

Abstract
The effect of the polypeptide environment on polyalanine-induced fibril formation was investigated with amyloidogenic fragments from PAPBN1, a nuclear protein controlling polyadenylation. Mutation-caused extensions of the natural 10 alanine sequence up to maximally 17 alanines result in fibril formation of PABPN1 and the development of the disease oculopharyngeal muscular dystrophy (OPMD). We explored the influence of fibril formation on the structure and function of a one-domain protein linked to the fibril-forming part of PABPN1. The well-characterized, stably folded, one-domain protein, cold-shock protein CspB from Bacillus subtilis, was fused either to the C terminus of the entire N-terminal domain of PABPN1 or directly to peptides consisting of 10 or 17 alanine residues. The fusion protein between the N-terminal domain of PABPN1 and CspB formed fibrils in which the structure and activity of CspB were retained. In the fibrils formed by fusions in which the polyalanine sequence was directly linked to CspB, CspB was unfolded. These results indicate that the folded conformation and the function of a protein domain can be maintained in amyloid-like fibrils, and that the distance between this domain and the fibril plays an important role.
Keywords: polyalanine, fibril formation, CspB, PABPN1, protein folding
Protein misfolding diseases receive increasing attention both from the field of drug design and basic science (for review, see Selkoe 2003; Scheibel and Buchner 2006). While a number of these diseases arise spontaneously or result from infections, congenital disorders are also well described (Soto et al. 2006). Well-known examples are familial forms of Morbus Alzheimer and Chorea Huntington (Lazo et al. 2005), diseases that are caused by point mutations in the gene for the amyloid precursor protein or trinucleotide expansions in the gene for huntingtin, respectively (for review, see Everett and Wood 2004). In the case of the latter, additional trinucleotide repeats result in the extension of a polyglutamine sequence. Less than 40 glutamines are considered to reflect the “healthy form” of the protein, and individuals possessing more extended polyglutamine segments develop the disease. A clear correlation between the length of the polyglutamine extension and disease onset and severity has been reported (Andrew et al. 1993; Borrell-Pages et al. 2006).
Besides polyglutamine extensions, expansions of polyalanine segments have been described to be associated with disease or developmental disorders (for review, see Albrecht and Mundlos 2005). However, in contrast to polyglutamine extensions, which can comprise more than 100 additional glutamines, pathogenic polyalanine extensions are much shorter and consist maximally of 15 additional alanine residues (for review, see Amiel et al. 2004). Hitherto, extensions of polyalanine sequences have been exclusively described in nucleic acid binding proteins, and more precisely, with only one exception, in transcription factors (Albrecht and Mundlos 2005). The only protein for which polyalanine-induced fibrillation has been confirmed in vivo and in vitro is the RNA-binding protein PABPN1, which controls the length of poly-A tails during mRNA processing (Wahle 1991; Wahle and Ruegsegger 1999).
Polyalanine extensions affecting the N-terminal domain of PABPN1 are associated with oculopharyngeal muscular dystrophy (OPMD), a late-onset disease (Brais et al. 1998; for recent review, see Abu-Baker and Rouleau 2007). Biopsy material from OPMD patients revealed intranuclear inclusions of a palisade-like appearance in muscle nuclei resembling amyloid-like deposits (Tomé et al. 1997; Calado et al. 2000). We showed previously that fibril formation of the N-terminal fragment of PABPN1 occurs in vitro, and that the propensity to form fibrils increases with the length of the polyalanine sequence (Scheuermann et al. 2003). However, it is still unclear whether fibrils are the cause for disease development: A study involving a transgenic fly model suggests that the RNA-binding domain of PABPN1 plays a major role in OPMD symptoms (Chartier et al. 2006). On the other hand, since the protein oligomerizes upon association with RNA, a defect in RNA binding likely reduces local concentrations of PABPN1 that may be required for fibril nucleus formation. An intact oligomerization domain of PABPN1 is crucial for disease development (Fan et al. 2001), which supports the hypothesis that intracellular threshold concentrations of PABPN1 influence fibril formation. Furthermore, the findings that OPMD symptoms can be mitigated by chemical and molecular chaperones point to a direct involvement of the fibrils in the disease development (Bao et al. 2002, 2004; Abu-Baker et al. 2003; Davies et al. 2005, 2006; Wang et al. 2005).
The molecular steps that occur during fibril formation of amyloidogenic proteins are currently intensively investigated. A protein's propensity to convert into fibrils cannot be predicted from its primary sequence, and the molecular transitions from the soluble to the fibrillar state(s) seem to differ from protein to protein. Recent reports suggest that the structures of fibrils depend on the fibrillation conditions (Tanaka et al. 2004; Petkova et al. 2005; Pedersen et al. 2006). In fact, the provocative hypothesis has been put forward that many, if not all, proteins can be converted into fibrils once the conditions suitable for fibril formation have been identified (Chiti et al. 1999a). However, proteins display pronounced differences in their propensities to convert to the fibrillar states, presumably depending on their native state stabilization energies, if (partial) unfolding precedes amyloid formation, or, in the case of unstructured proteins, on the kinetics of fibril formation (Chiti et al. 1999b).
PABPN1 can be roughly divided into three domains: The N-terminal domain with ca. 125 amino acids is rich in glutamic acid residues; the function of this domain is currently unknown. This domain is separated from the central RNP (ribonucleic protein) domain by a 34-amino acid comprising α-helical segment. Both the RNP domain and the C-terminal domain (259–306) contribute to RNA binding during polyadenylation (Kühn et al. 2003; for review, see Kühn and Wahle 2004). The N-terminal domain of PABPN1 is largely unstructured, and no tertiary contacts could be detected for this domain (Lodderstedt et al. 2008). Here, we asked whether the propensity of the N-terminal domain to form fibrils would be influenced by a fusion to a stably folded domain and, vice versa, whether fibrillation affects the structure and function of the linked folded domain. The activity of the RNP- and C-terminal domains of PABPN1 can, in principle, be assessed by complex in vitro polyadenylation assays, but a thermodynamic characterization of their stability is not established. Thus, for our investigations, we fused the cold-shock protein from Bacillus subtilis, CspB, C-terminally to the amyloidogenic sequences of PABPN1. CspB is a well-folded small protein with 67 residues, and its structure and stability are well characterized (Schindelin et al. 1993; Schnuchel et al. 1993; Schindler et al. 1995). Its native function can be assayed by its ability to bind with high affinity to oligonucleotides (Zeeb and Balbach 2003; Max et al. 2006; Zeeb et al. 2006), and its thermodynamic stability can be measured when it is fused to the N-terminal domain of PABPN1. In this work, we show that this fusion protein forms fibrils in which the stability and function of CspB are retained. When, however, only short oligo-alanine stretches of 10 or 17 residues are linked directly to CspB, fibrils still form, but CspB is destabilized and inactive in these fibrils.
