

Abstract
Inducible nitric oxide synthase (iNOS) protein is expressed in cardiac myocytes of patients and experimental animals with congestive heart failure (CHF). Here we show that iNOS expression plays a role in pressure overload induced myocardial chamber dilation and hypertrophy. In wild type mice, chronic transverse aortic constriction (TAC) resulted in myocardial iNOS expression, cardiac hypertrophy, ventricular dilation and dysfunction, and fibrosis, while iNOS deficient mice displayed much less hypertrophy, dilation, fibrosis and dysfunction. Consistent with these findings, TAC resulted in marked increases of myocardial ANP, 4-hydroxy-2-nonenal (4HNE, a marker of lipid peroxidation) and nitrotyrosine (a marker for peroxynitrite) in wild type mice but not in iNOS deficient mice. In response to TAC, myocardial eNOS and iNOS was expressed as both monomer and dimer in wild type mice, and this was associated with increased reactive oxygen species (ROS) production, suggesting that iNOS monomer was a source for the increased oxidative stress. Moreover, systolic overload induced Akt, mTOR and ribosomal protein S6 activation that was significantly attenuated in iNOS deficient mice. Furthermore, selective iNOS inhibition with 1400W (6mg/kg/hour) significantly attenuated TAC induced myocardial hypertrophy and pulmonary congestion. These data implicate iNOS in the maladaptative response to systolic overload, and suggest that selective iNOS inhibition or attenuation of iNOS monomer content might be effective for treatment of systolic overload-induced cardiac dysfunction.
Keywords: superoxide anion, peroxynitrite, iNOS monomer, mTOR
Introduction
Several investigators have demonstrated that iNOS protein is expressed in cardiac myocytes and endocardial endothelium of patients and animals with ventricular hypertrophy or congestive heart failure (CHF) regardless of cause 1-4. Thus, iNOS was co-expressed with TNF-α in cardiac myocytes from patients with dilated cardiomyopathy 2 and increased in several animal models of ventricular hypertrophy or CHF 5. Although unregulated NO production by iNOS has been proposed to exert negative effects on cardiomyocyte function, the effect of iNOS expression on ventricular hypertrophy and CHF in the in vivo heart is controversial. Thus, Heger et al 6 reported that over expression of iNOS in cardiac myocytes increased myocardial NOS activity and NO production, but had no effect on cardiac morphology or function. In contrast, Mungrue et al 7 reported that cardiac specific over expression of iNOS resulted in inflammatory cell infiltrate, left ventricular hypertrophy, dilation, fibrosis and contractile dysfunction. The level of iNOS expression in these transgenic mice would depend on the promoter activity, and the iNOS related phenotypes might vary depending on the level of myocardial iNOS expression. Furthermore, the effects of stress-induced iNOS expression in normal hearts may be different from that in the transgenic mice. Therefore, the present study examined the role of iNOS in the ventricular hypertrophy and CHF that develops in response to sustained pressure overload produced by transverse aortic banding (TAC) in mice with or without the iNOS gene. We provide the first evidence to our knowledge that iNOS deficiency (iNOS−/−) attenuates TAC-induced ventricular hypertrophy and CHF, and that iNOS expressed in response to systolic overload serves as a source for myocardial reactive oxygen species that contribute to left ventricular dilatation and hypertrophy.
Methods
Mice
Body weight and age (2-3 months old) matched male iNOS−/− (crossed back to C57BL/6J for 12 times) and wild type controls (C57BL/6J) were purchased from Jackson Laboratories. This study was approved by the Institutional Animal Care and Use Committee of University of Minnesota.
TAC-induced left ventricular hypertrophy
TAC was performed using the minimally invasive suprasternal approach described by Hu et al 8.
Selective iNOS inhibition with 1400W
To study the effect of selective iNOS inhibition on TAC-induced ventricular hypertrophy and dysfunction, adult male C57BL/6J mice were randomly divided into two groups immediately after TAC, and one group was treated with the selective iNOS inhibitor 1400W while the other group was treated with saline vehicle. 1400W was delivered at a constant dose of 6 mg/kg/hour via an Osmotic minipump (Alzet Model 2002). This dose 0f 1400W resulted in plasma 1400W concentrations 2.4-4.9 fold higher than the EC50 for tissue iNOS, and decreased plasma NOx generated by iNOS by 63-83% 9. This dose of 1400W had no effect on hemodynamic variables in normal animals, indicating lack of effect on constitutive NOS. We have found that male C57B6J mice develop ventricular dysfunction and pulmonary congestion in 4 weeks after moderate TAC (using a 26G needle), and develop ventricular dysfunction and pulmonary congestion in 2 weeks after severe TAC (using a 27G needle). Since the capacity of the minipump to deliver the required dose of 1400W was two weeks, in order to induce ventricular dysfunction in the mice within 2 weeks, in these animals we produced severe TAC by ligating the aorta over a 27G needle.
Echocardiography was performed when mice were anesthetized with 1.5% isoflurane by inhalation.
Western blots
NOS Protein content was analyzed using Western blots as previously described 10. Primary antibodies against iNOS, eNOS, nNOS, ANP, protein arginine methyltransferase 1 (PRMT1), 4-HNE, nitrotyrosine, total mTOR, phospho-mTOR, Akt, phospho-Akt, phosphor-S6 and total p70S6K were purchased from Transduction Laboratories, Santa Cruze Inc, Sigma, Upstate, or Cell Signaling Technology, respectively. Dimethylarginine dimethylaminohydrolase 1 (DDAH1) antibody was a gift from Dr. Kimmoto 10.
Measurement of reactive oxygen species (ROS)
Relative ROS production was determined by chemiluminescence of coelenterazine (4μM, Molecular Probe- 1) (See detail in Supplementary data) 11 and the red fluorescent dye dihydroethidium (DHE, 2 μM; Invitrogen).
MMP activity
In vitro gelatin lysis by MMP-2 and MMP-9 was assessed by zymography 12.
