

Abstract
Earlier studies have shown that Kaposi sarcomas contain cells infected with human herpesvirus (HHV) 6B, and in current studies we report that both AIDS-associated and classic-sporadic Kaposi sarcoma contain HHV-7 genome sequences detectable by PCR. To determine the distribution of HHV-7-infected cells relative to those infected with HHV-6, sections from paraffin-embedded tissues were allowed to react with antibodies to HHV-7 virion tegument phosphoprotein pp85 and to HHV-6B protein p101. The antibodies are specific for HHV-7 and HHV-6B, respectively, and they retained reactivity for antigens contained in formalin-fixed, paraffin-embedded tissue samples. We report that (i) HHV-7 pp85 was present in 9 of 32 AIDS-associated Kaposi sarcomas, and in 1 of 7 classical-sporadic HIV-negative Kaposi sarcomas; (ii) HHV-7 pp85 was detected primarily in cells bearing the CD68 marker characteristic of the monocyte/macrophage lineage present in or surrounding the Kaposi sarcoma lesions; and (iii) in a number of Kaposi sarcoma specimens, tumor-associated CD68+ monocytes/macrophages expressed simultaneously antigens from both HHV-7 and HHV-6B, and therefore appeared to be doubly infected with the two viruses. CD68+ monocytes/macrophages infected with HHV-7 were readily detectable in Kaposi sarcoma, but virtually absent from other normal or pathological tissues that harbor macrophages. Because all of the available data indicate that HHV-7 infects CD4+ T lymphocytes, these results suggest that the environment of the Kaposi sarcoma (i) attracts circulating peripheral lymphocytes and monocytes, triggers the replication of latent viruses, and thereby increases the local concentration of viruses, (ii) renders CD68+ monocytes/macrophages susceptible to infection with HHV-7, and (iii) the combination of both events enables double infections of cells with both HHV-6B and HHV-7.
In the last decade three hitherto unknown viruses designated human herpesviruses (HHV) 6A, 6B, and 7 have been shown to infect peripheral blood cells (1–3). Although HHV-6A and HHV-6B are closely related (4–6) they differ in human epidemiology and disease association. While HHV-6A has been isolated from a number of disease states, it has been associated with none (see refs. 7, 8). HHV-6B, a more common human pathogen, is associated with exanthem subitum of young children (9), and in immunocompromized adults it has been reported to cause more severe infections (10–13, see ref. 8). The virus can be recovered from T lymphocytes (1–3) and macrophages (14) and therefore is thought to remain in latent state in these cells. It can be induced in virologically negative lymphocyte cultures by infection with HHV-7 (15). Less is known about the association of HHV-7 infection with human diseases, despite the fact that the sequence of the entire genome has been reported recently (16). This virus causes widespread infections in young children of somewhat older age than those afflicted with HHV-6B, and on rare occasions it has been associated with exanthem subitum or febrile illness (17–21). The virus then persists in the body and is shed in the saliva (22–26). Mainly because HHV-7 has been isolated from peripheral blood lymphocytes and grows in CD4+ T lymphocytes (3, 27, 28), and by analogy with HHV-6B, it has been postulated that HHV-7 is harbored in latent form in T lymphocytes. In vitro the susceptibility to HHV-7 is restricted to CD4+ T lymphocytes (refs. 3 and 27, see ref. 29) and indeed the CD4 molecule appears to be a component of the viral receptor (30, 31). Attempts to infect terminally differentiated macrophages have not been successful (32).
Unlike HHV-6B, which has been detected in a variety of cells, tissues, and disease states (10–13, 33–37), cells productively infected with HHV-7 have been reported only in salivary glands (22–26). In an attempt to define the conditions that lead to productive infection in other tissues, we analyzed AIDS-associated and classic-sporadic Kaposi sarcoma specimens in the expectation that in these tissues the reactivation frequency would be higher than in other normal or pathological tissues. Indeed, numerous herpesviruses, including HHV-6 and human cytomegalovirus (HCMV), have been detected in Kaposi sarcoma lesions (ref. 38, see ref. 39). For HHV-6, antigenic expression was detected (38). For HCMV, expression of viral antigens is controversial (see ref. 39).
Research on HHV-7 has been impaired by difficulties in growing the virus in cell cultures and the scarcity of specific antibodies that differentiate between HHV-7 and HHV-6. The major proteins specific for HHV-7-infected cells were identified by means of mouse and rabbit polyvalent sera and a few mAb (40). Two mAbs (5E1 and 3B1) recognize a family of at least five antigenically related proteins with apparent Mr of 87,000, 85,000, 70,000, 61,000, and 57,000 (40). Of these the Mr 85,000 species designated pp85 is phosphorylated (41). The proteins of the pp85 complex are located to the virion tegument (41). Phosphoprotein 85 is the immunodominant HHV-7 antigen in the human population (42, 43).
