

Abstract
The pathogenesis of T cell-mediated diseases like rheumatoid arthritis (RA) has typically been explained in the context of the Th1-Th2 paradigm: the initiation/propagation by pro-inflammatory cytokines, and downregulation by Th2 cytokines. However, in our study based on the adjuvant-induced arthritis (AA) model of RA, we observed that Lewis (LEW) (RT.1l) rats at the recovery phase of AA showed the highest level of IFN-γ in recall response to mycobacterial heat-shock protein 65 (Bhsp65), whereas AA-resistant Wistar-Kyoto (WKY) (RT.1l) rats secreted high levels of IFN-γ much earlier following disease induction. However, no significant secretion of IL-10 or TGF-β was observed in either strain. Furthermore, pre-treatment of LEW rats with a peptide of self (rat) hsp65 (R465), which induced T cells secreting predominantly IFN-γ, afforded protection against AA and decreased IL-17 expression by the arthritogenic epitope-restimulated T cells. These results provide a novel perspective on the pathogenesis of autoimmune arthritis.
Keywords: Animal Models, Arthritis, Autoimmunity, Cytokines, Immune Regulation
INTRODUCTION
Rheumatoid arthritis (RA), multiple sclerosis (MS) and insulin-dependent diabetes mellitus (IDDM) represent three of the major human autoimmune disorders that are considered to be typical T cell-mediated diseases [1–7]. Until very recently, a simplified understanding of the pathogenesis of these diseases has invoked the damaging or pathogenic attribute of the T helper 1 (Th1) response versus the suppressive or regulatory function of the T helper 2 (Th2) response directed against specific disease-related antigens (Ags) [7–10]. This paradigm has been the cornerstone of the models of disease pathogenesis as well as therapeutic strategies for over two decades. However, the gradual accumulation of contradictory findings in some experimental model systems [7, 11–17] as well as the emerging role of interleukin-17 (IL-17) in the pathogenesis of inflammatory and autoimmune diseases [7, 18, 19] point to the need for revisiting the tenets of the Th1-Th2 paradigm in each of the major autoimmune diseases. Studies in models of experimental autoimmune encephalomyelitis (EAE) [7, 20, 21], collagen-induced arthritis (CIA) [14, 21, 22]}, type 1 diabetes (T1D) [15], uveitis [12], and myocarditis [13] have elaborated upon the disease-protective role of the pro-inflammatory cytokine, IFN-γ. In this study, we describe that IFN-γ is associated with protection against adjuvant arthritis (AA), which is considered to be a Th1-mediated disease. Our results validate and further enlarge the scope and implications of observations reported earlier in the AA model [23, 24].
Adjuvant arthritis, an experimental model of human rheumatoid arthritis (RA), can be induced in the Lewis (LEW) (RT.1l) rat by s.c. injection of heat-killed M. tuberculosis (Mtb) H37Ra [25–27]. Arthritic LEW rats raise T cell response to mycobacterial heat shock protein 65 (Bhsp65), and the T cells directed against the determinant region 180–188 and its longer variants are believed to represent the pathogenic subset of T cells [28–33]. AA is self-limiting with a gradual regression of inflammation. Unlike LEW rats, MHC-compatible Wistar-Kyoto (WKY) (RT.1l) rats are resistant to AA despite their ability to raise potent T cell response to Bhsp65/Mtb [29, 30]. The AA model has extensively been used over the past 5 decades for studies pertaining to disease pathogenesis and testing of new anti-arthritic agents. However, neither the cytokine profile of antigen (Bhsp65)-specific T cells at different phases of the disease nor the comparative cytokine responses of AA-susceptible versus AA-resistant rat strains have yet been fully defined.
In this study, we examined the temporal cytokine profiles of the Bhsp65-specific T cells in Mtb-immunized LEW and WKY rats, and further applied that information for immunomodulation of AA. Our results show that high levels of the pro-inflammatory cytokine IFN-γ are associated with recovery/resistance in AA, and that the IFN-γ induced by the disease-regulating peptide 465-479 of self (rat) hsp65 (R465) [34] downmodulates AA in part by suppressing IL-17. These results warrant a revisitation to the mechanisms underlying the pathogenesis of AA, which is considered to be a typical Th1-mediated disease.