Results
iNOS−/− attenuated moderate TAC-induced ventricular hypertrophy and dysfunction
Ventricular mass, lung weight, the ratio of ventricular weight to body weight, and the ratio of lung weight to body weight in iNOS−/− and wild type mice in response to TAC are shown in Figure 1 and Supplementary Table-1 (Table-S1). Under control conditions, ventricular weight, lung weight and the ratio of organ weight to body weight were not different between iNOS−/− and wild type mice (Figure 1). Although TAC for 28 days resulted in ventricular hypertrophy in both wild type and iNOS−/− mice, iNOS−/− significantly attenuated the TAC-induced increase of ventricular weight and the ratio of ventricular weight to body weight (Figure 1). As compared to wild type mice, a significantly attenuated increase of ventricular weight and the ratio of ventricular weight to body weight was also observed in iNOS−/− mice 8 days after TAC (data not shown). In addition, lung weight and the ratio of lung weight to body weight were significantly greater in wild type mice as compared to iNOS−/− mice 28 days after TAC, indicating increased pulmonary congestion in the wild type mice (Figure 1). There was no difference in mortality rate between iNOS−/− and wild type mice following TAC. To determine the degree of pressure overload produced by the TAC, systolic pressure proximal to the TAC site and the pressure gradient across the TAC site were determined in a group of wild type mice and KO mice immediately after the TAC procedure. The results showed that TAC produced a similar increase of LV systolic pressure and the pressure gradient across the aortic constriction (Supplementary Figure 3).
Figure 1.
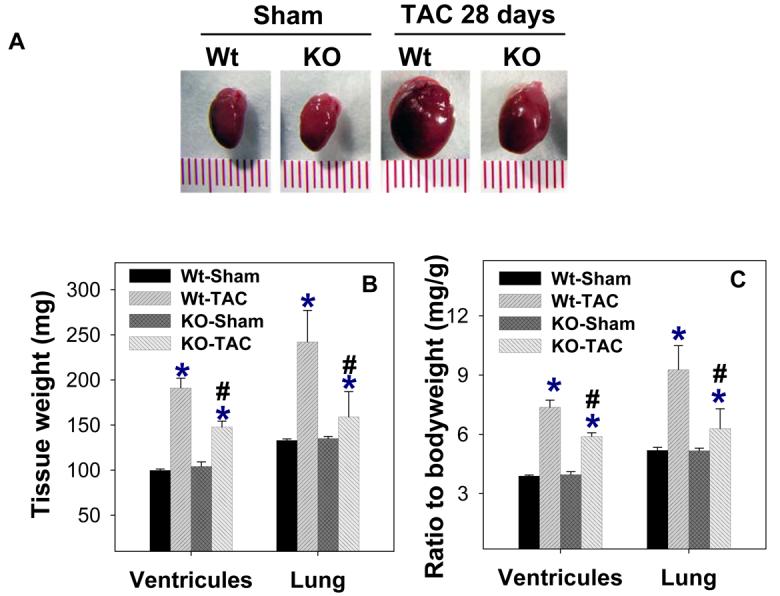
iNOS deletion attenuates ventricular hypertrophy and pulmonary congestion in response to TAC-induced pressure overload. A. Representative hearts from wild type and iNOS−/− mice 4 weeks after TAC or sham surgery (n=9 to 13). B. Tissue weights in each group; C. Ratio of tissue weight to body weight. Wt-TAC: Wild type mice after TAC for 4 weeks; KO-TAC: iNOS−/− mice after TAC for 4 weeks. *P<0.05 as compared to corresponding control; #p<0.05 as compared to Wt-TAC.
Echocardiographic imaging of the heart 28 days after TAC demonstrated significant increases of LV wall thickness, LV end systolic diameter (ESD) and LV end diastolic diameter (EDD) in both iNOS−/− and wild type mice in comparison with mice of similar body weight without TAC (Figure 2, Table-S1). However, TAC resulted in a significantly greater decrease of the left ventricular systolic shortening fraction and ejection fraction in wild type mice than in iNOS−/− mice (Figure 2, Table-S1).
Figure 2.


Echocardiograms demonstrating that iNOS deletion attenuated TAC-induced left ventricular hypertrophy and dysfunction. A. Representative M-mode echocardiograms demonstrating greater left ventricular dilatation in wild type mice as compared to iNOS−/− mice 4 weeks after TAC. B-G. Summary data from echocardiograms (n=9 to 12 per group) demonstrating that iNOS−/− attenuated the TAC-induced increase of LV end systolic diameter (C), LV wall thickness (F), LV fractional shortening (D) and LV ejection fraction (E). *P<0.05 as compared to corresponding control; #p<0.05 as compared to group of Wt-TAC.
iNOS−/− attenuates TAC-induced ventricular fibrosis
Histological staining of left ventricular tissue at 28 days after TAC demonstrated more interstitial fibrosis in wild type mice as compared to iNOS−/− mice (Figure 3A, Figure 3B). Under control conditions, the cross-sectional area of the cardiac myocytes was not different between wild type mice (227±10 μM2) and iNOS−/− mice (206 ±13 μM2). Myocyte hypertrophy occurred in both groups of animals in response to TAC, but the increase in myocyte cross-sectional area was significantly less in iNOS−/− mice (395±20 μM2) than in wild type mice (Wt: 464±31 μM², p<0.05) (Figure 3C, Figure 3D).
Figure 3.

Histological staining demonstrating that iNOS deletion attenuated TAC-induced myocardial fibrosis (A, B), cardiac myocyte hypertrophy (C, D), and the increase of myocardial MMP2 activity (E,F). MTS: Masson's trichrome staining, blue staining indicates fibrosis; WGA: Staining for wheat germ agglutinin with FITC-conjugated Flur-488 (Invitrogen), bright green staining indicates the area of the matrix and cell membrane. Summarized average data are from 4 representative mice per group. *p<0.05 compared to the corresponding control; #p<0.05 as compared to Wt-TAC.
TAC-induced alterations of eNOS, iNOS, nNOS, and ANP
Western blots demonstrated that iNOS protein was expressed in wild type mice at both 8 and 28 days after TAC (Figure-4). eNOS protein content was significantly increased 28 days after TAC in wild type mice, and iNOS−/− attenuated the TAC-induced induction of eNOS. Myocardial nNOS was unchanged after TAC.