We report that with the aid of mAb 5E1 we detected the presence of HHV-7 pp85 in CD68+ monocytes/macrophages present in Kaposi sarcoma lesions. This mAb is particularly suitable for in situ detection of HHV-7 productive infection inasmuch as it reacts with a conformation-independent epitope of an abundant, HHV-7-specific tegument protein. Inasmuch as the vast majority of the cells staining positively with mAb 5E1 belonged to the CD68+ lineage, and pp85 is a structural component of the virion, its presence in tumor-associated monocytes/macrophages within Kaposi sarcoma tissue indicates that HHV-7 causes a productive infection of these cells. Macrophages expressing the HHV-7 structural antigen were a relatively common feature in Kaposi sarcoma tissue, but could be detected only rarely in other normal or pathological tissues that harbor macrophages, e.g., lungs and liver (W.K., V.A., N.W., R.M., M.S., P. Mizandola, L. Menotti, and G.C.F., unpublished work). We interpret the results to suggest that susceptibility to infection with HHV-7 is altered in the environment of Kaposi sarcoma. We also report that in some of the HHV-7-positive specimens CD68+ monocytes/macrophages simultaneously expressed both the HHV-7 pp85 and the HHV-6B antigen p101.
MATERIALS AND METHODS
Cells and Viruses.
Cord blood mononuclear cells were grown as described (40). Briefly, cells were harvested by Ficoll gradient centrifugation and grown for 2 days in RPMI medium 1640 containing 10% inactivated fetal calf serum, 5 units of phytohemagglutinin, and 5 units of recombinant interleukin 2 before infection. The viruses used were HHV-6B(Z29) (2) and HHV-7(RK) (3). Progression of infection was monitored by immunofluorescence on cells fixed with cold methanol. All other details were previously described (40).
Antibodies.
The mAbs used in these studies were 5E1 specific for HHV-7 pp85 (40), commercially available mAb (no. 8535, Chemicon) specific for p101 of HHV-6B (44), mAb to HCMV (M757, Dako), mAb (Dako-CD68 KP1, no. M848) reacting with the KP1 epitope of CD68, a specific marker of macrophages, mAb M616 to factor VIII, mAb M823 to CD31, the latter four from Dako. The human papilloma virus antibody was a polyclonal antibody directed to L1 capsid proteins of the most known papillomaviruses (Dako LSAB, code no. N1547).
Tissue Specimens.
Thirty-two cutaneous Kaposi sarcoma specimens from 32 male AIDS patients (20 biopsy and 12 autopsy specimens) and seven tumor samples from seven HIV-seronegative male patients suffering from classic-sporadic Kaposi sarcoma were examined. All patients were from Switzerland. Additionally, seven histologically normal, autoptic specimens of parotid gland from HIV-seronegative patients were evaluated. All tissue specimens were routinely fixed in 10% buffered formalin and embedded in paraffin. Serial sections with a maximum thickness of 4 μm were stained with different antibodies. This allowed the identification of the same cell in at least two serial sections.
PCR Analyses.
DNA was extracted from deparaffinated tissues by proteinase K digestion according to standard procedures. Successful amplification of a β-globin fragment (268 bp in length) indicated that the samples were adequate for PCR analysis and that no inhibitors were present. To avoid contamination and product carryover, sterile materials were used throughout the procedure, and the microtome blade was cleaned extensively with xylene after cutting of each specimen. Occasionally, tissue specimens other than Kaposi sarcoma were included among the specimens being cut, and then processed for PCR; they resulted constantly negative, assuring that there was no contamination due to DNA carryover. DNA extraction, PCR, and gel electrophoresis were done in separate laboratories.
The primers HV7 and HV8 (28) were used to amplify HHV-7-specific DNA sequences. The cycling conditions were 30 sec for denaturation at 96°C, 30 sec for annealing of the primers at 58°C, and 40 sec extension at 72°C. After 35 cycles, an aliquot (5 μl) of the first PCR reaction was used as template for the nested PCR amplification (total volume of 50 μl). Internal primers designated HV10 and HV11 were used for the nested PCR amplification. The PCR conditions were the same as those listed for the primers HV7 and HV8. The amplification product (124 bp) was subjected to electrophoresis on a 2% agarose gel and stained with ethidium bromide.
HHV-6 specific sequences were detected by means of a nested PCR described in detail elsewhere (45). The amplification product (92 bp) was analyzed on a 2% agarose gel and stained with ethidium bromide. An aliquot of the nested PCR product was digested with the restriction enzyme HinfI to distinguish between HHV-6 type A and B (45). The specificity of the amplification products was confirmed by direct sequence analysis. Positive and negative controls were included as a routine for both HHV-7 and HHV-6. The positive controls consisted of DNA extracted from HHV-7- and HHV-6-infected cord blood mononuclear cells, respectively. The negative controls consisted of PCR reactions containing all reagents without DNA, or DNA from uninfected cord blood mononuclear cells.
Immunohistochemistry (IHC).
IHC was done according to standard procedures. Tissue sections were deparaffinized with xylene and briefly digested with pronase (0, 1% for 7 min). The endogenous peroxidase was blocked by incubation with 1% H2O2 in methanol. The sections were exposed to primary antibodies for 90 min or overnight at room temperature. After rinsing several times with PBS, the sections were reacted with an avidin-biotin peroxidase kit (Vecta-stain ABC kit; Vector Laboratories). Peroxidase detection was done with 3-amino-9- ethyl carbazole or neufuchsin as chromagen, according to the manufacturer’s protocol. Finally, sections were counterstained with 1% hematoxylin.
Computerized Image Analysis.