Results and Discussion
In fusions with the N-terminal domain of PABPN1, CspB is correctly folded
Previous in vitro analyses of the polyalanine-induced fibrillation of PABPN1 were performed with the N-terminal fragment only, because the full-length protein forms amorphous aggregates, which obscures fibril formation (Scheuermann et al. 2003; Lodderstedt et al. 2007). The N-terminal fragment starts with 10 alanine residues immediately after the start methionine, and comprises the first 125 residues of wild-type PABPN1 (Fig. 1; Scheuermann et al. 2003). In previous work, we investigated the biophysical properties of three variants of the N-terminal fragment, one with 10 alanines corresponding to the wild-type sequence (N-WT), one with 17 alanines (N−(+7)Ala) (as the most extreme extension found in OPMD patients), and one in which the alanine segment had been deleted (N-ΔAla). This variant never formed fibrils. During these studies, circumstantial evidence was obtained that the N-terminal domain of PABPN1 lacks a stably folded tertiary structure (Lodderstedt et al. 2008).
Figure 1.

Schematic representation of the fusions analyzed in this work. The constructs were generated in a way that CspB contained the start methionine in the fusions with the entire N-terminal of PABPN1, but lacked the start methionine in the direct fusions.
In order to explore how the folding state of the neighboring polypeptide environment influences alanine-induced fibril formation, we fused CspB, a well-characterized, stably folded single domain protein (Schindelin et al. 1993; Schnuchel et al. 1993; Schindler et al. 1995) to five different versions of the amyloidogenic part of PABPN1. In these constructs CspB was linked to the wild-type version of the N-terminal domain (N-WT), to versions that contained 17 instead of the natural 10 alanine stretch (N−(+7)Ala), or in which the 10 alanines had been deleted (N-ΔAla). In addition, 10 or 17 alanines were fused directly to CspB (Fig. 1). These fusion proteins were recombinantly produced in Escherichia coli and purified as described in Materials and Methods.
The structures of the fusion proteins were analyzed by CD spectroscopy. The chimeras consisting of N-WT or N-ΔAla fused to CspB showed identical spectra indicating similar if not identical secondary-structural elements (Fig. 2A). The fusion protein consisting of N−(+7)Ala and CspB showed an increased amount of α-helical structure compared to the proteins in which CspB was fused to N-WT or N-ΔAla. This increase in α-helical structure originates probably from the seven additional alanine residues, since oligo-alanine stretches have a high helix propensity (Scholtz and Baldwin 1992; Miller et al. 2002). In fact, for the N-terminal domain of PABPN1 alone, an increase in helicity was found when the natural sequence was extended by seven alanines (Scheuermann et al. 2003). The measured CD spectrum of N−(+7)Ala-CspB is very well represented by the sum of the CD spectra of the separate components (N−(+7)Ala and CspB [Fig. 2B]). This provides good evidence that, in the fusion protein, both partners retain the same conformation as in isolation.
Figure 2.

Far-UV CD analysis of the fusion proteins and the separate fusion partners. (A) CD spectra of the fusion proteins of the N-terminal domains of PABPN1 and CspB: N-WT–CspB, solid line; N−(+7)Ala–CspB, dashed line, and N-ΔAla–CspB dotted line. Protein concentrations were 0.8 mg/mL. (B) CD spectra of the N−(+7)Ala–CspB fusion and the single fusion components: N−(+7)Ala–CspB, solid line; CspB, dotted line; N−(+7)Ala, dashed line; addition spectrum of N−(+7)Ala and CspB, dotted-dashed line. Protein concentrations were 0.8 mg/mL for N−(+7)Ala–CspB, 1.2 mg/mL for CspB, and 1.3 mg/mL for N−(+7)Ala.
CspB binds to stretches of single-stranded DNA and RNA (Graumann and Marahiel 1994). Binding can be monitored very well by using an oligonucleotide with seven deoxythymidines (dT7) as a substrate. Upon binding of dT7, the fluorescence of Trp8 of CspB becomes quenched (Lopez et al. 1999, 2001; Lopez and Makhatadze 2000; Zeeb et al. 2006). Thus, dT7 provides a sensitive tool to probe the native structure of CspB. Fluorescence titrations with the dT7 oligonucleotide showed that CspB binds to dT7 with equal efficiency as an isolated protein or when fused to the N-terminal domain of PABN1 (Fig. 3). The almost identical K D values and binding stoichiometries of free and fused CspB (Table 1) confirm that in the soluble forms of all five fusion proteins, CspB is fully folded and functional (see below).
Figure 3.

Binding of dT7 to the fusion proteins between the N-terminal domains of PABPN1 and CspB: N-WT–CspB, black circles; N−(+7)Ala–CspB, filled circles; N-ΔAla–CspB, open circles; CspB, open diamonds. The corresponding K D values are listed in Table 1. Measurements were carried out as described in Materials and Methods. For all measurements a protein concentration of 3 μM was used.
Table 1.
KD values upon dT7 binding of CspB in fusions with the N-terminal domains of PABPN1 and of CspB in direct fusions with polyalanine sequences

The N-terminal domain of PABPN1 is devoid of Trp residues. Therefore, the fluorescence of Trp8 of the CspB moiety of the fusion proteins could be used as a sensitive and selective probe to determine the thermodynamic stability of the CspB moiety by unfolding studies. The urea-induced unfolding and refolding of CspB in the fusion proteins resulted in virtually identical unfolding transition curves (Fig. 4), which gave ΔG(H2O) and m values very similar to those determined previously for isolated CspB (Table 2). Taken together, these data show that the function, structure, and stability of CspB are fully retained in the fusion proteins.
Figure 4.

Urea-induced unfolding transitions of the fusions of the N-terminal domains of PABPN1 with CspB: N-WT–CspB, black circles, solid line; N−(+7)Ala–CspB, gray circles, dashed line; N-ΔAla–CspB, open circles, dotted line. The corresponding ΔG (H2O) and m values are indicated in Table 2. Measurements were carried out in 0.1 M Na-cacodylate/HCl, pH 7 at 25°C.
Table 2.