Figure 4.
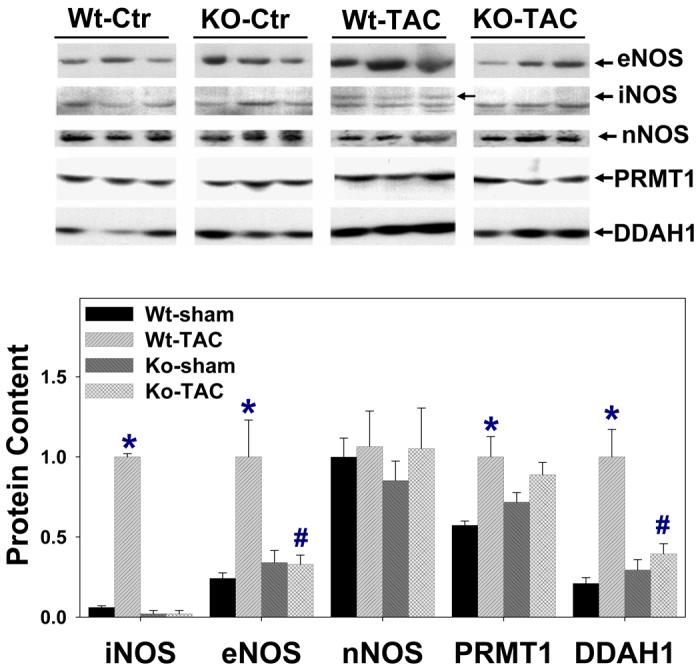
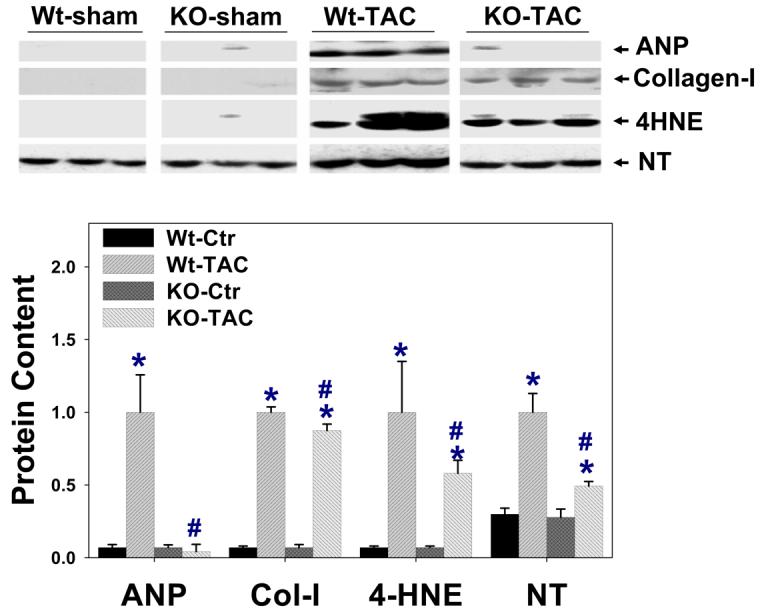
Alterations of myocardial eNOS, iNOS, nNOS, DDAH-1, PRMT-1, ANP, 4-HNE, collagen-I, and NT protein content in wild type mice and iNOS−/− mice (n=6 per group). iNOS was detected only in wild type mice after TAC. *P<0.05 as compared to the corresponding control; #p<0.05 as compared to Wt-TAC.
Dimethylarginine dimethylaminohydrolase 1 (DDAH1), and protein arginine methyltransferase (PRMT1) regulate NO availability by either degrading or synthesizing the endogenous NOS inhibitor, asymmetric dimethylarginine (ADMA). Therefore, the protein contents of DDAH1 and PRMT1 were determined. Interestingly, both DDAH1 and PRMT1 were increased in wild type mice after TAC (Figure-4), while iNOS−/− attenuated the TAC-induced induction of DDAH1.
In addition, iNOS−/− attenuated TAC-induced increase of myocardial ANP (Figure-4), consistent with the finding of less ventricular hypertrophy and CHF in the iNOS−/− mice. Myocardial nitrotyrosine and 4HNE were increased in both wild type and iNOS−/− mice after TAC (Figure-4), but the increases were significantly less in iNOS−/− mice than in wild type mice, implying lower oxidative stress in the iNOS−/− mice.
Myocardial fibrosis and MMP activity
After TAC, iNOS−/− hearts developed less fibrosis as compared with wild type mice (Figure 3). Furthermore, iNOS−/− mice had significantly lower myocardial MMP2 activity as demonstrated by zymography (Figure 3). Although myocardial collagen-1 was increased in both wild type and iNOS−/− mice 28 days after TAC, the TAC-induced increase of collagen-I expression was significantly less in the iNOS−/− mice (Figure 4).
iNOS−/− attenuates TAC-induced Akt-mTOR-S6 activation
As increased oxidative stress can activate Akt, and activation of mTOR and ribosomal protein S6 (S6) regulates cell growth, total and activated Akt, mTOR and S6 were determined. TAC caused significant increases of phospho-AktSer473, phospho-mTORSer2488, phospho-S6Ser235/236 and phospho-ErkThr202/204 (Erk-42kDa), while total Akt was unchanged (Figure 5). iNOS deletion attenuated the TAC-induced increases of AktSer473, mTORSer2488, S6Ser235/236 and ErkThr202/204 (Figure 5).
Figure 5.