METHODS
Animals
Male, 4–6 wk old Lewis (LEW/Hsd) (RT.1l) and Wistar-Kyoto (WKY/NHsd) (RT.1l) rats were purchased from Harlan Sprague-Dawley (HSD) (Indianapolis, IN and Madison, WI, respectively). The animals were housed in the vivarium of the University of Maryland School of Medicine (Baltimore, MD) and were handled in accordance with the guidelines of the institutional animal care and use committee (IACUC).
Antigens/Mitogen/Cytokine
The recombinant Bhsp65 was expressed and purified in our laboratory as described in detail elsewhere [34, 35]. Specific peptides containing the amino acid (a.a.) sequences of Bhsp65 and Rhsp65 were obtained from Macromolecular Resources and Global Peptide Services (both at Fort Collins, CO) [34, 35]. These include Bhsp65 peptides 180-188 (B180), 177-191 (B177), 333-379 (B333); Rhsp65 peptide 465-479 (R465); and HEL peptide 65-78 (HEL65). Tuberculin purified protein derivative (PPD) was purchased from Mycos Research (Fort Collins, CO). Hen eggwhite lysozyme (HEL) and Concanavalin A (Con A) were obtained from Sigma-Aldrich Co. (St. Louis, MO).
Induction and evaluation of AA
Rats were immunized s.c. at the base of the tail with 200 μL (5 mg/mL) of heat-killed M. tuberculosis H37Ra (Mtb) (Difco, Detroit, MI) suspended in mineral oil (Sigma-Aldrich). Beginning d 7 after immunization, these rats were scored daily for signs of arthritis. The severity of arthritis in each paw was graded on the basis of erythema and swelling of the paw on a scale of 0 to 4 [29, 35]. The highest arthritic score was 4 for each paw, with the maximum score of 16 per rat. Furthermore, the phases of AA were labeled as follows: incubation (Inc), onset (Ons), peak (Pk), and recovery (Rec) phase.
Lymph node cell (LNC) proliferation assay
The draining lymph nodes (para-aortic, inguinal, and popliteal) of arthritic LEW rats were harvested at the pre-defined phase of AA. LNC of WKY rats immunized with Mtb were harvested at the time points corresponding to different phases of AA in LEW rats. Thereafter, a single-cell suspension of LNC was prepared, and the cells were washed thrice with HBSS (Invitrogen, Frederick, MD) [29, 35]. These LNC were cultured at 37°C in 95% air and 5% CO2 in a flat-bottom 96-well plate at a concentration of 2.5 × 105 cells/well in HL-1 serum-free medium (Ventrex Laboratories, Portland, ME) supplemented with 2 mM L-glutamine, 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate, with or without antigen. Con A was used as a positive control, whereas HEL served as a negative control. The results of [3H]-thymidine (International Chemical and Nuclear, Irvine, CA) incorporation in cells were expressed either as counts per minute (cpm) or as a stimulation index (S.I. = cpm of cells cultured with antigen/cpm of cells in medium alone). LNC of each rat in a group were tested separately in replicates and then the results were pooled together.
Collection of supernatant from LNC culture and testing for cytokines by ELISA
As the pathogenesis of autoimmune arthritis involves T cells, B cells, and myeloid lineage cells [36–38], we tested bulk LNC to assess the overall disease-relevant cytokine milieu within the draining nodes in vivo during the course of AA. The draining LNC of Mtb-immunized LEW and WKY rats were harvested at defined time points post-injection (Inc, Ons, Pk, and Rec phase) and cultured in a 96-well plate as described above. These LNC were then restimulated with the appropriate antigen in vitro, and the culture supernates were collected at 48 or 72 h thereafter [35]. These supernates were either tested immediately or stored at −20°C for testing at a later time point using ELISA kits for detection of IFN-γ and IL-10 (all from Biosource, Camarillo, CA), or for TGF-β1 (R&D Systems, Minneapolis, MN). The detection limit (pg/mL) of assays for IFN-γ, IL-10, and TGF-β1 was 13, 10, and 4.2, respectively. (Other investigators [31, 33] and us (Moudgil, K.D. and Kim, E, personal observation) have found the detection of rat IL-4 rather difficult, and therefore, relied on IL-10 instead.) The assays were performed following the manufacturer’s instructions, and the results were expressed as pg/mL. For comparison between groups, background cytokine levels were deducted from antigen-induced cytokines and the results were expressed as Δ pg/mL (pg/mL of cytokine from cells cultured with antigen – pg/mL of cytokine from cells in medium alone) [35, 39]. For each rat in a group, LNC culture supernates in replicates were tested separately and then the results were pooled together.