Ektachrome slides of serial sections were scanned using a high-resolution film scanner (Kodak RFS 3570) and further processed with Adobe Photoshop software on a Macintosh Power PC 8100. An area representing approximately 60% related to the original image size was processed for image analysis. The images then were artificially colored. Images of sections reacted with anti-HHV-7 mAb were colored in greenish background, resulting in a brown-colored immunoreactive product (see Figs. 4 and 5B). Images of sections reacted with anti-CD68 antibody or anti-HHV-6 B antibody were artificially colored in blue-reddish background, resulting in a red immunoreactive signal (Figs. 4 and 5A). The images then were overlaid in transparent layers, allowing the identification of colocalization of two immunohistochemical signals within the same cell present in two serial sections.
Figure 4.

Immunohistochemical detection of HHV-7 pp85 in AIDS-associated Kaposi sarcoma, its localization to macrophage/monocytes positive for CD68 marker. Staining of HHV-7 pp85 antigen with mAb 5E1 (B, arrowheads). Staining with an anti-CD68 antibody (A, arrowheads). (C) Overlaid serial sections from A and B show localization of HHV-7 pp85 to CD68+ macrophages (arrowheads).
Figure 5.

Immunohistochemical colocalization of HHV-7 pp85 with the HHV-6B-specific antigen p101. (A)Staining with mAb to HHV-6B p101. (B) Staining of HHV-7 pp85 antigen with mAb 5E1 (arrows). (C) Overlaid serial sections from A and B show colocalization of HHV-7 pp85 and HHV-6B p101 antigens, marked in part with arrows.
RESULTS
Specificity of the mAbs and Reactivity with Histological Samples.
Preliminary experiments were carried out to ascertain that mAbs 5E1 to HHV-7 pp85 and mAb 8535 to HHV-6B p101 react specifically with the respective virus and do not display crossreactivity to the heterologous virus. Previously, the specificity of mAb 8535 was determined relative to HHV-6 variants (42). Its specificity with respect to HHV-7 is unknown. Cord blood mononuclear cells infected with HHV-7(RK) or HHV-6B(Z29) were reacted by immunofluorescence with either antibody. Lysates of cord blood mononuclear cells uninfected or infected with each virus were subjected to denaturing electrophoresis, transferred to nitrocellulose sheets, and reacted with the antibodies. The results are shown in Fig. 1. By both analyses the two mAbs showed specificity to the respective virus and failed to crossreact with the heterologous virus. The results confirm and extend previous data on the specificity of the antibodies and further show that selectivity was maintained in an immunostaining reaction.
Figure 1.

(A) Immunofluorescence staining of cord blood mononuclear cells infected with HHV-7(RK) (A, B) or HHV-6B(Z29) (C, D) reacted with mAb 5E1 to HHV-7 pp85 (A, C) or mAb 8535 to HHV-6B p101 (B, D). (B) Immunoblot of lysates of cord blood mononuclear cells uninfected, infected with HHV-6B(Z29), or HHV-7(RK)-reacted with mAb 5E1 (lanes 1–3) or mAb 8535 (lanes 4–6). The migration position of molecular weight (MW) markers is shown at left. In both immunofluorescence and immunoblot reactions note the specificity of the antibodies to the virus to which they were derived, and the lack of crossreactivity to the heterologous virus.
To ascertain if mAb 5E1 is suitable for in situ detection of HHV-7 antigen expression in routinely formalin-fixed paraffin-embedded histological samples, we analyzed by IHC human salivary glands, the only human tissue known to secrete virus in healthy individuals (22, 23, 26). As shown in Fig. 2, in salivary gland tissues clusters of acini reacting with the mAb to HHV-7 pp85 were readily detected, indicating that the antibody was able to react with and retained its specificity to denatured HHV-7 antigen contained in formalin-fixed paraffin-embedded tissues.
Figure 2.
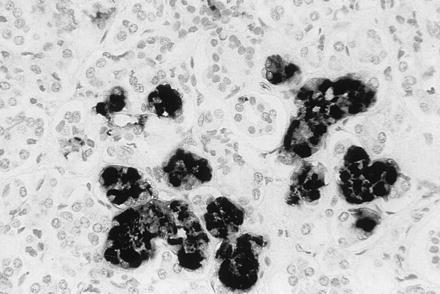
Immunohistochemical staining of a section of human parotid gland with mAb 5E1. The HHV-7 antigen pp85 is detectable in some acini of the salivary gland.
Detection of HHV-7 and HHV-6 DNA Sequences in Kaposi Sarcoma Specimens.
Previously, one of our laboratories reported on the presence of HHV-6B in 26 Kaposi sarcoma samples (38). This prompted us to search for the presence of HHV-7 in 32 samples, inclusive of the 26 samples already analyzed for HHV-6 (38). In the first analysis, the samples were analyzed for the presence of HHV-7 by PCR and IHC by two independent workers and evaluated independently. HHV-7 DNA sequences were detected in 6 of 32 (19%) cutaneous AIDS-associated Kaposi sarcoma samples and in 1 of 7 (14%) samples of cutaneous Kaposi sarcoma of classic-sporadic type by nested PCR (Fig. 3, and Table 1). In comparative assays, HHV-6 DNA sequences were detected in 10 of 32 (31%) of AIDS-associated Kaposi sarcoma lesions, and 2 of 7 (29%) classical-sporadic Kaposi sarcoma (Table 1). Results on the detection of HHV-6 DNA relative to 26 of the 32 samples (38) are given here cumulatively for comparison. Restriction enzyme analyses identified the HHV-6 as type B (HHV-6B) in all positive Kaposi sarcoma lesions.