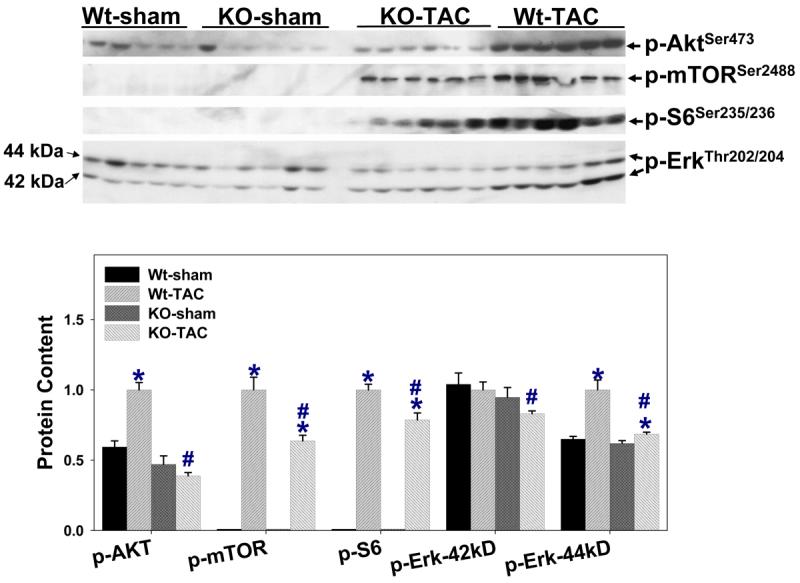
iNOS deletion attenuated the TAC-induced increases of phospho-AktSer473, phospho-mTORSer2488, phospho-S6Ser235/236 and phospho-ErkThr202/204. *P<0.05 as compared to the corresponding control; #p<0.05 as compared to Wt-TAC.
Expression of eNOS and iNOS monomer and dimer
NOS monomer produces superoxide anion, while NOS dimer generates NO. Since evidence of increased myocardial oxidative stress was observed in the wild type mice after TAC, and iNOS was robustly expressed in the wild type mice early after TAC, relative myocardial iNOS and eNOS monomer and dimer were determined in wild type mice 8 days after TAC by using undenatured gel. As shown in Figure 6, iNOS was present as both monomer and dimer in the wild type mice after TAC (18±2% monomer). Myocardial eNOS monomer was undetectable in sham mice, but was present in the iNOS−/− mice (39±1% monomer) and wild type mice (38±3% monomer) after TAC.
Figure 6.
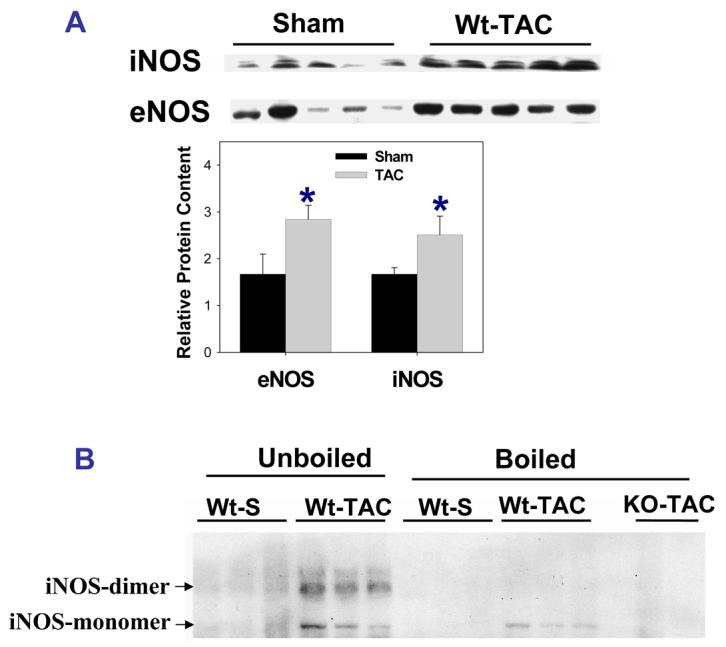
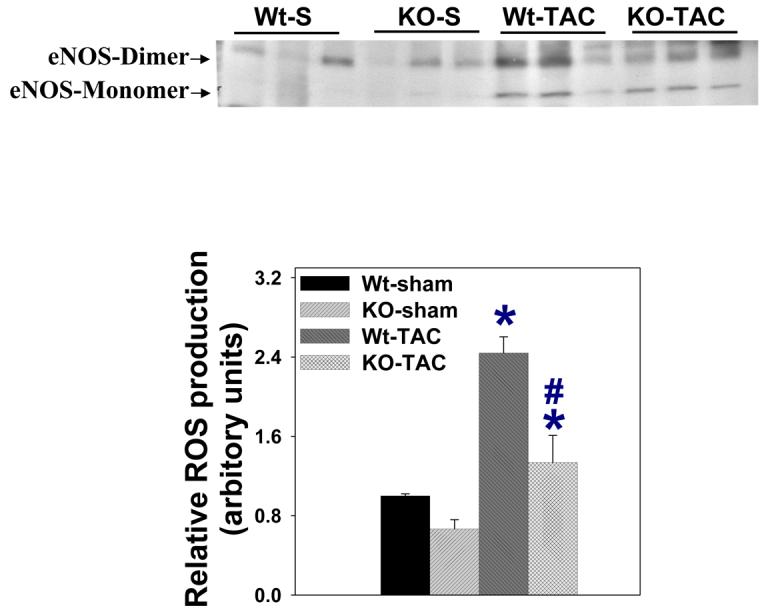
A. Western blot showing increased myocardial eNOS and iNOS expression in wild type mice in response to 8-days severe TAC. B. Both iNOS monomer and iNOS dimer were detected in wild type mice 8 days after TAC in unboiled conditions, while iNOS dimer was diminished after boiling the sample for 15 minutes. C. eNOS expressed as both monomer and dimer in wild type mice and iNOS−/−mice 8 days after TAC in unboiled conditions. D. Attenuated ROS production in iNOS−/− mice. #p<0.05 as compared to Wt-TAC.
iNOS−/− attenuated the TAC-induced increase of reactive oxygen species (ROS)
To determine whether the finding of NOS monomer was associated with increased ROS generation, superoxide anion content was determined with a chemiluminescence assay in myocardial tissue extracts. TAC resulted in increases of superoxide anion in both wild type and iNOS deficient mice (Figure 5D), but iNOS−/− partially attenuated TAC-induced myocardial superoxide anion production (Figure 5D). In separate samples obtained from wild type mice after TAC, both the selective iNOS inhibitor 1400W (decreased to 75±10% after 1400W treatment) or the nonselective NOS inhibitor L-NAME (decreased to 62 ±13% after L-NAME treatement) attenuated myocardial superoxide production. These findings support the notion that NOS uncoupling contributed to myocardial superoxide production. Intracellular ROS generation was also estimated with red DHE (typically nuclear localization) staining in frozen sections; the results demonstrated that TAC caused an increase of ROS production, and that iNOS−/− partially attenuated the TAC-induced superoxide production (Figure S2).