Measurement of cytokines by ribonuclease protection assay (RPA) and real-time PCR
RNA was isolated from antigen-primed or control LNC by Trizol reagent (Invitrogen, Frederick, MD). The purity and quantity of RNA was estimated by reading the absorbance of purified RNA in a spectrophotometer at 260 and 280 nm. A) RPA: RPA was then performed using rat template sets (rCK1, 2, and 3) according to the manufacturer’s instructions (BD Pharmingen, San Diego, CA) [40, 41]. Probes labeled with α-32P-uridine triphosphate (8 × 105 cpm) were hybridized with total RNA (10 μg) prepared from LNC. The hybridized RNA were treated with RNase A and T1, and then run on a 5% urea-polyacrylamide-bisacrylamide (19: 1) gel. The expression of cytokines was quantified by phosphor imaging using L32 (a housekeeping gene: ribosomal protein L32) as a standard, and the results were presented as ‘normalized total pixel’. B) Real-time PCR: RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA), and the resulting cDNA was tested in a quantitative real-time PCR assay using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and ABI Prism 7900HT cycler (Applied Biosystems, Foster City, CA). Appropriate primers (synthesized at the Biopolymer Core Facility, UMB, Baltimore, MD) using the Primer Express 2.0 Program (Applied Biosystems, Foster City, CA) were designed for the detection of mRNAs for IL-17, IL-23, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The expression of cytokines was standardized against GAPDH expression, and the results were presented as ‘relative gene expression.’ The sequence of PCR primers was as follows: IL-17, forward: ttccatccatgtgcctgatg; IL-17, reverse: ctcggcgtttggacacact; IL-23, forward: aggtctcaaggacaacagcca; IL-23, reverse: aaggctcccctgtgaagatgt.
Pre-treatment of LEW rats with R465 and its effect on subsequent AA
Antigen (R465 or HEL) was emulsified in IFA and then used for s.c. immunization (200 ug/rat) of LEW rats. After 2 wk, these rats were injected with heat-killed Mtb for the induction of AA as described above. Thereafter, all rats were scored regularly for the severity of arthritis. The difference in the arthritic scores of the experimental (R465-treated) and control (HEL-treated) group of rats were analyzed statistically.
Adoptive transfer of Ag-primed LNC into LEW rats
LEW rats were immunized s.c. at the base of the tail either with peptide R465 in IFA (experimental group) or with peptide 65-78 of HEL (HEL65) in IFA (control group). After 2 wk, the draining LNC were harvested and cultured (3 × 106 cells/mL) for 48 h in complete RPMI-1640 (RPMI-1640 medium supplemented with 10% heat-inactivated FBS (Life Technologies, Rockville, MD), 1% L-glutamine, 1% penicillin-streptomycin, and 2-ME (5 × 10−5 M)) in the presence of Con A (2.5 μg/mL) [35]. Thereafter, these cells were collected, washed thoroughly, and transferred i.v. into naïve LEW rats at 1 × 108 cells/rat. On the day of cell transfer, the recipient rats and naïve rats (additional control) were immunized with Mtb (1 mg/rat) s.c. for induction of AA, and rats were then observed regularly for signs of arthritis. The severity of the disease was graded as described above.
Statistical analysis
Student-t test was used to test the statistical significance of the differences observed among various test and control groups. Either one- or two-tailed assuming equal or unequal variance (determined by the F test) was used as appropriate for the data. Nonparametric Wilcoxon-rank sum test was employed to compare the arthritic scores of any two groups of rats over the entire disease course. In all these tests, p < 0.05 was considered to be significant, except for two-tailed Student-t test for which p < 0.025 served as the cut-off value.
RESULTS
IFN-γ secretion level is highest during Rec phase of AA in LEW rats, but at Inc phase after Mtb challenge in AA-resistant WKY rats
To examine the temporal cytokine secretion profile in response to Bhsp65 during the course of AA, the draining LNC of Mtb-injected LEW rats were harvested at different time points corresponding to the Inc, Ons, Pk, and Rec phases of the disease as described above, and the supernates of LNC restimulated in vitro with the appropriate Ag were collected and tested for various cytokines by ELISA. In parallel, supernates of LNC harvested from Mtb-immunized WKY (AA-resistant) rats at the time points corresponding to different phases of AA in the LEW rat were also tested. The antigens tested include native Bhsp65 and two overlapping peptides of Bhsp65 (B177 and B180), which represent the arthritogenic epitope of Bhsp65 [29, 31, 32]. These two peptides are crossreactive at the T cell level, but the longer peptide is a better immunogen as well as a recall antigen compared to the shorter peptide.