Thermodynamic analysis of CspB derived from urea-induced unfolding and refolding experiments

Fusions of the N-terminal fragments of PABPN1 with CspB form fibrils in which CspB is active
Previous studies showed for the N-terminal fragments of PABPN1 polyalanine-dependent fibril formation (Scheuermann et al. 2003; Lodderstedt et al. 2007). Thus, we asked whether fibril formation would occur also when folded CspB is fused to the C terminus of the N-terminal domains of PABPN1. Fibril formation was monitored by ANS fluorescence (data not shown), and the existence of fibrils was confirmed by electron microscopy (Fig. 5). Fibril formation required the presence of the polyalanine stretch, since only N−(+7)Ala–CspB and N-WT–CspB, but not N-ΔAla–CspB or CspB alone, formed fibrils under these conditions. The presence of CspB seemed to retard fibril formation (data not shown), but this effect could not be analyzed in a quantitative fashion, because the kinetics of fibril formation varied to some extent for different protein preparations.
Figure 5.

Electron micrographs of fibrillized fusions of the N-terminal domains and CspB. (A) N−(+7)Ala–CspB after incubation for 103 d. (B) N-WT–CspB after incubation for 137 d. The fibrillation conditions were 5 mM KH2PO4, 100 mM NaCl, pH 7.5 at 37°C. The protein concentration was 0.3 mM.
To investigate whether fibril formation affects the folded conformation of CspB, fibrils were sedimented, washed to remove nonfibrillar protein, and the functional state of the cold-shock protein was tested by its binding to dT7. The results in Figure 6 demonstrate that even in the fibrils, CspB is still functional. The quantitative analysis of the binding isotherms (Fig. 6A), measured by fluorescence quenching of Trp8, revealed a 20% decrease upon dT7 binding and a 30% decrease of the binding stoichiometry compared to the corresponding unfibrillized species. Comparable results were obtained when fibrils were analyzed in a sedimentation experiment in the presence of dT7. In this assay, fibrils were incubated with increasing concentrations of dT7 and then sedimented via ultracentrifugation. The quantification of the remaining amount of dT7 in the supernatant resulted in a comparable dT7 binding stoichiometry as in the fluorescence studies (Fig. 6B). Thus, these results show that the majority of the CspB molecules retain their functional state in the fibrillar state. In a reciprocal fashion, this also shows that the folded structure of CspB does not interfere with the conformational changes that occur during the conversion from the soluble to the fibrillar form of the PABPN1 moiety. It remains unclear whether the slight decrease in binding-active CspB molecules is a consequence of fibril formation or simply due to the long incubation times at 37°C that are required for fibril formation.
Figure 6.

dT7 binding of N−(+7)Ala–CspB fibrils. (A) The dT7 binding assay with N−(+7)Ala–CspB fibrils in comparison to the soluble form: N−(+7)Ala–CspB, black circles; N−(+7)Ala–CspB fibrils, open circles. The protein concentration was 3 μM. (B) Titration of dT7. N−(+7)Ala–CspB fibrils (7 μM) were centrifuged after incubation with increasing concentrations of dT7. Quantifications were performed as described in Materials and Methods.
In fibrils originating from direct fusions of polyalanine sequences and CspB, the Csp moiety is unfolded
Next, we examined how fibril formation and the conformation of CspB are affected when it is fused directly to a stretch of alanine residues, without the N-terminal domain of PABPN1 in between. Two fusion proteins were created, in which the N terminus of CspB was linked directly with stretches of 10 or 17 alanines: 10Ala–CspB and 17Ala–CspB, respectively. We refer to them as “direct fusions” in the following (Fig. 1). Both variants were recombinantly expressed in E. coli and purified according to the purification protocol for the fusions of the N-terminal fragments with CspB. Polyalanine-induced structural changes in the soluble forms were analyzed by far UV-CD measurements, which indicated that the two fusion proteins show increased α-helical structures compared to CspB (Fig. 7). This result was expected as α-helical contributions due to alanine residues have been reported for peptides and the N-terminal domain of PABPN1 previously (Miller et al. 2001; 2002; Scheuermann et al. 2003). The native structure of CspB in the fusions was verified by near UV-CD analysis and fluorescence measurements (data not shown). More importantly, the native structure of CspB in these fusion proteins was confirmed by its ability to bind to dT7 (Fig. 8). As in the case of the fusions between the entire N-terminal fragments of PABPN1 and CspB, the direct fusion proteins bound dT7 with affinities and stoichiometries similar to those of free CspB (Table 1).
Figure 7.

Far-UV CD spectra of the direct fusions and free CspB. CspB, solid line; 10Ala–CspB, dashed line; 17Ala–CspB, dotted line. Protein concentrations were 1.2 mg/mL for CspB, 1.0 mg/mL for 17Ala–CspB, and 1.3 mg/mL for 10 Ala–CspB.
Figure 8.

The dT7 binding of the direct fusions and free CspB. CspB, black diamonds, solid line; 10Ala–CspB, gray circles, dashed line; 17Ala–CspB, open circles, dotted line. Derived K D values are given in Table 1. Measurements were carried out as described for Figure 6.
The analysis of the thermodynamic stabilities of 10Ala–CspB and 17Ala–CspB by urea-induced unfolding–refolding transitions revealed that the direct fusion with polyalanine stretches decreased the thermodynamic stability of the CspB moiety (Fig. 9; Table 2). The ΔG (H2O) value for its unfolding is reduced from 12 kJ/mol (free CspB) to ca. 6 kJ/mol in the fusion proteins. Furthermore, the slight decreases of the m values in the fusion proteins indicate a loss in cooperativity (Table 2). Taken together, the direct fusions of polyalanine sequences with CspB display correctly folded CspB, though with a reduced thermodynamic stability.
Figure 9.

Urea-induced unfolding transitions of the direct fusions and free CspB. CspB, black circles, solid line; 10Ala–CspB, gray circles, dashed line; 17Ala–CspB, open circles, dotted line. The corresponding ΔG (H2O) and m values are given in Table 2. Measurements were carried out in 5 mM KH2PO4, 100 mM NaCl, pH 7.5 at 25°C.