Figure 3.

Representative examples of PCR products amplified from DNA extracted form Kaposi sarcoma samples, with primers specific for HHV-7. Patients A2 and A3, AIDS-associated Kaposi sarcoma. Patient C2, classic-sporadic Kaposi sarcoma. The positive control represents DNA amplified from HHV-7-infected cord blood mononuclear cells. Negative control represents DNA amplified from uninfected cord blood mononuclear cells. The size of amplified products is given in base pairs.
Table 1.
Frequency of detection of HHV-7 and HHV-6 viral antigens by IHC and of the viral DNAs by PCR in Kaposi sarcoma lesions
| Type of Kaposi sarcoma | Number of patients | HHV-7
|
HHV-6B*
|
HHV-7 + HHV-6B/ HHV-7† | ||
|---|---|---|---|---|---|---|
| PCR | IHC | PCR | IHC | |||
| AIDS | 32 | 6 (19) | 9 (28) | 10 (31) | 12 (37) | 3/6 |
| Classical | 7 | 1 (14) | 1 (14) | 2 (29) | 2 (29) | 1/1 |
Percent of positive samples is indicated by parentheses.
Data relative to 26 samples from ref. 37. Previous and current HHV-6 data are given here cumulatively for comparison purpose.
Fraction of dual HHV-6 + HHV-7 positive samples relative to the HHV-7 PCR-positive samples.
Expression of HHV-7 and HHV-6 Antigens.
The immunohistochemical analysis of Kaposi sarcoma specimens with mAb 5E1 revealed the presence of HHV-7 antigen in 9 of 32 (28%) AIDS-associated Kaposi sarcoma samples and in 1 of 7 (14%) classic-sporadic Kaposi sarcoma samples (Table 1). The HHV-7 antigen localized to the cytoplasm of cells located preferentially in areas surrounding pre-existing and newly formed vessels within the tumor, or diffusely scattered over the tumor tissue without distinct pattern (Fig. 4B). These cells did not appear to be endothelial, spindle-shaped tumor cells, or T lymphocytes. Previously, the Kaposi sarcoma cells expressing HHV-6 were identified as CD68+ cells (38). To determine whether the cells that expressed the HHV-7 antigen also belonged to the same lineage, serial sections were stained with a mAb to CD68 antigen, a marker of cells of the macrophage/monocyte lineage, and with mAb 5E1 to HHV-7 pp85. By computerized image analysis, the majority of the cells harboring HHV-7 antigen were identified as CD68+ monocytes/macrophages (Fig. 4C). The number of samples that scored positive for pp85 expression was slightly higher than that positive for HHV-7 DNA (28% vs. 19%). Because DNA for PCR analyses was obtained from paraffinated tissues, the relatively higher sensitivity of the immunohistochemical assays most likely reflected a relatively lower stability of DNA in samples subjected to formalin fixation, paraffin embedding, deparaffination, and subsequent processing for DNA extraction.
The specimens analyzed for the presence of HHV-7 pp85 also were tested for immunohistochemical detection of the HHV-6B-specific p101 (44). A positive reaction was detected in 12 of 32 AIDS-associated Kaposi sarcoma samples (37%) and in 2 of 7 (29%) classic-sporadic Kaposi sarcoma (Table 1). As previously reported (38), the protein was detected preferentially in cells in perivascular areas classified as CD68+ monocytes/macrophages on the basis of their reactivity with anti-CD68 antibodies. In essence, the distribution of cells infected with HHV-6B was similar to that of cells infected with HHV-7. Although the number of samples from classical Kaposi sarcoma was relatively small, for both HHV-7 and HHV-6 the frequency of DNA and antigen detection did not appear to differ significantly between AIDS-associated and classic-sporadic Kaposi sarcoma.
The authenticity and specificity of the immunohistochemical reaction was supported by two series of results. The first centers on the overall agreement between the results obtained with PCR and IHC assays. For HHV-7, 5 of 6 PCR-positive AIDS-associated Kaposi sarcoma specimens and the single specimen from classical Kaposi sarcoma also were positive by IHC. For HHV-6, the 10 PCR-positive AIDS-associated Kaposi sarcoma specimens and the two samples from classical sporadic Kaposi sarcoma were also positive by IHC. The second concerned the specificity of the staining reaction, which rested on the following tests. The Kaposi sarcoma samples that stained positively with antibodies to HHV-7 or HHV-6 did not show a positive immunohistochemical reaction when reacted with mAbs to herpes simplex virus glycoprotein D, human cytomegalovirus, and to a polyclonal antibody to human papillomavirus. Furthermore, liver tissue specimens, reacted with mAb 5E1 at the same time as the Kaposi sarcoma specimens, constantly showed a negative reaction, and served as internal negative control. Finally, staining of the Kaposi sarcoma specimens with mAbs to factor VIII (von Willebrand factor) and to CD31 showed a positive reaction on endothelial cells, as expected, and not on CD68+ cells. Altogether these controls ruled out that staining of CD68+ cells resulted from a false-positive unspecific binding of antibodies.