There was little or no detectable ex vivo secretion of IFN-γ by LNC throughout the period of observation post-Mtb immunization of both LEW and WKY rats (data not shown). However, following restimulation with Bhsp65, there was a gradual increase in IFN-γ secretion by LNC of LEW rats with time post-Mtb injection, while an inverse profile was observed in WKY rats (Fig. 1). The IFN-γ levels secreted in response to B177 and B180 increased with time post-Mtb injection in LEW rats with the level for B177 higher than that for B180, and these levels were much higher than the minimal secretion observed in WKY rats. Taken together, AA-susceptible LEW rats paradoxically secreted predominantly pro-inflammatory cytokine IFN-γ during the Rec phase, when clinical signs of arthritis were in gradual regression, while AA-resistant WKY rats secreted high levels of the same cytokine during the Inc phase. These results suggest a positive association between the levels of IFN-γ and recovery/protection from arthritis.
Figure 1. The level of IFN-γ secreted by LNC of Mtb-immunized rats was highest during Rec phase in LEW rats, but at Inc phase in WKY rats.

The draining LNC were harvested from LEW (□) and WKY (■) rats at different time points after Mtb injection as described in detail under Materials and Methods. These LNC were cultured for 48 h in a 96-well plate with or without the addition of any exogenous antigen. The supernates were then collected and analyzed for IFN-γ by ELISA. The results are shown as pg/mL (mean ± SEM). (*, p < 0.05 and **, p ≤ 0.025, compared with cytokine secretion at Inc phase for the same rat strain; +, p ≤ 0.05, and ++, p ≤ 0.025, compared between the two rat strains at the corresponding phase of AA.) The results shown in Figures 1 and 2 are from 3 or more rats at each time point in the course of AA (Inc = incubation phase; Ons = onset phase; Pk = peak phase; and Rec = recovery phase).
The levels of IL-10 are relatively lower during Rec/Inc phase in LEW/WKY rats, respectively than that of the ex vivo levels suggesting inhibition by Th1 cytokines
We also examined the secretion of immunoregulatory/anti-inflammatory cytokines, IL-10 and TGF-β, during the course of AA. The profile of ex vivo IL-10 secretion by LNC of LEW rats followed that of the clinical course of AA (Fig. 2). Compared to the ex vivo secretion, IL-10 secretion in response to Bhsp65 was suppressed at both Ons and Pk phases in LEW rats, but only at Ons phase in WKY rats. This low IL-10 secretion (Fig. 2) contrasted with the high IFN-γ secretion (Fig. 1), suggesting the inhibition of IL-10 secretion by the Th1 cytokine. However, the pattern of IL-10 secretion in response to B177 and B180 was similar to that of ex vivo secretion for LEW/WKY rats. These results show that no significant IL-10 secretion was induced by these Bhsp65 peptides. In comparison to IL-10, there was no significant secretion of TGF-β in LEW or WKY rats either ex vivo or following Ag recall at different time points following Mtb immunization (data not shown).
Figure 2. The secretion of IL-10 by LNC of Mtb-primed LEW and WKY rats is inhibited after in vitro restimulation with Bhsp65.