In fusions with the entire N-terminal domain of PABPN1, CspB did not inhibit the conversion to the fibrillar state. Thus, we asked whether fibril formation would also occur with the direct fusions. By electron microscopy initially mostly amorphous aggregates instead of the typical fibrillar structures were detected (Fig. 10A,C). Upon prolonged incubation for several months, however, fibrils were observed with 10Ala–CspB (Fig. 10B). In the case of 17Ala–CspB, only in some spots of the EM grids fibrillar structures could be detected (Fig. 10D, arrow). Though fibril formation of this fusion protein was assayed under various temperatures, protein concentrations, and the addition of salts, “pure” fibrils, as with the other fusion proteins, were never observed here. In order to investigate whether fibril formation can be induced by seeds, fibrils of 10Ala–CspB were fragmented and added to both soluble 10Ala–CspB and 17Ala–CspB. In both samples, increases of ANS fluorescence over time were recorded (Fig. 10G). EM analysis revealed that seeding was possible only with 10Ala–CspB (Fig. 10E) while with 17Ala–CspB, fibrillar structures could never be discerned unambiguously (Fig. 10F). The occurrence of increases in ANS fluorescence with samples of 17Ala–CspB is probably due to nonfibrillar aggregates. In contrast, the addition of N−(+7)Ala–CspB seeds to 10Ala–CspB and 17Ala–CspB resulted, in both cases, in fibrillar structures (data not shown). The fact that 10Ala–CspB formed fibrils more readily than 17Ala–CspB was surprising, and can currently only be explained by the higher hydrophobic interactions in the nonfibrillar aggregates of 17Ala–CspB.
Figure 10.

Electron micrographs of the direct fusions. (A,B) 10Ala–CspB after incubation times of 50 and 77 d. (C,D) 17Ala–CspB after incubation times of 50 and 77 d, respectively. 17Ala–CspB was incubated at a protein concentration of 0.5 mM at 30°C, 10Ala–CspB at a protein concentration of 0.7 mM at 37°C. (E) Samples after seeding of 10Ala–CspB and (F) of 17Ala–CspB with 10Ala–CspB seeds (0.1%) after an incubation time of 68 d. 10Ala–CspB was incubated at a protein concentration of 0.5 mM at 37°C and 17Ala–CspB of 0.4 mM at 30°C. (G) Time-dependent ANS fluorescence increases for (E) and (F). 10Ala–CspB (triangles), 17Ala–CspB (circles), open symbols represent seeded reactions, closed symbols the nonseeded reactions.
Next, the structure and function of CspB in the fibrils of 10Ala–CspB were analyzed by tryptophan fluorescence spectroscopy. Upon excitation at 295 nm, the emission maximum was at 340 nm while the maximum of the soluble form was at 352 nm, a result that implies that Trp8 of CspB might be less solvent accessible in the fibrils than in the soluble form (Fig. 11). Addition of dT7 to the fibril sample did not decrease the fluorescence of Trp8, indicating that, in these fibrils, CspB is no longer able to bind dT7. To confirm that CspB activity is not lost due to the long incubation periods but due to unfolding in the fibril, the supernatant of the fibrillation sample, which usually contains a small amount of unfibrillized protein, was tested for dT7 binding. At least 50% of the CspB molecules of the supernatant were active (data not shown).
Figure 11.

Test for dT7 binding of 10Ala–CspB fibrils. Fluorescence spectra of 10Ala–CspB, fibrils (solid line); 10Ala–CspB fibrils in the presence of 5 μM dT7 (dashed line); 10Ala–CspB in the soluble state (dotted line). Protein concentration was 2 μM, details are described in Materials and Methods; AU, arbitrary units.
Taken together, our results demonstrate that a β-cross-structure and a folded and functional protein domain can coexist in fibrils provided that the fibrillar structure is not perturbed by the folded domain and vice versa. When CspB is separated from the polyalanine stretch by the N-terminal domain of PABPN1, it remains correctly folded and functional in the fibrils. In contrast, when it is fused directly to the polyalanine sequence, it loses its native structure and function. The presence of native domains in fibrils is not unprecedented, and has been shown for Sup35p, for example. In fibrils of full-length Sup35p, the GTPase activity of the C-terminal domain was retained (Krzewska et al. 2007). Similarly, the Ure2 protein is natively folded in the prion state (Bai et al. 2004). Furthermore, when the activities of four C-terminally linked partner proteins in fusions with amyloidogenic Ure2p domains were tested in the fibrillar state, two of them retained their full, and the other two, reduced activity indicating that the fibrillar state can tolerate natively folded domains in the neighborhood (Baxa et al. 2002, 2003). Toleration of an enzymatically active, native-like state has been demonstrated for fibrils arising from domain swapped RNase A (Sambashivan et al. 2005).
We assume that in the fusions with the entire N-terminal domain of PABPN1, CspB retains its folded and functional state in fibrils probably because the major part of the N-terminal segment is flexible and not integrated into the fibrillar core. Thus, it represents a spacer between the β-cross-structure of the fibril and CspB. Studies to determine which part of the N-terminal domain is buried in the β-cross-structure are underway.
In fibrils of the direct fusions, CspB loses conformational stability, presumably because the spacer sequence is removed. Interference of the polyalanine sequence with the native structure of CspB in the soluble form was excluded by spectroscopic analysis and dT7 binding. Still, the presence of the N-terminal polyalanine sequence reduced the thermodynamic stability of CspB. The altered thermodynamic stability probably explains the aggregate formation of these fusions upon incubation. Possibly, with time, the aggregates may convert into fibrillar species. Further experiments have to be performed to clarify whether fibrillation and aggregation are independent or associated processes. In addition, it remains to be tested whether the conversion of the aggregates into fibrils proceeds via folded or unfolded CspB intermediates.
This biochemical analysis of fibril formation employed artificial fusion proteins, but still some tentative conclusion can be drawn with regard to the development of the OPMD disease. The folded state of CspB in fibrils of the fusions with the N-terminal domains could indicate that also in fibrils of PABPN1, the RNP domain, lying C-terminal to the N-terminal domain may retain its biological activity in OPMD tissue. In this case, loss of PABPN1 function might not be the cause for the symptoms observed in OPMD patients.
Materials and Methods
Recombinant constructs
For the fusions encoding the N-terminal part of PABPN1 and CspB, the two DNA fragments were amplified by the following primers: forward or external primer I (complementary to the sequence encoding the N-terminal domain of PABPN1): 5′-CCGCGCGGCAGCCATATGGCAGCA-3′ for N−(+7)Ala and N-WT; 5′-CCGCGCGGCAGCCATATGGCAGGA-3′ for N-ΔAla. NdeI restriction sites are underlined. For reverse primer (reverse complementary to the sequence encoding the C-terminal part of the PABPN1 fragment and providing an overlap with the CspB coding sequence = fusion primer): 5′-CCATTTTACTTTACCTTCTAACATTCGAGCTTTTATTGCTTCTAA-3′. For amplification of the CspB coding part the reverse complementary sequence of the fusion primer and the reverse or external primer II: 5′-CGGGATCCTTACGCTTCTTTAGTAACGTT-3′ containing a BamHI restriction site (underlined) were used. After amplification, the overlapping fragments were combined in a fusion PCR and amplified with both external primers. The fusion constructs were restricted with NdeI and BamHI and cloned into pET15b (Novagen).