Myocardial eNOS and iNOS distribution
Under control conditions, eNOS was mainly distributed in capillaries and endothelial cells of larger blood vessels, with a similar expression pattern in wild type and iNOS−/− mice (Figure 7). Interestingly, although eNOS was still highly expressed in endothelial cells of blood vessels after TAC, an apparent increase of eNOS expression was observed in areas of fibrosis and in cardiac myocytes around the fibrotic areas (Figure 7). This TAC-induced eNOS induction was attenuated iNOS−/− mice (Figure 7). Immunostaining demonstrated that iNOS was broadly expressed in cardiac myocytes and connective tissue in wild type mice after TAC (Figure-S3). The TAC-induced myocardial iNOS expression pattern in wild type mice was similar to the pattern of iNOS expression in mice following LPS stimulation.
Figure-7.

Myocardial eNOS was predominantly expressed in vascular endothelial cells in normal heart, and TAC increased eNOS expression in myocardial area with fibrosis and some adjacent cardiac myocytes in wild type mice. Wheat germ agglutinin staining (green) indicates areas of matrix, blood vessels and cell membrane. Blue staining indicates the cell nuclei; red staining indicates eNOS. Samples used for staining were obtained 8 days after TAC or sham surgery.
1400W attenuated TAC-induced ventricular hypertrophy and dysfunction
Since iNOS deficiency attenuated the TAC-induced ventricular hypertrophy and dysfunction, we examined whether selective iNOS inhibition with 1400W would have similar protective effects on TAC-induced ventricular hypertrophy in wild type mice. 1400W significantly attenuated the TAC-induced ventricular hypertrophy (Figure 8B), pulmonary congestion (Figure 8B) and ventricular dysfunction (Figure 8E, Figure 8F). 1400W also significantly attenuated the TAC-induced myocardial fibrosis (Figure 8J, Figure 8K). 1400W did not affect heart rate or LV wall thickness (Figure 8), and had no effect on mean aortic pressure (86±3 mmHg in mice treated with 1400W vs. 83 ±5 mmHg in mice without 1400W) or LV systolic pressure (98±3.7 in 1400W treated mice vs. 102±3.6 mmHg in untreated mice). 1400W had no effect on ventricular function and fibrosis in sham mice.
Figure 8.
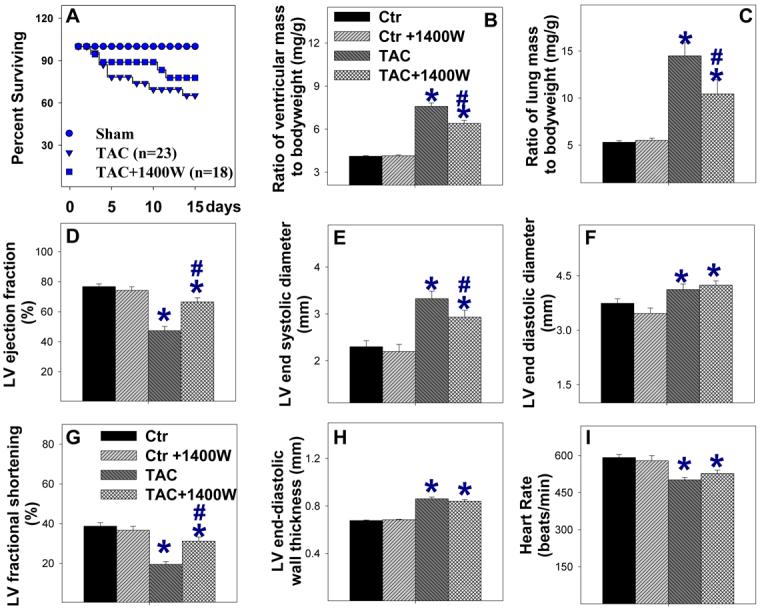
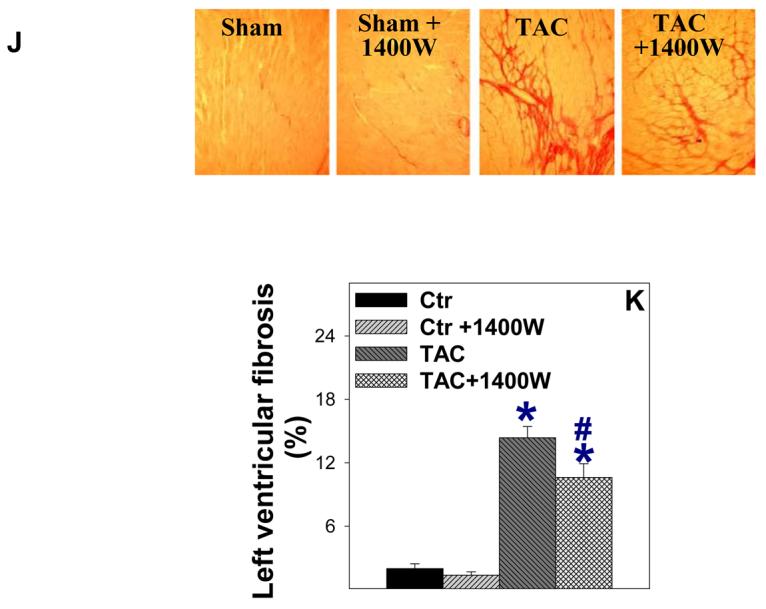
1400W attenuated TAC-induced cardiac death (A), ventricular hypertrophy (B), pulmonary congestion (C), LV dysfunction (D, G), ventricular dilation (E), and left ventricular fibrosis (J, K) in wild type mice.