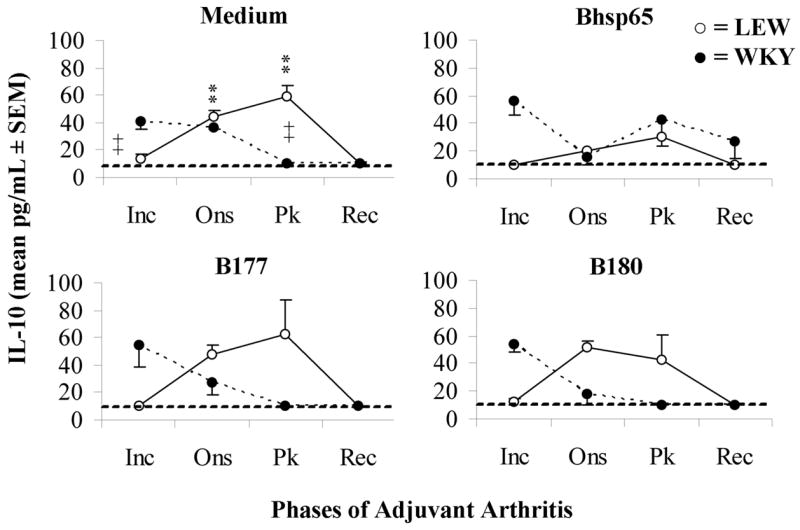
LNC from LEW (○) and WKY (●) rats were harvested at different time points after Mtb injection, and then cultured with Ag as described in the legend to Fig. 1. The supernates were collected and analyzed for IL-10 by ELISA. The results are presented as pg/mL (mean ± SEM). (*, p < 0.05 and **, p ≤ 0.025, compared with cytokine secretion at Inc phase for the same rat strain; +, p ≤ 0.05, and ++, p ≤ 0.025, compared between the two rat strains at the corresponding phase of AA.)
As described above, LEW and WKY rats immunized with Mtb reveal different profiles of cytokine responses. This difference in these two rat strains reflects a difference in the qualitative functional attributes of the Mtb-primed, antigen-recalled T cells rather than a quantitative difference in their response to Bhsp65 because the LEW and WKY rats immunized with Mtb gave comparable levels of proliferative responses to Bhsp65 and its pathogenic determinant B177 (Fig. 3). These results indirectly also suggest comparable levels of cellular priming events in the draining lymph nodes of Mtb-immunized LEW and WKY rats.
Figure 3. LEW and WKY rats raise comparable levels of response to Bhsp65 following Mtb challenge.
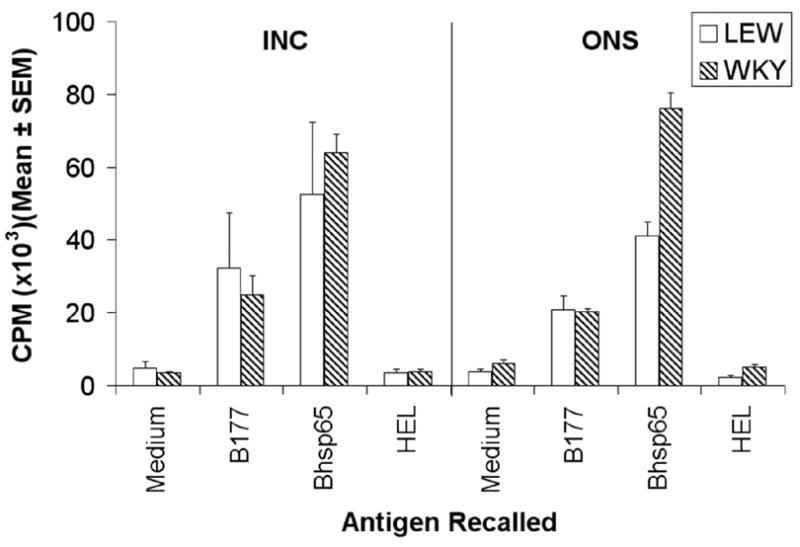
The draining LNC were harvested from LEW and WKY rats (n= 4+) at time points corresponding to the incubation (Inc) and onset (Ons) phases of AA after Mtb injection as described under ‘Materials and Methods’. These LNC were tested in a proliferation assay using the native Bhsp65 and its pathogenic epitope B177. HEL was used as the control antigen. The results are presented as cpm (mean ± SEM). The cpm values for the positive control (Con A) were as follows: LEW rats, Inc = 258,306 ± 6,046, Ons = 238,004 ± 9,036; and WKY rats, Inc = 218,598 ± 6,547, Ons = 248,075 ± 7,536.
Pre-treatment of LEW rats with AA-modulating C-terminal determinant 465-479 of Rhsp65 (R465) enhances Th1 cytokine expression but downregulates IL-17 expression by Mtb-immunized LNC in response to B177
Considering that the IFN-γ secretion profile of LNC re-stimulated with Bhsp65 or its pathogenic epitope B177/B180 correlated with reduced severity of AA, we reasoned that IFN-γ might induce protection against AA. To avoid potential toxicity, we explored a more practical approach for immunomodulation than the use of IFN-γ per se, namely, using a peptide of Rhsp65, R465-479 (R465), which we have previously shown to downmodulate the course of AA [34]. However, in that earlier study, neither the ability of R465-primed lymphoid cells to transfer protection against AA nor the cytokine profile of R465-reactive cells was determined. Therefore, we first examined these two aspects of R465, and then tested the influence of R465 pre-treatment on the immune response to the disease-related Ags to seek some insight into the AA-protective effect of R465. Prior to performing the adoptive transfer experiment, we independently validated the previously reported [34] AA-protective effect of the R465 peptide preparation used in this study (Fig. 4A).