Coinfection of CD68+ Monocytes/Macrophages with HHV-6 and HHV-7.
Comparative analyses of the Kaposi sarcoma specimens by PCR and IHC indicated that three AIDS-associated Kaposi sarcoma samples (two autoptic and one bioptic) and one classical-sporadic Kaposi sarcoma bioptic sample scored positive simultaneously for HHV-6 and HHV-7. To determine the distribution of cells infected with these viruses, serial sections were reacted with antibodies to HHV-7 pp85 and HHV-6 p101 (Fig. 5 A–C). Coinfected cells were observed in all four samples. Remarkably, in these specimens the proportion of CD68+ monocytes/macrophages showing double-infection was very high. An example of cells doubly infected with HHV-6 and HHV-7 is shown in Fig. 5C.
DISCUSSION
We report two significant observations that alter our perception of susceptibility of human cells to infection by herpesviruses that establish productive and latent infections in humans. First, we show evidence that a structural protein of HHV-7 was present in CD68+ cells of monocyte/macrophage lineage. Second, we report that in a number of Kaposi sarcoma tissues, CD68+ cells of monocyte/macrophage lineage contained proteins reacting with antibodies specific for HHV-6 and HHV-7 and therefore these cells appear to be doubly infected with both viruses. The significance of these results stems from the following considerations.
Our studies were done with two mAbs specific for HHV-6 and HHV-7, respectively. The HHV-7-specific antibody reacts with the proteins of the pp85 complex located in the virion tegument encoded by U14 gene (40–42). The specificity of the antibody rests on the evidence that it reacted only with the HHV-7 protein (Fig. 1), it detected HHV-7 antigen in acini of human salivary glands (Fig. 2), and that some of the Kaposi sarcomas contained cells that reacted only with the anti-HHV-7 antibody and not with the anti-HHV-6B antibody, and vice versa. The antibody to HHV-6 p101 also is directed to a selective epitope of HHV-6 (44), and, as shown here, does not crossreact with HHV-7-infected cells (Fig. 1). Specificity and authenticity of the staining reactions were supported by lack of reactivity to a number of unrelated antibodies (to herpes simplex virus gD, HCMV, human papillomavirus, and endothelial cell markers), and the overall correlation between PCR-positive and IHC-positive samples. Contrary to other studies that detected the mere presence of viral DNA sequences in Kaposi sarcoma (HCMV, HHV-6, and human papillomavirus; refs. 46–49), our studies were designed to detect HHV-7 and HHV-6 gene expression and, by extension, viral replication. Under this respect they differ also from studies that failed to detect gene expression from other viruses (HCMV and human papillomavirus) (50–52).
Numerous studies have shown that HHV-7 infects exclusively CD4+ T-lymphocytes (refs. 3, 27, and 28; see ref. 29). The virus also is excreted in saliva, and viral DNA is detectable in salivary glands (22–26). Attempts to infect in vitro terminally differentiated macrophages have not been successful (32), and therefore the finding that CD68+ monocytes/macrophages in Kaposi sarcoma lesions are infected was unexpected. Relevant to the interpretation of these results is that cells of monocyte/macrophage lineage infected with HHV-7 were readily detectable in the environment of the Kaposi sarcoma, while very infrequent in other normal or pathological tissues that harbor macrophages, e.g., lung and liver (W.K., V.A., N.W., R.M., M.S., P. Mizandola, L. Menotti, and G.C.F., unpublished work). Failure to detect HHV-7-infected CD68+ monocytes/macrophages in other tissues argues against latently or persistently infected monocytes being commonly present in individuals and migrating into Kaposi sarcoma lesion. Given the relatively short lifetime of macrophages, the possibility that macrophages infected elsewhere migrate to Kaposi sarcoma also is unexpected. Altogether the data suggest that CD68+ monocytes/macrophages become infected within the Kaposi sarcoma lesions itself. We propose that the particular environment of Kaposi sarcoma tissue, rich in cytokines and chemokines (53), may extend the range of HHV-7-susceptible cells to CD68+ monocytes/macrophages. Either coreceptors normally not expressed on their surfaces might be induced, or permissiveness at a step subsequent to virus entry might be enhanced. Kaposi sarcoma lesions are known to express a variety of cytokines and chemokines, and the hypothesis that some of these alter the susceptibility of the cells in their immediate environment may not be far-fetched. Remarkably, such an extended tropism is observed in the human host and is not an artefact occurring in cell culture.
Herpes viruses are known to establish latent infection in transformed cells in vivo. Superinfection of a latently infected cell lineage with a second virus would be uncommon but not unprecedented. For example, AIDS-related body cavity lymphomas were shown to carry both Epstein–Barr virus and HHV-8, the recently discovered herpesvirus associated with Kaposi sarcoma (54, 55). The presence of two distinct viruses in presumably untransformed cells in human tissue is extremely unusual based on the fact that the number of cells and their distribution in the body renders double-infection extremely rare. To our knowledge, double in vivo infection of the same untransformed cell has been reported very rarely, namely between HIV and herpes simplex virus type 1 in keratinocytes and macrophages (56), and, at a very low frequency, between HIV and HCMV (57). Given that double-infection in the human host is an extremely unlikely possibility, the presence of doubly infected cells in some Kaposi sarcoma lesions is likely to reflect an increased probability of double-infection in situ, resulting from a high viral load.