Discussion
The major new findings of this study are that chronic systolic overload resulted in expression of myocardial iNOS as both monomer and dimer in wild type mice, and this was associated with greater ventricular hypertrophy, dilation, fibrosis and dysfunction as compared to iNOS−/− mice. Consistent with these findings, selective iNOS inhibition with 1400W significantly attenuated the TAC-induced ventricular hypertrophy and dysfunction in wild type mice. Expression of iNOS after TAC was associated with greater increases of myocardial nitrotyrosine, 4HNE, ANP, and phosphorylation of Akt, mTOR, S6 in wild type mice as compared to iNOS−/− mice. The finding of myocardial iNOS monomer, a structural unit that generates superoxide anion, suggests that iNOS is a significant source of superoxide anion in the overloaded or failing heart. The finding that iNOS deletion protected against TAC-induced ventricular hypertrophy and CHF, in association with decreased myocardial nitrotyrosine and 4HNE, suggests that selective iNOS inhibition might potentially be an effective strategy for treatment of myocardial dysfunction and congestive heart failure in the chronically overloaded heart.
iNOS effects on ventricular remodeling/fibrosis and CHF
Although no previous reports have directly examined the effect of iNOS deletion on pressure overload-induced ventricular hypertrophy and CHF, several investigators have studied the effect of iNOS deficiency on ventricular remodeling after myocardial infarction 5, 13-15. Studies from 3 different groups have demonstrated that iNOS deficiency caused mildly or moderately decreased infarct-induced mortality, improved ventricular function, reduced myocardial nitrotyrosine content, reduced plasma nitrate, and decreased programmed cell death during both the acute and chronic phases of myocardial infarction 5, 14-15, suggesting a detrimental effect of iNOS expression. In addition, using conditional cardiac specific transgenic mice, Mungrue et al found that overexpression of human iNOS in cardiac myocytes resulted in increased myocardial peroxynitrite, myocardial fibrosis, ventricular hypertrophy, congestive heart failure and cardiac sudden death 7, indicating that high level overexpression of iNOS can induce ventricular hypertrophy and CHF. A recent study from our laboratory demonstrated that selective pharmacologic iNOS inhibition significantly improved left ventricular contractility and myocardial oxygen consumption in end-stage pacing-induced canine heart failure 3, indicating that iNOS inhibition can acutely improve ventricular function in failing hearts. It should be noted, however, that there are conflicting reports in which iNOS deficiency failed to reduce myocardial infarct-induced ventricular dysfunction or mortality13. The differing results regarding the role of iNOS in ventricular remodeling may be related to variations in infarct size, the stage of heart failure and different genetic backgrounds of the mice 5, 13-15. The differing results obtained from two cardiac specific transgenic lines of iNOS overexpression may be due to variations of promoter efficiency in these transgenic lines 6-7.
The TAC-induced iNOS expression in the wild type mice in the present study, in conjunction with less severe ventricular hypertrophy, dilation, fibrosis and dysfunction in the iNOS−/− mice in response to systolic overload, is consistent with previous reports that iNOS can exert detrimental effects on the heart. The finding that iNOS−/− only partially attenuated the TAC-induced myocardial ROS production and ventricular dysfunction is not unexpected, since previous reports have demonstrated that other factors such as eNOS uncoupling 16, and increases of NADPH oxidase 17 and xanthine oxidase18 can contribute to increased oxidative stress in the failing heart. The finding that iNOS deficiency and selective iNOS inhibition with 1400W attenuated left ventricular remodeling and dysfunction suggests that selective iNOS inhibition might be a useful approach for treating systolic overload-induced ventricular hypertrophy and CHF.
eNOS and iNOS expression in hypertrophied or failing hearts
Increased iNOS expression and activity have been documented in myocardial specimens from patients and animals with ventricular hypertrophy 1 and congestive heart failure 1-4. In the present study, the finding of a faint iNOS band in the wild type hearts 8 days after sham surgery was likely the result of tissue trauma from the sham surgery. The expression of myocardial iNOS protein in hearts of wild type mice both 8 days and 28 days after TAC is consistent with previous reports of iNOS expression in hypertrophied and failing hearts 1-2, 4, although the mechanism for upregulation of iNOS expression after TAC is not totally clear. The relatively higher expression of iNOS and eNOS protein early after TAC is consistent with a previous report in which iNOS and eNOS expression peaked 3-5 days after TAC 4. The increased expression of eNOS after TAC is consistent with previous reports that eNOS protein was increased in hearts with congestive heart failure 4, 10 or ventricular hypertrophy in response to pressure overload4, 19. The increased eNOS expression in cardiac myocytes near the area with fibrosis is consistent with a previous report in cardiomyopathy samples obtained from mice with mutations of gamma-sarcoglycan or delta-sarcoglycan 20. In the context of a recent study demonstrating that eNOS uncoupling contributes to TAC-induced myocardial oxidative stress and ventricular remodeling16, and the fact that myocardial eNOS monomer was present after TAC, the protective effect of iNOS−/− on TAC induced ventricular remodeling may relate to an attenuation of eNOS induction. It should be noted that the effect of eNOS on TAC induced cardiomyopathy is controversial; although one study demonstrated that eNOS deficiency profoundly attenuated TAC-induced ventricular hypertrophy and CHF, two other studies reported that eNOS deficiency exacerbated ventricular hypertrophy produced by mild TAC19, 21.
DDAH1 is an enzyme that degrades the endogenous NOS inhibitor ADMA, while PRMT1 is an enzyme that regulates ADMA production. In a canine model of pacing induced heart failure, we recently found that myocardial DDAH activity was decreased, while myocardial PRMT1 and DDAH1 protein contents were unchanged, after the development of CHF 10. Interestingly, in the present study TAC resulted in significant increases of both PRMT1 and DDAH1 in the wild type mice, suggesting model or strain dependent responses of these proteins.