Figure 4. Both the pretreatment with Rhsp65 peptide 465-479 (R465) and the adoptive transfer of R465-primed LNC into naïve LEW rats downmodulates the severity of AA.
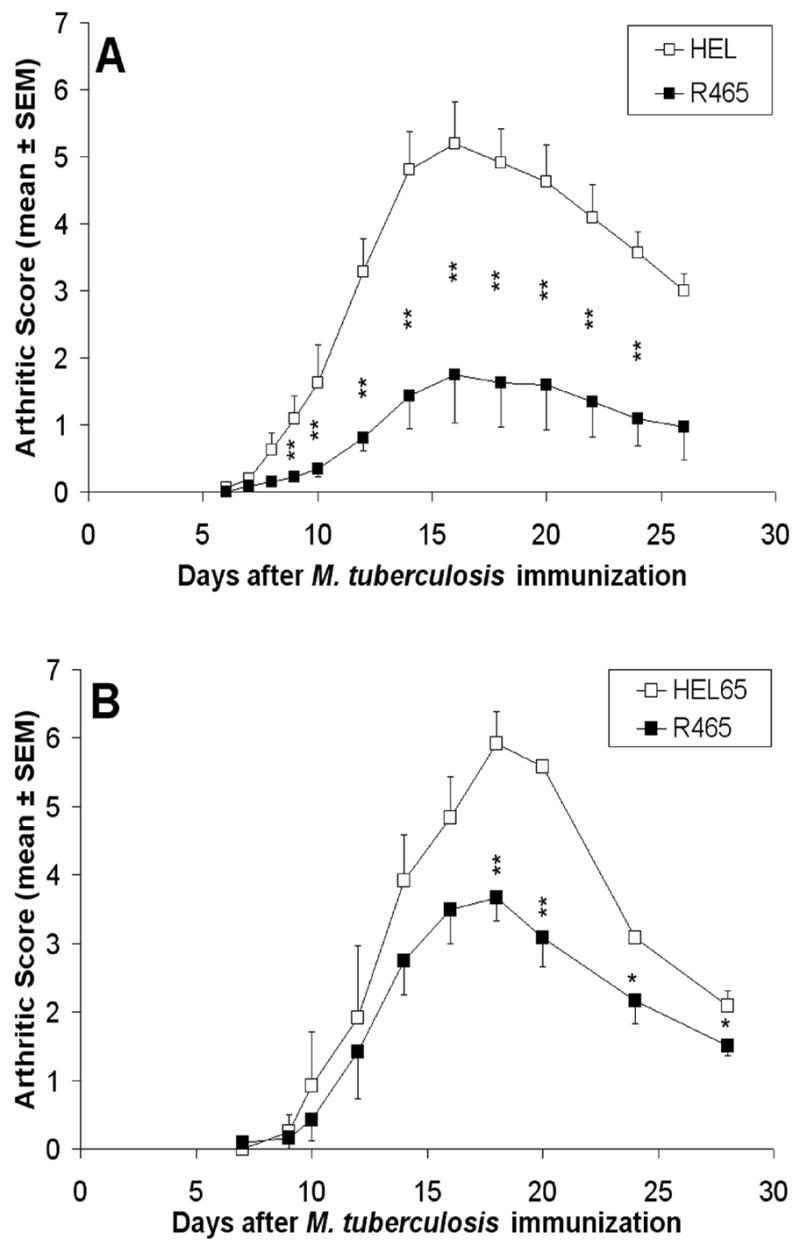
A cohorteach of LEW rats (experimental group) was immunized with R465, HEL, or HEL peptide 65-78 (HEL65), each emulsified in IFA. After 2 wk, some of these rats (A; n= 8 for each group) were injected with Mtb and then followed regularly and graded for signs of AA, whereas others (B; n= 3 for each group) were euthanized and their draining LNC were harvested and cultured with Con A (2.5 μg/mL) for 48 h for adoptive transfer. For the latter, 1x108 cells from R465- or HEL65-immunized rats were washed thoroughly and injected i.v. into the tail vein of naïve LEW recipients, followed by Mtb injection s.c. (*, p < 0.05 and **, p ≤ 0.025, when scores of the R465 group were compared with those of the HEL/HEL65 group. The difference in arthritic scores of the two respective rat groups was also significant (p < 0.05) by Wilcoxon rank sum test.)