In summary, while several alternative hypotheses could explain the new findings reported in this paper, namely the extended host range of HHV-7 and double HHV-6B/HHV-7 infection, the one best supported by our knowledge of the biology of Kaposi sarcoma is as follows. The environment of Kaposi sarcoma functions to attract circulating monocytes and lymphocytes carrying latent HHV-6B or HHV-7, activates and enhances replication of latent viruses, and renders the invading monocytes/macrophages susceptible to infection with HHV-7. The net result would be a high viral load in the environment of the lesion and infection of monocytes/macrophages to a level detectable by IHC. Whether HHV-6 and HHV-7, once reactivated from latency, in turn, might enhance cytokine secretion by macrophages, modulate HIV-1 (58) expression, and thus act as cofactors in the maintenance and progression of the lesion (39, 59) remains to be ascertained. Furthermore, if the presence of cells infected with at least HHV- 7, and possibly both HHV-6 and HHV-7, is indeed the consequence of the mechanism proposed above, the question arises whether the presence of other viruses found in Kaposi sarcoma lesions, including HHV-8, is also due to such a mechanism.
Acknowledgments
The work done at the University of Bologna was supported by the following grants: AIDS Project from Istituto Superiore di Sanità, BIOMED2 BMH4 CT95 1016 from UE, Target Project in Genetic Engineering from Consiglio Nazionale delle Ricerche, Associazione Italiana per la Ricerca sul Cancro, MURST 40 and 60%. The studies performed at the University Hospital of Zurich were financially supported by Gertrud-Rueegg-Stiftung, Zurich.
ABBREVIATIONS
- HHV
human herpesvirus
- HCMV
human cytomegalovirus
- IHC
immunohistochemistry
Note Added in Proof
Recently Sirianni et al. (60) reported that cultures of circulating Kaposi sarcoma-like spindle cells with markers of both macrophages and endothelial cells carry HHV-8 sequences, and suggested that these cells may localize into tissues and participate in the formation of Kaposi sarcoma lesions. As the cells positive by IHC for HHV-7 and HHV-6B were identified for reactivity with antibodies to the CD68 marker, it is possible that the cells designated above as DC68+ monocytes/macrophages are related to the circulating cells described by Sirianni et al.
References
- 1.Salahuddin S Z, Ablashi D V, Markham P D, Josephs S F, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarski B, Gallo R C. Science. 1986;234:596–601. doi: 10.1126/science.2876520. [DOI] [PubMed] [Google Scholar]
- 2.Lopez C, Pellett P, Stewart J, Goldsmith C, Sanderlin K, Black J, Warfield D, Feorino P. J Infect Dis. 1988;157:1271–1273. doi: 10.1093/infdis/157.6.1271. [DOI] [PubMed] [Google Scholar]
- 3.Frenkel N, Schirmer E C, Wyatt L S, Katsafanas G, Roffman E, Danovich R M, June C H. Proc Natl Acad Sci USA. 1990;87:748–752. doi: 10.1073/pnas.87.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyatt L S, Balachandran N, Frenkel N. J Infect Dis. 1990;162:852–857. doi: 10.1093/infdis/162.4.852. [DOI] [PubMed] [Google Scholar]
- 5.Ablashi D V, Balachandran N, Josephs S F, Hung C L, Kruger G R F, Kramarski B, Salahuddin S Z, Gallo R C. Virology. 1991;184:545–552. doi: 10.1016/0042-6822(91)90424-a. [DOI] [PubMed] [Google Scholar]
- 6.Gompels U A, Nicholas J, Lawrence G, Jones M, Thomson B J, Martin M E D, Efstathiou S, Craxton M, Macaulay H A. Virology. 1995;209:29–51. doi: 10.1006/viro.1995.1228. [DOI] [PubMed] [Google Scholar]
- 7.Ablashi D V, Agut H, Berneman Z, Campadelli-Fiume G, Carrigan D, et al. Arch Virol. 1993;129:363–366. [Google Scholar]
- 8.Pellett P, Black J B. In: Fields Virology. 3rd Ed. Fields B N, Knipe D, Howley P, editors. Philadelphia: Lippincott; 1996. pp. 2587–2608. [Google Scholar]
- 9.Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, Kurata T. Lancet. 1988;i:1065–1067. doi: 10.1016/s0140-6736(88)91893-4. [DOI] [PubMed] [Google Scholar]
- 10.Corbellino M, Lusso P, Gallo R C, Parravacini C, Galli M, Moroni M. Lancet. 1993;342:1242. doi: 10.1016/0140-6736(93)92226-j. [DOI] [PubMed] [Google Scholar]