Fusions encoding CspB preceded by alanine sequences (direct fusions) were generated by deletion of the sequence encoding the N-terminal part of PABPN1 between the alanine and CspB encoding sequence of the N-terminal PABPN1–CspB constructs. The deletion was performed via the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) with DNA constructs encoding for the N-terminal PABPN1–CspB fusions as templates. The forward primer had the sequence: 5′-GCAGCAGCAGCAGGAGCAGCATTAGAAGGTAAAGTAAAATGG-3′. The reverse primer had the reverse complementary sequence. PCR products were treated with DpnI and transformed in E. coli (BL21 [DE3]).
Recombinant protein expression and purification
Recombinant DNA constructs were expressed in E. coli (strain BL 21 [DE3]) that had been transformed in addition with the plasmid pUBS520 providing additional copies for a rare tRNA in E. coli (Brinkmann et al. 1989). Cultivation was performed in shake flasks. Cells were induced with 1 mM IPTG at OD600 = 0.5–0.8 and harvested after 4 h. Cell pellets were resuspended at a ratio of 0.25 g/mL in 50 mM Tris/HCl, pH 8, containing protease inhibitor complete, EDTA-free (Roche). Cells were disrupted by high-pressure treatment with Micron Lab 40 (APV Homogeniser) reaching a maximal pressure of 1200 bar. Cell debris were sedimented by centrifugation. The supernatant was incubated for 30 min at 50°C and recentrifuged. The protein solution was dialyzed against 6 M GdmCl, 50 mM Tris/HCl, pH 8, centrifuged and loaded on a Ni-NTA column (His Bind Resin, Novagen) (V = 50 mL). Washing and refolding of CspB was performed by using 50 mM Tris/HCl, 20 mM imidazole, pH 8. The washing step was stopped when a stable UV-absorbance signal (280 nm) was observed. Elution was achieved by a continuous gradient from 20 to 250 mM imidazole within three column volumes. During elution, 5 mM EDTA was added to the samples. The pooled fractions containing the fusions were concentrated with PEG 35,000 (Fluka) and further purified via gel filtration (HiLoad Superdex 75 prep grade, Amersham Biosciences). Purified proteins were dialyzed against 5 mM KH2PO4, 100 mM NaCl, pH 7.5 and stored at –80°C.
Determination of the protein concentration
Protein concentrations of the soluble species were calculated by the absorbance at 280 nm. The respective molar extinction coefficients were determined as 6990 M−1 cm−1 for the fusions consisting of the N-terminal domains and CspB and as 5500 M−1 cm−1 for the fusion consisting of polyalanine sequences directly coupled to CspB (Gill and von Hippel 1989). The accuracy of the determinations was confirmed by an amino acid analysis (carried out by the Center for Protein Analytics, FH Bingen).
Protein concentrations of fibrils were calculated by determination of the amount of the unique tryptophan residue (in CspB) under denaturing conditions (fibrils of the fusion of the N-terminal domain of PABPN1 and CspB could not be solubilized by 6 M guanidinium hydrochloride [GdmCl], which leads only to the denaturation of CspB moiety) (Pajot 1976). Fibrils were diluted in 2 mL of 6 M GdmCl, 5 mM KH2PO4, 100 mM NaCl, pH 7.5. Tryptophan was added in 1-μL steps from a 982-μM stock solution, and the sample was gently stirred for 1 min. Fluorescence was recorded with a Jobin Yvon Fluoromax2 equipped with a thermostatic cell holder connected to a circulating water bath. Measurements were performed at 20°C. The excitation of tryptophan fluorescence was carried out at 295 nm. Spectra were accumulated three times and averaged. Tryptophan emission was monitored at 361 nm. Fluorescence signals were buffer- and volume-change corrected. With increases of the tryptophan concentrations, a linear increase of the fluorescence intensity was observed. The intersection point of the curve with the Y-axis was used for the determination of the initial concentration of tryptophan in the sample that could be correlated with the amount of protein in the fibril. Calibration was performed with soluble proteins and resulted in correction factors of 0.64 and 0.77, for the fusions consisting of the N-terminal domains and CspB and the direct fusions, respectively.
Circular dichroism (CD) and fluorescence spectroscopy
CD spectra were recorded with a Jasco J810 spectropolarimeter in 0.1 mm cuvettes in 5 mM KH2PO4, 100 mM NaCl, pH 7.5 at 25°C. The spectra were measured 10 times and averaged. Data were collected in 0.1-nm steps. Spectra were buffer corrected. Tryptophan fluorescence was recorded with a Jobin Yvon Fluoromax2 equipped with a thermostatted cell holder. Measurements were performed in 5 mM KH2PO4, 100 mM NaCl, pH 7.5 at 20°C. The excitation wavelength was 295 nm at a bandwidth of 5 nm. Spectra were recorded in 1-cm cuvettes at a bandwidth of 5 nm and accumulated three times. Data were averaged and buffer corrected.
dT7 Binding assay
The single-stranded DNA fragment dT7 5′-TTTTTTT-3′ was purchased from Thermo Electron GmbH (Germany). Concentrations of dT7 were calculated by the absorbance at 260 nm. The extinction coefficient of dT7 at 260 nm was determined as 58,800 M−1 cm−1 according to Wallace and Miyada (1987). Binding was measured by fluorescence (see above). All measurements were performed at 20°C. Proteins were diluted to final concentrations of 1–3 μM in a volume of 2 mL. The assay buffer was 5 mM KH2PO4, 100 mM NaCl, pH 7.5. dT7 was added in steps of 1 μL from a 400-μM stock solution. Thus, the dilution effect was below 2.25%. After adding dT7 to protein solutions, samples were gently stirred for 1 min. The excitation wavelength was 295 nm. A slit width of 5 nm was applied. Spectra were accumulated three times, averaged, and buffer corrected. Fluorescence quench calculations were based on changes at 352 nm. Intensity changes with increasing dT7 concentrations revealed hyperbolic binding isotherms, which were analyzed according to Equation 1 (Lohman and Bujalowski 1991; Eftink 1997):
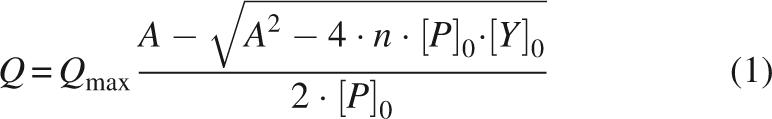 |
with A = K D + [P]0 + n [Y]0, where Q is the quenching of the intrinsic fluorescence intensity of the Trp8; Q max represents the maximum quenching upon complete saturation of the protein with dT7; [P]0 and [Y]0 are the overall protein and dT7 concentrations; n is the stoichiometry of the protein–ssDNA complex (number of protein molecules bound to one molecule dT7), and K D is the equilibrium dissociation constant of the complex.