Contribution of iNOS to oxidative stress in hypertrophied and failing hearts
Myocardial hypertrophy and failure are associated with increased superoxide anion (O2−) production 22, and accumulation of oxidized lipid and protein products such as nitrotyrosine and 4-HNE 5. We recently found that the SOD mimetic M40401 enhanced endothelium-dependent coronary vasodilation and increased LV dP/dtmax in dogs with pacing-induced CHF 10, implying that increased O2− production is partially responsible for coronary endothelial dysfunction and ventricular dysfunction in the failing heart. Oxygen free radicals are linked to fibrosis and matrix turnover involving the activation of MMPs 23. Overexpressing glutathione peroxidase 24, or administering BH4 to decrease myocardial O2− production 16 decreased myocardial MMP abundance. In the present study, the decreased myocardial oxidative stress in the iNOS deficient mice was associated with decreased MMP1 content, supporting the notion that oxidative stress affects myocardial matrix turnover.
NOS uncoupling
NOS can produce NO, O2− or peroxynitrite. This unique property is a consequence of the dimeric nature of the enzyme, in which the two subunits are able to function independently 25. NOS optimally exists as a homodimer that generates NO and L-citrulline from L-arginine. However, when exposed to oxidant stress, or when deprived of its reducing cofactor BH4 or substrate L-arginine, NOS can uncouple to the monomeric form that generates O2− rather than NO 16,26-28. BH4 is required for iNOS dimerisation 29 and also stabilizes nNOS and eNOS dimers. Thus, a decrease of BH4 or unregulated NO production by iNOS to decrease L-arginine availability can cause NOS uncoupling 25, 30. Using purified iNOS protein, several studies have demonstrated that iNOS can produce both NO and O2−, and that deficiency of L-arginine or BH4 will induce iNOS to produce O2− 27,30, indicating that iNOS can be uncoupled. In addition, in cultured macrophages depletion of cytosolic L-arginine triggered O2− production by iNOS that was blocked by a NOS inhibitor, suggesting that iNOS uncoupling was responsible for the O2− production 26.
In the cardiovascular system, most studies of NOS uncoupling have focused on eNOS. Thus, uncoupled eNOS produces O2− 31 in apoE deficient mice, and BH4 improves the endothelial dysfunction associated with hypercholesterolemia, atherosclerosis or hypertension 32-33. Takimoto et al recently demonstrated that TAC resulted in a decrease of plasma BH4, an increase of myocardial eNOS monomer content, and increased O2− production 16. Moreover, administration of BH4 reduced the eNOS monomer content and decreased myocardial O2− production and TAC-induced cardiomyopathy, indicating that eNOS uncoupling contributed to the TAC-induced ventricular dysfunction 16. In the present study, iNOS deletion reduced the evidence of TAC-induced myocardial oxidative stress, indicating that iNOS contributed to oxidative stress in the wild type mice, either directly through iNOS uncoupling or by iNOS–dependent eNOS uncoupling. Based on the finding that myocardial iNOS was expressed as both monomer and dimer, and iNOS deletion attenuated the evidence of oxidative stress in animals exposed to TAC, it can be concluded that iNOS uncoupling contributed to the increased oxidative stress in the wild type mice. However, iNOS might also decrease intracellular BH4 and L-arginine availability to eNOS and thereby induce eNOS uncoupling. The relative contributions of iNOS uncoupling vs. iNOS-dependent eNOS uncoupling in the TAC-induced increase of myocardial oxidative stress merit further investigation.
Akt, mTOR and S6 activation in response to oxidative stress
Akt phosphorylation can regulate ventricular hypertrophy 34, and oxidative stress has been reported to activate Akt 35 by regulating PTEN. Akt was reported to cause downstream activation of mTOR and p70S6K, and previous reports have demonstrated that inhibition of mTOR signaling with rapamycin attenuated TAC-induced ventricular hypertrophy 36-37 and caused regression of cardiac hypertrophy produced by TAC 38. Interestingly, a recently study reported that activation of Akt-mTOR and NFkappaB participate in the development of ventricular hypertrophy, while the antioxidant pyrrolidine dithiocarbamate attenuated NFkappaB, Akt and p70S6K activation and TAC-induced ventricular hypertrophy 37. Ribosomal protein S6 is a downstream target of p70S6K, and S6 phosphorylation increases the translation of a subset of mRNAs that promote protein synthesis. The finding of increased phospho-Akt and phospho-S6 in our study is consistent with the previous reports 16,37. Our finding that iNOS deletion attenuated TAC-induced myocardial oxidative stress, phospho-Akt, phospho-mTOR, phospho-S6 and ventricular hypertrophy supports the notion that oxidative stress exacerbates systolic overload-induced Akt-mTOR activation and ventricular hypertrophy.
Limitations
The pressure gradient across the aortic constriction was not measured in the present study. However, care was taken to insure that the identical TAC procedure was performed by the same surgeon who was blinded to the genotype of the mice. Since iNOS−/− attenuated the TAC-induced increase of eNOS expression, and eNOS monomer was present in wild type mice exposed to TAC, we were unable to determine whether the protective effect of iNOS−/− was directly due to the absence of iNOS or secondary to an attenuation of TAC-induced eNOS induction. We did not measure plasma BH4 levels in the present study. However, using an almost identical TAC model, a recent study reported that TAC resulted in a significant decrease of plasma BH4 in C57/B6J mice, and that administration of BH4 attenuated TAC-induced myocardial eNOS monomer formation and oxidative stress, suggesting that a decrease of BH4 after TAC may contribute to NOS monomer formation 16. An additional limitation is that the iNOS −/− and wild type mice were not littermates. Finally, because of the relatively large amount of tissue required for assay of myocardial NOS activity, this determination was not performed in the present study.
Supplementary Material
Acknowledgements
The authors wish to acknowledge the guidance, support and encouragement of Dr. Robert J. Bache which contributed importantly to the successful completion of this study.
Sources of Funding
This study was supported by NHLBI Grants HL71790 and HL21872 from the National Institute of health. Dr. Zhang is supported by a Scientist Development Award from the American Heart Association.