- 11.Knox K, Carrigan D R. Lancet. 1994;343:577–578. doi: 10.1016/s0140-6736(94)91524-5. [DOI] [PubMed] [Google Scholar]
- 12.Drobyski W R, Dunne W M, Burd E M. J Infect Dis. 1993;167:735–739. doi: 10.1093/infdis/167.3.735. [DOI] [PubMed] [Google Scholar]
- 13.Okuno T, Higashi K, Shiraki K. Transplantation. 1990;49:519–522. doi: 10.1097/00007890-199003000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Kondo K, Kondo T, Okuno T, Takahashi M, Yamanishi K. J Gen Virol. 1991;72:1401–1408. doi: 10.1099/0022-1317-72-6-1401. [DOI] [PubMed] [Google Scholar]
- 15.Katsafanas G C, Schirmer E C, Wyatt L, Frenkel N. Proc Natl Acad Sci USA. 1996;93:9788–9792. doi: 10.1073/pnas.93.18.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicholas J. J Virol. 1996;70:5975–5989. doi: 10.1128/jvi.70.9.5975-5989.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark D A, Freeland M L, Mackie L K, Jarrett R F, Onions D E. J Infect Dis. 1993;168:251–252. doi: 10.1093/infdis/168.1.251. [DOI] [PubMed] [Google Scholar]
- 18.Wyatt L S, Rodriguez W J, Balachandran N, Frenkel N. J Virol. 1991;65:6260–6265. doi: 10.1128/jvi.65.11.6260-6265.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshikawa T, Asano Y, Kobayashi I, Nakashima T, Yazaki T, Suga S, Ozaki T, Wyatt L S, Frenkel N. J Med Virol. 1993;41:319–323. doi: 10.1002/jmv.1890410412. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K, Kondo T, Torigoe S, Okada S, Mukai T, Yamanishi K. J Pediatr. 1994;125:1–5. doi: 10.1016/s0022-3476(94)70113-x. [DOI] [PubMed] [Google Scholar]
- 21.Portolani M, Cermelli C, Mirandola P, Di Luca D. J Med Virol. 1995;45:282–283. doi: 10.1002/jmv.1890450307. [DOI] [PubMed] [Google Scholar]
- 22.Wyatt L S, Frenkel N. J Virol. 1992;66:3206–3209. doi: 10.1128/jvi.66.5.3206-3209.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Black J B, Inoue N, Kite-Powell K, Zaki S, Pellett P E. Virus Res. 1993;29:91–98. doi: 10.1016/0168-1702(93)90128-a. [DOI] [PubMed] [Google Scholar]
- 24.Kidd I M, Clark D A, Aitkhaled M, Griffiths P D, Emery V C. J Infect Dis. 1996;174:396–401. doi: 10.1093/infdis/174.2.396. [DOI] [PubMed] [Google Scholar]
- 25.Sada E, Yasukawa M, Ito C, Takeda A, Shiosaka T, Tanioka H, Fujita S. J Clin Microbiol. 1996;34:2320–2321. doi: 10.1128/jcm.34.9.2320-2321.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Luca D, Mirandola P, Ravaioli T, Dolcetti R, Frigatti A, Bovenzi P, Sighinolfi L, Monini P, Cassai E. J Med Virol. 1995;45:462–468. doi: 10.1002/jmv.1890450418. [DOI] [PubMed] [Google Scholar]
- 27.Frenkel N, Wyatt L S. Dev Biol Stand. 1992;7:259–265. [PubMed] [Google Scholar]
- 28.Berneman Z N, Ablashi D V, Li G, Eger-Fletcher M, Reitz M S, Jr, Hung C L, Brus I, Komaroff A L, Gallo R C. Proc Natl Acad Sci USA. 1992;89:10552–10556. doi: 10.1073/pnas.89.21.10552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lusso P, Gallo R C. Immunol Today. 1995;16:67–71. doi: 10.1016/0167-5699(95)80090-5. [DOI] [PubMed] [Google Scholar]
- 30.Lusso P, Secchiero P, Crowley R W, Garzino-Demo A, Berneman Z N, Gallo R C. Proc Natl Acad Sci USA. 1994;91:3872–3876. doi: 10.1073/pnas.91.9.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasukawa M, Inoue Y, Ohminami H, Sada E, Miyake K, Tohyama T, Shimada T, Fujita S. J Virol. 1997;71:1708–1712. doi: 10.1128/jvi.71.2.1708-1712.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowley R W, Secchiero P, Zella D, Cara A, Gallo R C, Lusso P. J Immunol. 1996;156:2004–2008. [PubMed] [Google Scholar]
- 33.Di Luca D, Mirandola P, Ravaioli T, Bigoni B, Cassai E. Infect Agents Dis. 1996;5:203–214. [PubMed] [Google Scholar]
- 34.Chen M, Popescu N, Woodworth C, Berneman Z, Corbellino M, Lusso P, Ablashi D V, Di Paolo J A. J Virol. 1994;68:1173–1178. doi: 10.1128/jvi.68.2.1173-1178.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Challoner P B, Smith K T, Parker J D, MacLeod D L, Coulter S N, Rose T M, Schultz E R, Bennett J L, Garber R L, Chang M, Schad P A, Stewart P M, Nowinski R C, Brown J P, Burmer G C. Proc Natl Acad Sci USA. 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lusso P, Malnati M, Garzino-Demo A, Crowley R W, Long E O, Gallo R C. Nature (London) 1993;362:458–462. doi: 10.1038/362458a0. [DOI] [PubMed] [Google Scholar]
