang SL, Moini B, Ingelfinger JR: Angiotensin II increases Pax-2 expression in fetal kidney cells via the AT2 receptor. J Am Soc Nephrol 15: 1452–1465, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Hsieh TJ, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Chan JS: High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology 143: 2975–2985, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Hsieh TJ, Fustier P, Wei CC, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Fantus IG, Hamet P, Chan JS: Reactive oxygen species blockade and action of insulin on expression of angiotensinogen gene in proximal tubular cells. J Endocrinol 183: 535–550, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Sachetelli S, Liu Q, Zhang SL, Liu F, Hsieh TJ, Brezniceanu ML, Guo DF, Filep JG, Ingelfinger JR, Sigmund CD, Hamet P, Chan JS: RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int 69: 1016–1023, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Zhang SL, Chen X, Wei CC, Filep JG, Tang SS, Ingelfinger JR, Chan JS: Insulin inhibits dexamethasone effect on angiotensinogen gene expression and induction of hypertrophy in rat kidney proximal tubular cells in high glucose. Endocrinology 143: 4627–4635, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Brenner BM, Garcia DL, Anderson S: Glomeruli and blood pressure: Less of one, more the other? Am J Hypertens 1: 335–347, 1988 [DOI] [PubMed] [Google Scholar]
- 42.Brenner BM, Mackenzie HS: Nephron mass as a risk factor for progression of renal disease. Kidney Int Suppl 63: S124–S127, 1997 [PubMed] [Google Scholar]
- 43.Mackenzie HS, Brenner BM: Fewer nephrons at birth: A missing link in the etiology of essential hypertension? Am J Kidney Dis 26: 91–98, 1995 [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie HS, Lawler EV, Brenner BM: Congenital oligonephropathy: The fetal flaw in essential hypertension? Kidney Int Suppl 55: S30–S34, 1996 [PubMed] [Google Scholar]
- 45.Chi MM, Pingsterhaus J, Carayannopoulos M, Moley KH: Decreased glucose transporter expression triggers BAX-dependent apoptosis in the murine blastocyst 12. J Biol Chem 275: 40252–40257, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Keim AL, Chi MM, Moley KH: Hyperglycemia-induced apoptotic cell death in the mouse blastocyst is dependent on expression of p53 10. Mol Reprod Dev 60: 214–224, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Leunda-Casi A, Genicot G, Donnay I, Pampfer S, De Hertogh R: Increased cell death in mouse blastocysts exposed to high D-glucose in vitro: implications of an oxidative stress and alterations in glucose metabolism 4. Diabetologia 45: 571–579, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Moley KH, Chi MM, Knudson CM, Korsmeyer SJ, Mueckler MM: Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways 14. Nat Med 4: 1421–1424, 1998 [DOI] [PubMed] [Google Scholar]
- 49.Pampfer S, Donnay I: Apoptosis at the time of embryo implantation in mouse and rat 19. Cell Death Differ 6: 533–545, 1999 [DOI] [PubMed] [Google Scholar]
- 50.Pampfer S, Cordi S, Vanderheyden I, Van Der Smissen P, Courtoy PJ, Van Cauwenberge A, Alexandre H, Donnay I, De Hertogh R: Expression and role of Bcl-2 in rat blastocysts exposed to high D-glucose 11. Diabetes 50: 143–149, 2001 [DOI] [PubMed] [Google Scholar]
- 51.Ekblom P, Miettinen A, Virtanen I, Wahlstrom T, Dawnay A, Saxen L: In vitro segregation of the metanephric nephron 4. Dev Biol 84: 88–95, 1981 [DOI] [PubMed] [Google Scholar]
- 52.Piscione TD, Rosenblum ND: The molecular control of renal branching morphogenesis: Current knowledge and emerging insights 3. Differentiation 70: 227–246, 2002 [DOI] [PubMed] [Google Scholar]
- 53.Chen YW, Liu F, Tran S, Zhu Y, Hebert MJ, Ingelfinger JR, Zhang SL: Reactive oxygen species and nuclear factor-kappa B pathway mediate high glucose-induced Pax-2 gene expression in mouse embryonic mesenchymal epithelial cells and kidney explants. Kidney Int 70: 1607–1615, 2006 [DOI] [PubMed] [Google Scholar]
- 54.Kumar D, Robertson S, Burns KD: Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem 259: 67–70, 2004 [DOI] [PubMed] [Google Scholar]
- 55.Brezniceanu ML, Liu F, Wei CC, Tran S, Sachetelli S, Zhang SL, Guo DF, Filep JG, Ingelfinger JR, Chan JS: Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int 71: 912–923, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA: Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol 281: F345–F356, 2001 [DOI] [PubMed] [Google Scholar]