References
- 1.Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH, Cooke JP, McKenna WJ, Fowler MB. Expression of inducible nitric oxide synthase in human heart failure. Circulation. 1996;93:1087–1094. doi: 10.1161/01.cir.93.6.1087. [DOI] [PubMed] [Google Scholar]
- 2.Habib FM, Springall DR, Davies GJ, Oakley CM, Yacoub MH, Polak JM. Tumour necrosis factor and in dilated cardiomyopathy. Lancet. 1996;347:1151–1155. doi: 10.1016/s0140-6736(96)90610-8. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Traverse JH, Du R, Hou M, Bache RJ. Nitric oxide modulates myocardial oxygen consumption in the failing heart. Circulation. 2002;106:273–9. doi: 10.1161/01.cir.0000021120.90970.b9. [DOI] [PubMed] [Google Scholar]
- 4.Nadruz W, Jr, Lagosta VJ, Moreno H, Jr, Coelho OR, Franchini KG. Simvastatin prevents load-induced protein tyrosine nitration in overloaded hearts. Hypertension. 2004;43:1060–6. doi: 10.1161/01.HYP.0000124252.43470.2c. [DOI] [PubMed] [Google Scholar]
- 5.Liu YH, Carretero OA, Cingolani OH, Liao TD, Sun Y, Xu J, Li LY, Pagano PJ, Yang JJ, Yang XP. Role of Inducible Nitric Oxide Synthase in Cardiac Function and Remodeling in Mice with Heart Failure Due to Myocardial Infarction. Am J Physiol Heart Circ Physiol. 2005;289:H2616–23. doi: 10.1152/ajpheart.00546.2005. [DOI] [PubMed] [Google Scholar]
- 6.Heger J, Godecke A, Flogel U, Merx MW, Molojavyi A, Kuhn-Velten WN, Schrader J. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ Res. 2002;90:93–9. doi: 10.1161/hh0102.102757. [DOI] [PubMed] [Google Scholar]
- 7.Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T, Schulz R, Butany J, Stewart DJ, Husain M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest. 2002;109:735–43. doi: 10.1172/JCI13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–9. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 9.Thomsen LL, Scott JM, Topley P, Knowles RG, Keerie AJ, Frend AJ. Selective inhibition of inducible nitric oxide synthase inhibits tumor growth in vivo: studies with 1400W, a novel inhibitor. Cancer Res. 1997;57:3300–4. [PubMed] [Google Scholar]
- 10.Chen Y, Li Y, Zhang P, Traverse JH, Hou M, Xu X, Kimoto M, Bache RJ. Dimethylarginine dimethylaminohydrolase and endothelial dysfunction in failing hearts. Am J Physiol Heart Circ Physiol. 2005;289:H2212–9. doi: 10.1152/ajpheart.00224.2005. [DOI] [PubMed] [Google Scholar]
- 11.Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, Kitakaze M, Liao JK. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–37. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem. 2005;280:19230–42. doi: 10.1074/jbc.M411373200. [DOI] [PubMed] [Google Scholar]
- 13.Jones SP, Greer JJ, Ware PD, Yang J, Walsh K, Lefer DJ. Deficiency of iNOS does not attenuate severe congestive heart failure in mice. Am J Physiol Heart Circ Physiol. 2005;288:H365–70. doi: 10.1152/ajpheart.00245.2004. [DOI] [PubMed] [Google Scholar]
- 14.Feng Q, Lu X, Jones DL, Shen J, Arnold JMO. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- 15.Sam F, Sawyer DB, Xie Z, Chang DLF, Ngoy S, Brenner DA, Siwik DA, Singh K, Apstein CS, Colucci WS. Mice lacking inducible nitric oxide synthase have improved left ventricular contractile function and reduced apoptotic cell death late after myocardial infarction. Circ Res. 2001;89:351–356. doi: 10.1161/hh1601.094993. [DOI] [PubMed] [Google Scholar]
- 16.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–31. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cave A, Grieve D, Johar S, Zhang M, Shah AM. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2327–34. doi: 10.1098/rstb.2005.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res. 2005;66:444–53. doi: 10.1016/j.cardiores.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Heydemann A, Huber JM, Kakkar R, Wheeler MT, McNally EM. Functional nitric oxide synthase mislocalization in cardiomyopathy. J Mol Cell Cardiol. 2004;36:213–23. doi: 10.1016/j.yjmcc.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Ichinose F, Bloch KD, Wu JC, Hataishi R, Aretz HT, Picard MH, Scherrer-Crosbie M. Pressure overload-induced LV hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am J Physiol Heart Circ Physiol. 2004;286:H1070–5. doi: 10.1152/ajpheart.00940.2003. [DOI] [PubMed] [Google Scholar]
- 22.Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, Utsumi H, Machida Y, Egashira K, Takeshita A. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86:152–157. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- 23.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 24.Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H, Takeshita A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2004;109:544–9. doi: 10.1161/01.CIR.0000109701.77059.E9. [DOI] [PubMed] [Google Scholar]
- 25.Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43:521–31. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 26.Xia Y, Zweier JL. Direct measurement of nitric oxide generation from nitric oxide synthase. Proc Natl Acad Sci U S A. 1997;94:12705–10. doi: 10.1073/pnas.94.23.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93:6770–4. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–54. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 29.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–6. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 30.Xia Y, Roman LJ, Masters BS, Zweier JL. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem. 1998;273:22635–9. doi: 10.1074/jbc.273.35.22635. [DOI] [PubMed] [Google Scholar]
- 31.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–8. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 32.Cosentino F, Patton S, d'Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, Malinski T, Luscher TF. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest. 1998;101:1530–7. doi: 10.1172/JCI650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, Luscher T, Rabelink T. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99:41–6. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 35.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–10. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–70. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- 37.Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, Kelley JL, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL, Li C. Attenuation of cardiac hypertrophy by inhibiting both mTOR and NFkappaB activation in vivo. Free Radic Biol Med. 2005;39:1570–80. doi: 10.1016/j.freeradbiomed.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 38.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–5. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 39.Kalinowski L, Dobrucki LW, Brovkovych V, Malinski T. Increased nitric oxide bioavailability in endothelial cells contributes to the pleiotropic effect of cerivastatin. Circulation. 2002;105:933–8. doi: 10.1161/hc0802.104283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.