- 37.Di Luca D, Dolcetti R, Mirandola P, De Re V, Secchiero P, Carbone A, Boiocchi M, Cassai E. J Infect Dis. 1994;170:211–215. doi: 10.1093/infdis/170.1.211. [DOI] [PubMed] [Google Scholar]
- 38.Kempf W, Adams V, Pfaltz M, Briner J, Schmid M, Moos R, Hassam S. Hum Pathol. 1995;26:914–919. doi: 10.1016/0046-8177(95)90016-0. [DOI] [PubMed] [Google Scholar]
- 39.Kempf W, Adams V. Biochem Mol Med. 1996;58:1–12. doi: 10.1006/bmme.1996.0025. [DOI] [PubMed] [Google Scholar]
- 40.Foà-Tomasi L, Avitabile E, Ke L, Campadelli-Fiume G. J Gen Virol. 1994;75:2719–2727. doi: 10.1099/0022-1317-75-10-2719. [DOI] [PubMed] [Google Scholar]
- 41.Stefan, A., Secchiero, P., Baechi, T., Kempf, W. & Campadelli-Fiume, G. (1997) J. Virol. 71, in press. [DOI] [PMC free article] [PubMed]
- 42.Foà-Tomasi L, Fiorilli M P, Avitabile E, Campadelli-Fiume G. J Gen Virol. 1996;77:511–518. doi: 10.1099/0022-1317-77-3-511. [DOI] [PubMed] [Google Scholar]
- 43.Black J B, Schwarz T F, Patton J L, Kite-Powell K, Pellett P E, Wiersbitzky S, Bruns R, Muller C, Jager G, Stewart J A. Clin Diagn Lab Immunol. 1996;3:79–83. doi: 10.1128/cdli.3.1.79-83.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pellett P, Sanchez-Martinez D, Dominguez G, Black J B, Anton E, Greenamoyer C, Dambaugh T R. Virology. 1993;195:521–553. doi: 10.1006/viro.1993.1403. [DOI] [PubMed] [Google Scholar]
- 45.Aubin J T, Collandre H, Candotti D, Ingrand D, Rouzioux C, Burgard M, Richard S, Huraux J-M, Agut H. J Clin Microbiol. 1991;29:367–372. doi: 10.1128/jcm.29.2.367-372.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jahan N, Razzaque A, Greenspan J, Conant M A, Josephs S F, Nakamura S, Rosenthal L J. AIDS Res Hum Retroviruses. 1989;5:225–231. doi: 10.1089/aid.1989.5.225. [DOI] [PubMed] [Google Scholar]
- 47.Andersen C B, Karkov J, Bjerregaard B, Visfeldt J. Acta Pathol Microbiol Immunol Scand. 1991;99:893–897. [PubMed] [Google Scholar]
- 48.Huang Y Q, Li J J, Rush M G, Poisz B J, Nicolaides A, Jacobson M, Zhang W G, Coutavas E, Abbott M A, Friedman-Kien A E. Lancet. 1992;339:515–518. doi: 10.1016/0140-6736(92)90338-4. [DOI] [PubMed] [Google Scholar]
- 49.Bovenzi P, Mirandola P, Secchiero P, Strumia R, Cassai E, Di Luca D. Lancet. 1993;341:1288–1289. doi: 10.1016/0140-6736(93)91198-u. [DOI] [PubMed] [Google Scholar]
- 50.Cerimele D, Scappaticci S, Cattaneo E, Cottoni F. Arch Dermatol Res. 1983;79:79–80. doi: 10.1007/BF00510052. [DOI] [PubMed] [Google Scholar]
- 51.Biggar R J, Dunsmore N, Kurman R J, Shah K V, Kordor J, Cottoni F, Hatzakis A, Gigase P L. Lancet. 1992;339:1604–1605. doi: 10.1016/0140-6736(92)91866-7. [DOI] [PubMed] [Google Scholar]
- 52.Civantos J, Penneys N, Ziegels-Weissman J. AIDS Res. 1984;1(2):121–125. doi: 10.1089/aid.1.1983.1.121. [DOI] [PubMed] [Google Scholar]
- 53.Ensoli B, Nakamura S, Salahuddin S Z, Biberfeld P, Larson L, Beaver B, Wong-Staal F, Gallo R C. Science. 1988;243:223–226. doi: 10.1126/science.2643161. [DOI] [PubMed] [Google Scholar]
- 54.Chang Y, Cesarman E, Pessin M S, Lee F, Culpepper J, Knowles D M, Moore P S. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 55.Cesarman E, Chang Y, Moore P S, Said J, Knowles D M. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 56.Heng M C Y, Yang Heng S, Allen S G. Lancet. 1994;343:255–258. doi: 10.1016/s0140-6736(94)91110-x. [DOI] [PubMed] [Google Scholar]
- 57.Bertram S, Hufert F T, Van Lunzen J, von Laer D. J Med Virol. 1996;49:283–288. doi: 10.1002/(SICI)1096-9071(199608)49:4<283::AID-JMV5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 58.Ensoli B, Lusso P, Schachter F, Josephs S F, Rappaport J, Negro F, Gallo R C, Wong-Staal F. EMBO J. 1989;8:3019–3027. doi: 10.1002/j.1460-2075.1989.tb08452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinkovics J G, Szakacs J, Györkey F. In: Drugs of Abuse, Immunity and AIDS. Friedman H, Klein T W, Spector S, editors. New York: Plenum; 1992. [Google Scholar]
- 60.Sirianni M C, Uccini S, Angeloni A, Faggioni, Cottoni F, Ensoli B. Lancet. 1997;349:255. doi: 10.1016/s0140-6736(05)64866-0. [DOI] [PubMed] [Google Scholar]