In another set of experiments relating to adoptive transfer, we observed that the injection of R465-primed LNC into naïve LEW rats down-modulated the severity of AA (p<0.05) (Fig. 4B), further validating the results of peptide (R465)-induced protection (Fig. 4A) against subsequent AA. Moreover, the cytokine profile of these R465-primed cells showed an enhanced expression of IFN-γ, TNF-α, and TNF-β compared to that of the medium background or of the HEL-restimulated, control LNC (Fig. 5A), whereas IL-4, IL-5, and TGF-β1 were not detected (data not shown), and IL-10 expression was approximately three-fold lower than that of the pro-inflammatory cytokines. The results of ELISA for cytokine secretion by R465-primed LNC also showed a four-fold higher expression of IFN-γ compared to IL-10 (Fig. 5B). Thus, R465-primed LNC secreted predominantly pro-inflammatory cytokines.
Figure 5. (A) R465-primed LNC revealed a predominantly Th1 cytokine profile.

LEW rats were immunized with R465 in IFA. After 2 wk, their draining LNC were harvested and cultured either with R465 or with control Ag (HEL) for 24 h for cytokine testing by RPA. LNC cultured in medium alone (Medium) served as baseline controls. Total RNA was purified from LNC and further tested as described under Materials and Methods. The results of RPA were presented as ‘Normalized Total Pixel’ after cytokine densitometric readings were normalized with a housekeeping gene, L32. (B) RPA results validated by ELISA for cytokines. LEW rats were immunized with R465/IFA and their LNC were cultured as described in section ‘A’. After antigenic restimulation with R465 or HEL65, the culture supernates were tested in ELISA for cytokines. The results were expressed as pg/ml. (C, D) Pre-treatment of naïve LEW rats with R465 enhanced Th1 cytokine expression by B177-reactive LNC following Mtb challenge. LEW rats were immunized either with PBS/IFA (C) or with R465/IFA (D). After 2 wk, all these rats were challenged with Mtb. Nine days thereafter, the draining LNC were harvested and cultured with the indicated Ag for use in RPA as described in section ‘A’. The results of RPA are shown as ‘Normalized Total Pixel’.
We then determined the influence of R465 pre-treatment of LEW rats on the cytokine response to the arthritogenic epitope B177 of Bhsp65 following Mtb injection (test group) (Fig. 5D), and compared this with the response to B177 in rats that received PBS/IFA pre-treatment followed by Mtb injection (control group) (Fig. 5C). The results showed that R465 pretreatment led to increase in the expression of IFN-γ, TNF-α, and TNF-β in response to the pathogenic determinant B177 compared to the control group. Furthermore, an increase was also noted for IL-1α and IL-1β expression. Thus, pro-inflammatory cytokine secretion was upregulated following pre-treatment with R465, and it was associated with downmodulation of AA instead of aggravation of the disease.
It is increasingly being realized that IL-17 and IL-23 play an important role in inflammation and tissue-specific autoimmune diseases such as RA and multiple sclerosis (MS) [18]. To further define the AA-protective mechanisms induced by R465, we tested the expression, if any, of IL-17 by B177-reactive LNC and its modulation by R465. B177-restimulation of LNC of PBS-pretreated, Mtb-immunized LEW rats revealed a much higher level (23.2) of the relative gene expression for IL-17 compared to that (6.4) of control LNC cultured in medium alone (Fig. 6). However, no such increase was observed following restimulation of LNC with the control peptide B333 or with native Bhsp65. Interestingly, R465-pretreatment of LEW rats prior to Mtb challenge led to a significant suppression of the IL-17 gene expression in response to B177-restimulation. In contrast to IL-17, no significant change in IL-23 gene expression was observed in LNC upon restimulation with B177 or Bhsp65 (data not shown). Thus, the modulation of AA by R465 involves differential modulation of Th1 cytokines (Fig. 5) and IL-17 (Fig. 6).
Figure 6. R465 pre-treatment of LEW rats decreased IL-17 expression by B177-reactive cells.