dT7 Sedimentation assay
Fibrils at a concentration of 7 μM were incubated under gentle shaking with increasing concentrations of dT7 at 20°C for 30 min. Subsequently, the samples were centrifuged for 30 min at 35,000 rpm (OptimaTM TLX ultracentrifuge). The concentration of unbound dT7 in the supernatant was quantified by the absorbance at 260 nm. The binding stoichiometry was determined by the intersection of the slope corresponding to the increase of unbound dT7 with the baseline caused by CspB binding.
Fibril formation and fibril analysis by ANS (8-anilino-1-naphtalene sulfonic acid) fluorescence
Proteins were dissolved to final concentrations of 0.3–0.7 mM and incubated at 30°C for 17Ala–CspB and at 37°C for 10Ala–CspB, and the fusions of the N-terminal domains and CspB. The buffer was 5 mM KH2PO4, pH 7.5 containing 100 mM NaCl. Traces of NaN3 were added to the reaction. Seeded reactions contained 0.1% (w/v) 10Ala–CspB seeds. For determining ANS fluorescence, samples were briefly mixed and aliquots were diluted into 50 μM ANS in 5 mM KH2PO4, 100 mM NaCl, pH 7.5 at final concentrations of 5 μM. Fluorescence was recorded at an emission wavelength of 480 nm upon excitation at 370 nm with a Jobin Yvon Fluoromax2 at 20°C. Experiments were performed in 1-cm cuvettes with an excitation and emission slit width of 5 nm.
Urea-induced unfolding and refolding transitions
Urea (Biochemika Ultra from Fluka) concentrations were determined by refraction (Pace 1986). Fusions of the N-terminal domains and CspB were diluted to concentrations of 2.5 μM in 0.1 M Na-cacodylate/HCl pH 7, the direct fusions and free CspB were diluted to concentrations of 3.0 μM in 5 mM KH2PO4, 100 mM NaCl, pH 7.5, containing various urea concentrations. Denaturation was allowed to proceed for 1 h at 25°C. Fluorescence was recorded at an emission wavelength of 345 nm upon excitation at 295 nm with a Jobin Yvon Fluoromax2 at 25°C. Experiments were performed in 1-cm cuvettes with excitation and emission band widths of 5 nm each. The unfolding transitions were analyzed by assuming a two-state transition between the folded (N) and unfolded (U) conformation. For obtaining the thermodynamic parameters ΔG(H2O) and m values, nonlinear least-square fits (Santoro and Bolen 1988) were performed.
Miscellaneous
For separation of fibrils from remaining soluble species, fibril solutions were centrifuged at 260,000g (OptimaTM TLX ultracentrifuge) for 1 h, washed in 5 mM KH2PO4, 100 mM NaCl, pH 7.5 containing 6 M GdmCl for the fusions of the N-terminal domain and 2 M GdmCl for the direct fusions. After recentrifugation, fibrils were washed in 5 mM KH2PO4, 100 mM NaCl, pH 7.5, again centrifuged, and resuspended in the same buffer.
The seed and sample preparations for electron microscopy were carried out as described (Lodderstedt et al. 2007).
Acknowledgments
We thank Jochen Balbach for advice with dT7 binding assays, and Rainer Rudolph and Elmar Wahle for critical comments on the manuscript. This work was funded by the Deutsche Forschungsgemeinschaft (DFG) through Graduiertenkolleg 1026 (Conformational transitions in macromolecular interactions).
Footnotes
Reprint requests to: Elisabeth Schwarz, Institut für Biochemie und Biotechnologie, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Strasse 3, 06120 Halle, Germany; e-mail: Elisabeth.Schwarz@biochemtech.uni-halle.de; fax: 49-345-55-27-013.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073276308.