- 57.Carey RM, McGrath HE, Pentz ES, Gomez RA, Barrett PQ: Biomechanical coupling in renin-releasing cells. J Clin Invest 100: 1566–1574, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gomez RA, Tufro-McReddie A, Norwood VF, Harris M, Pentz ES: Renin-angiotensin system: Kidney growth and development. Exp Nephrol 2: 130–1574, 1994 [PubMed] [Google Scholar]
- 59.Mezzano S, Droguett A, Burgos ME, Ardiles LG, Flores CA, Aros CA, Caorsi I, Vio CP, Ruiz-Ortega M, Egido J: Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int Suppl S64–S70, 2003 [DOI] [PubMed]
- 60.Srinivas S, Goldberg MR, Watanabe T, D'Agati V, al-Awqati Q, Costantini F: Expression of green fluorescent protein in the ureteric bud of transgenic mice: A new tool for the analysis of ureteric bud morphogenesis. Dev Genet 24: 241–251, 1999 [DOI] [PubMed] [Google Scholar]
- 61.Watanabe T, Costantini F: Real-time analysis of ureteric bud branching morphogenesis in vitro. Dev Biol 271: 98–108, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Cui S, Li C, Ema M, Weinstein J, Quaggin SE: Rapid isolation of glomeruli coupled with gene expression profiling identifies downstream targets in Pod1 knockout mice. J Am Soc Nephrol 16: 3247–3255, 2005 [DOI] [PubMed] [Google Scholar]
- 63.Zhang SL, Chen YW, Tran S, Chenier I, Hebert MJ, Ingelfinger JR: Reactive oxygen species in the presence of high glucose alter ureteric bud morphogenesis. J Am Soc Nephrol 18: 2105–2115, 2007 [DOI] [PubMed] [Google Scholar]
- 64.Cuezva JM, Burkett ES, Kerr DS, Rodman HM, Patel MS: The newborn of diabetic rat. I. Hormonal and metabolic changes in the postnatal period. Pediatr Res 16: 632–637, 1982 [DOI] [PubMed] [Google Scholar]
- 65.Kanwar YS, Akagi S, Nayak B, Sun L, Wada J, Xie P, Thakur A, Chugh SS, Danesh FR: Renal-specific oxidoreductase biphasic expression under high glucose ambience during fetal versus neonatal development. Kidney Int 68: 1670–1683, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Eriksson U, Andersson A, Efendic S, Elde R, Hellerstrom C: Diabetes in pregnancy: Effects on the foetal and newborn rat with particular regard to body weight, serum insulin concentration and pancreatic contents of insulin, glucagon and somatostatin. Acta Endocrinol (Copenh) 94: 354–364, 1980 [DOI] [PubMed] [Google Scholar]
- 67.Oh W, Gelardi NL, Cha CJ: Maternal hyperglycemia in pregnant rats: Its effect on growth and carbohydrate metabolism in the offspring. Metabolism 37: 1146–1151, 1988 [DOI] [PubMed] [Google Scholar]
- 68.Kervran A, Guillaume M, Jost A: The endocrine pancreas of the fetus from diabetic pregnant rat. Diabetologia 15: 387–393, 1978 [DOI] [PubMed] [Google Scholar]
- 69.Aerts L, Sodoyez-Goffaux F, Sodoyez JC, Malaisse WJ, Van Assche FA: The diabetic intrauterine milieu has a long-lasting effect on insulin secretion by B cells and on insulin uptake by target tissues. Am J Obstet Gynecol 159: 1287–1292, 1988 [DOI] [PubMed] [Google Scholar]
- 70.Oh W, Gelardi NL, Cha CJ: The cross-generation effect of neonatal macrosomia in rat pups of streptozotocin-induced diabetes. Pediatr Res 29: 606–610, 1991 [DOI] [PubMed] [Google Scholar]
- 71.Weibel ER, ed.: Numerical density: Shape and size of particles. In: Stereological Methods, Vol. 2 Theoretical Foundations, London, Academic Press, 1980, pp 149–152
- 72.Bertram JF: Counting in the kidney. Kidney Int 59: 792–796, 2001 [DOI] [PubMed] [Google Scholar]
- 73.Brezniceanu ML, Wei CC, Zhang SL, Hsieh TJ, Guo DF, Hebert MJ, Ingelfinger JR, Filep JG, Chan JS: Transforming growth factor-beta 1 stimulates angiotensinogen gene expression in kidney proximal tubular cells. Kidney Int 69: 1977–1985, 2006 [DOI] [PubMed] [Google Scholar]
- 74.Chen X, Zhang SL, Pang L, Filep JG, Tang SS, Ingelfinger JR, Chan JS: Characterization of a putative insulin-responsive element and its binding protein(s) in rat angiotensinogen gene promoter: Regulation by glucose and insulin. Endocrinology 142: 2577–2585, 2001 [DOI] [PubMed] [Google Scholar]
- 75.Wei CC, Guo DF, Zhang SL, Ingelfinger JR, Chan JS: Heterogenous nuclear ribonucleoprotein F modulates angiotensinogen gene expression in rat kidney proximal tubular cells. J Am Soc Nephrol 16: 616–628, 2005 [DOI] [PubMed] [Google Scholar]