References
- Abu-Baker, A., Rouleau, G.A. Oculopharyngeal muscular dystrophy: Recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim Biophys Acta. 2007;1772:173–185. doi: 10.1016/j.bbadis.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Abu-Baker, A., Messaed, C., Laganiere, J., Gaspar, C., Brais, B., Rouleau, G.A. Involvement of the ubiquitin–proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2003;12:2609–2623. doi: 10.1093/hmg/ddg293. [DOI] [PubMed] [Google Scholar]
- Albrecht, A., Mundlos, S. The other trinucleotide repeat: Polyalanine expansion disorders. Curr. Opin. Genet. Dev. 2005;15:285–293. doi: 10.1016/j.gde.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Amiel, J., Trochet, D., Clement-Ziza, M., Munnich, A., Lyonnet, S. Polyalanine expansions in human. Hum. Mol. Genet. 2004;13(Review issue 2):R235–R243. doi: 10.1093/hmg/ddh251. [DOI] [PubMed] [Google Scholar]
- Andrew, S.E., Goldberg, Y.P., Kremer, B., Telenius, H., Theilmann, J., Adam, S., Starr, E., Squitieri, F., Lin, B., Kalchman, M.A., et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat. Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- Bai, M., Zhou, J.M., Perrett, S. The yeast prion protein Ure2 shows glutathione peroxidase activity in both native and fibrillar forms. J. Biol. Chem. 2004;279:50025–50030. doi: 10.1074/jbc.M406612200. [DOI] [PubMed] [Google Scholar]
- Bao, Y.P., Cook, L.J., O'Donovan, D., Uyama, E., Rubinsztein, D.C. Mammalian, yeast, bacterial, and chemical chaperones reduce aggregate formation and death in a cell model of oculopharyngeal muscular dystrophy. J. Biol. Chem. 2002;277:12263–12269. doi: 10.1074/jbc.M109633200. [DOI] [PubMed] [Google Scholar]
- Bao, Y.P., Sarkar, S., Uyama, E., Rubinsztein, D.C. Congo red, doxycycline, and HSP70 overexpression reduce aggregate formation and cell death in cell models of oculopharyngeal muscular dystrophy. J. Med. Genet. 2004;41:47–51. doi: 10.1136/jmg.2003.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxa, U., Speransky, V., Steven, A.C., Wickner, R.B. Mechanism of inactivation on prion conversion of the Saccharomyces cerevisiae Ure2 protein. Proc. Natl. Acad. Sci. 2002;99:5253–5260. doi: 10.1073/pnas.082097899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxa, U., Taylor, K.L., Wall, J.S., Simon, M.N., Cheng, N., Wickner, R.B., Steven, A.C. Architecture of Ure2p prion filaments: The N-terminal domains form a central core fiber. J. Biol. Chem. 2003;278:43717–43727. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages, M., Zala, D., Humbert, S., Saudou, F. Huntington's disease: From huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brais, B., Bouchard, J.P., Xie, Y.G., Rochefort, D.L., Chretien, N., Tome, F.M., Lafreniere, R.G., Rommens, J.M., Uyama, E., Nohira, O., et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- Brinkmann, U., Mattes, R.E., Buckel, P. High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene. 1989;85:109–114. doi: 10.1016/0378-1119(89)90470-8. [DOI] [PubMed] [Google Scholar]
- Calado, A., Tome, F.M., Brais, B., Rouleau, G.A., Kühn, U., Wahle, E., Carmo-Fonseca, M. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum. Mol. Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- Chartier, A., Benoit, B., Simonelig, M. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J. 2006;25:2253–2262. doi: 10.1038/sj.emboj.7601117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti, F., Webster, P., Taddei, N., Clark, A., Stefani, M., Ramponi, G., Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. 1999a;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti, F., Taddei, N., White, P.M., Bucciantini, M., Magherini, F., Stefani, M., Dobson, C.M. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 1999b;6:1005–1009. doi: 10.1038/14890. [DOI] [PubMed] [Google Scholar]
- Davies, J.E., Wang, L., Garcia-Oroz, L., Cook, L.J., Vacher, C., O'Donovan, D.G., Rubinsztein, D.C. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 2005;11:672–677. doi: 10.1038/nm1242. [DOI] [PubMed] [Google Scholar]
- Davies, J.E., Sarkar, S., Rubinsztein, D.C. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2006;15:23–31. doi: 10.1093/hmg/ddi422. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. Fluorescence methods for studying equilibrium macromolecule–ligand interactions. Methods Enzymol. 1997;278:221–257. doi: 10.1016/s0076-6879(97)78013-3. [DOI] [PubMed] [Google Scholar]
- Everett, C.M., Wood, N.W. Trinucleotide repeats and neurodegenerative disease. Brain. 2004;127:2385–2405. doi: 10.1093/brain/awh278. [DOI] [PubMed] [Google Scholar]
- Fan, X., Dion, P., Laganiere, J., Brais, B., Rouleau, G.A. Oligomerization of polyalanine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death. Hum. Mol. Genet. 2001;10:2341–2351. doi: 10.1093/hmg/10.21.2341. [DOI] [PubMed] [Google Scholar]
- Gill, S.C., von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Graumann, P., Marahiel, M.A. The major cold shock protein of Bacillus subtilis CspB binds with high affinity to the ATTGG- and CCAAT sequences in single stranded oligonucleotides. FEBS Lett. 1994;338:157–160. doi: 10.1016/0014-5793(94)80355-2. [DOI] [PubMed] [Google Scholar]
- Hill, D.T., Payne, V.W.E., Rogers, J.W., Kown, S.R., Tomé, F.M.S., Chateau, D., Helbling-Leclerc, A., Fardeau, M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul. Disord. 1997;7(Suppl 1):S63–S69. doi: 10.1016/S0960-8966(97)00085-0. [DOI] [PubMed] [Google Scholar]
- Krzewska, J., Tanaka, M., Burston, S.G., Melki, R. Biochemical and functional analysis of the assembly of full-length Sup35p and its prion-forming domain. J. Biol. Chem. 2007;282:1679–1686. doi: 10.1074/jbc.M608110200. [DOI] [PubMed] [Google Scholar]
- Kühn, U., Wahle, E. Structure and function of poly(A) binding proteins. Biochim. Biophys. Acta. 2004;1678:67–84. doi: 10.1016/j.bbaexp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Kühn, U., Nemeth, A., Meyer, S., Wahle, E. The RNA binding domains of the nuclear poly(A)-binding protein. J. Biol. Chem. 2003;278:16916–16925. doi: 10.1074/jbc.M209886200. [DOI] [PubMed] [Google Scholar]
- Lazo, N.D., Maji, S.K., Fradinger, E.A., Bitan, G., Teplow, D.B. The amyloid β-protein. In: Sipe J., editor. Amyloid proteins; The β-sheet conformation and disease. Wiley-VCH Verlag GmbH; Germany: 2005. pp. 385–491. [Google Scholar]
- Lodderstedt, G., Hess, S., Hause, G., Scheuermann, T., Scheibel, T., Schwarz, E. Effect of oculopharyngeal muscular dystrophy-associated extension of seven alanines on the fibrillation properties of the N-terminal domain of PABPN1. FEBS J. 2007;274:346–355. doi: 10.1111/j.1742-4658.2006.05595.x. [DOI] [PubMed] [Google Scholar]
- Lodderstedt, G., Sachs, R., Faust, J., Bordusa, F., Kühn, U., Golbik, R., Kerth, A., Wahle, E., Balbach, J., Schwarz, E. Hofmeister salts and potential therapeutic compounds accelerate in vitro fibril formation of the N-terminal domain of PABPN1 containing a disease-causing alanine extension. Biochemistry. 2008;47:2181–2189. doi: 10.1021/bi701322g. [DOI] [PubMed] [Google Scholar]
- Lohman, T.M., Bujalowski, W. Thermodynamic methods for model-independent determination of equilibrium binding isotherms for protein–DNA interactions: Spectroscopic approaches to monitor binding. Methods Enzymol. 1991;208:258–290. doi: 10.1016/0076-6879(91)08017-c. [DOI] [PubMed] [Google Scholar]
- Lopez, M.M., Makhatadze, G.I. Major cold shock proteins, CspA from Escherichia coli and CspB from Bacillus subtilis, interact differently with single-stranded DNA templates. Biochim. Biophys. Acta. 2000;1479:196–202. doi: 10.1016/s0167-4838(00)00048-0. [DOI] [PubMed] [Google Scholar]
- Lopez, M.M., Yutani, K., Makhatadze, G.I. Interactions of the major cold shock protein of Bacillus subtilis CspB with single-stranded DNA templates of different base composition. J. Biol. Chem. 1999;274:33601–33608. doi: 10.1074/jbc.274.47.33601. [DOI] [PubMed] [Google Scholar]
- Lopez, M.M., Yutani, K., Makhatadze, G.I. Interactions of the cold shock protein CspB from Bacillus subtilis with single-stranded DNA. Importance of the T base content and position within the template. J. Biol. Chem. 2001;276:15511–15518. doi: 10.1074/jbc.M010474200. [DOI] [PubMed] [Google Scholar]
- Max, K.E., Zeeb, M., Bienert, R., Balbach, J., Heinemann, U. T-rich DNA single strands bind to a preformed site on the bacterial cold shock protein Bs-CspB. J. Mol. Biol. 2006;360:702–714. doi: 10.1016/j.jmb.2006.05.044. [DOI] [PubMed] [Google Scholar]
- Miller, J.S., Kennedy, R.J., Kemp, D.S. Short, solubilized polyalanines are conformational chameleons: Exceptionally helical if N- and C-capped with helix stabilizers, weakly to moderately helical if capped with rigid spacers. Biochemistry. 2001;40:305–309. doi: 10.1021/bi0019500. [DOI] [PubMed] [Google Scholar]
- Miller, J.S., Kennedy, R.J., Kemp, D.S. Solubilized, spaced polyalanines: A context-free system for determining amino acid α-helix propensities. J. Am. Chem. Soc. 2002;124:945–962. doi: 10.1021/ja011726d. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- Pajot, P. Fluroescence of proteins in 6-M guanidine hydrochloride. A method for the quantitative determination of tryptophan. Eur. J. Biochem. 1976;63:263–269. doi: 10.1111/j.1432-1033.1976.tb10228.x. [DOI] [PubMed] [Google Scholar]
- Pedersen, J.S., Dikov, D., Flink, J.L., Hjuler, H.A., Christiansen, G., Otzen, D.E. The changing face of glucagon fibrillation: Structural polymorphism and conformational imprinting. J. Mol. Biol. 2006;355:501–523. doi: 10.1016/j.jmb.2005.09.100. [DOI] [PubMed] [Google Scholar]
- Petkova, A.T., Leapman, R.D., Guo, Z., Yau, W.M., Mattson, M.P., Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Sambashivan, S., Liu, Y., Sawaya, M.R., Gingery, M., Eisenberg, D. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature. 2005;437:266–269. doi: 10.1038/nature03916. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M., Bolen, D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- Scheibel, T., Buchner, J. Protein aggregation as a cause for disease. Handb. Exp. Pharmacol. 2006;172:199–219. doi: 10.1007/3-540-29717-0_9. [DOI] [PubMed] [Google Scholar]
- Scheuermann, T., Schulz, B., Blume, A., Wahle, E., Rudolph, R., Schwarz, E. Trinucleotide expansions leading to an extended poly-L-alanine segment in the poly (A) binding protein PABPN1 cause fibril formation. Protein Sci. 2003;12:2685–2692. doi: 10.1110/ps.03214703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, H., Marahiel, M.A., Heinemann, U. Universal nucleic acid-binding domain revealed by crystal structure of the B. subtilis major cold-shock protein. Nature. 1993;364:164–168. doi: 10.1038/364164a0. [DOI] [PubMed] [Google Scholar]
- Schindler, T., Herrler, M., Marahiel, M.A., Schmid, F.X. Extremely rapid protein folding in the absence of intermediates. Nat. Struct. Biol. 1995;2:663–673. doi: 10.1038/nsb0895-663. [DOI] [PubMed] [Google Scholar]
- Schnuchel, A., Wiltscheck, R., Czisch, M., Herrler, M., Willimsky, G., Graumann, P., Marahiel, M.A., Holak, T.A. Structure in solution of the major cold-shock protein from Bacillus subtilis . Nature. 1993;364:169–171. doi: 10.1038/364169a0. [DOI] [PubMed] [Google Scholar]
- Scholtz, J.M., Baldwin, R.L. The mechanism of α-helix formation by peptides. Annu. Rev. Biophys. Biomol. Struct. 1992;21:95–118. doi: 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Soto, C., Estrada, L., Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2006;31:150–155. doi: 10.1016/j.tibs.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Tanaka, M., Chien, P., Naber, N., Cooke, R., Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- Tomé, F.M., Chateau, D., Helbling-Leclerc, A., Fardeau, M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul. Disord. 1997;7:S63–S69. doi: 10.1016/s0960-8966(97)00085-0. [DOI] [PubMed] [Google Scholar]
- Wahle, E. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- Wahle, E., Ruegsegger, U. 3′-End processing of pre-mRNA in eukaryotes. FEMS Microbiol. Rev. 1999;23:277–295. doi: 10.1111/j.1574-6976.1999.tb00400.x. [DOI] [PubMed] [Google Scholar]
- Wallace, R.B., Miyada, C.G. Oligonucleotide probes for the screening of recombinant DNA libraries. Methods Enzymol. 1987;152:432–442. doi: 10.1016/0076-6879(87)52050-x. [DOI] [PubMed] [Google Scholar]
- Wang, Q., Mosser, D.D., Bag, J. Induction of HSP70 expression and recruitment of HSC70 and HSP70 in the nucleus reduce aggregation of a polyalanine expansion mutant of PABPN1 in HeLa cells. Hum. Mol. Genet. 2005;14:3673–3684. doi: 10.1093/hmg/ddi395. [DOI] [PubMed] [Google Scholar]
- Zeeb, M., Balbach, J. Single-stranded DNA binding of the cold-shock protein CspB from Bacillus subtilis: NMR mapping and mutational characterization. Protein Sci. 2003;12:112–123. doi: 10.1110/ps.0219703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeb, M., Max, K.E., Weininger, U., Low, C., Sticht, H., Balbach, J. Recognition of T-rich single-stranded DNA by the cold shock protein Bs-CspB in solution. Nucleic Acids Res. 2006;34:4561–4571. doi: 10.1093/nar/gkl376. [DOI] [PMC free article] [PubMed] [Google Scholar]