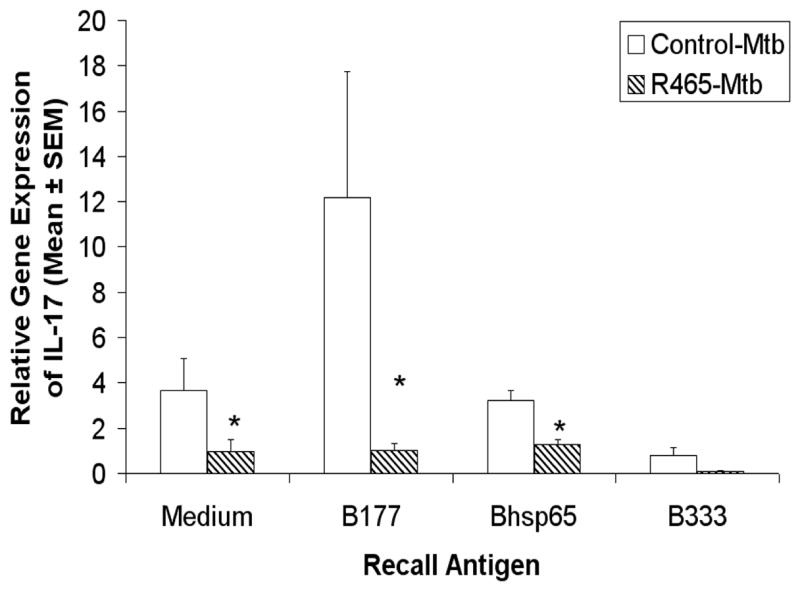
Following the immunization scheme described in section B of Figure 5, the draining LNC of LEW rats were harvested and cultured with Ag for use in real-time PCR. The results of IL-17 expression (n = 8) were standardized against GAPDH expression and presented as ‘Relative Gene Expression.’ *, p < 0.05, when compared with the respective PBS control.
DISCUSSION
In this study, we examined the Ag-specific cytokine responses to Bhsp65, B177, and B180 in AA-susceptible LEW rats versus AA-resistant WKY rats following a potentially arthritogenic challenge with Mtb. The overlapping peptides B177 and B180 contain the arthritogenic epitope 180-188 of Bhsp65 [29, 31, 32]. As Bhsp65 contains multiple T cell epitopes [28–33], the cytokine profile of LNC of LEW/WKY rats recalled with Bhsp65 is expected to show differences compared to those obtained using B177/B180 as recall antigens. In addition, the overlapping peptides B177 and B180 are expected to show subtle differences in cytokine profiles owing to their differences in immunogenicity and avidity for T cell recall response [29, 31, 32]. Furthermore, although both LEW and WKY rats raise potent proliferative T cell response to B177/B180 following Mtb challenge [29], their cytokine responses to these two epitopes have not been tested before. Considering that LEW rats are AA-susceptible, whereas WKY rats are AA-resistant [29, 42], differences in cytokine secretion in response to B177 and/or B180 in the two rat strains are not unexpected. Furthermore, LEW and WKY rats differentially respond to multiple epitopes of Bhsp65 after Mtb immunization [29], therefore, one or more of the multiple epitopes of Bhsp65 besides B177/B180 can contribute to the cytokines secreted in recall response to native Bhsp65 in LEW as well as WKY rats.
LEW rats secreted predominantly Th1 cytokine IFN-γ at the highest level during the recovery phase of AA, while WKY rats secreted the same cytokine at the maximum level relatively early after Mtb challenge, when LEW rats neither showed signs of clinical arthritis nor produced any significant amounts of IFN-γ. In contrast, no significant secretion of IL-10 or TGF-β was observed in LEW or WKY rats. These results suggest that the pro-inflammatory cytokine IFN-γ rather than the typical immunoregulatory/anti-inflammatory cytokines plays a major role in recovery from, as well as resistance against, AA.
We observed that AA in the LEW rat was downmodulated by a Th1-inducing peptide (R465) of self hsp65, both by the pretreatment with R465 in IFA and by the adoptive transfer of R465-primed T cells. The pro-inflammatory cytokine transcript levels following immunization with R465 were increased by approximately 50% (for TNF-α and TNF-β to more than 200% (for IFN-γ) above the controls, while the levels of anti-inflammatory cytokine transcripts were either not detected (for IL-4, IL-5 and TGF-β1) or were much lower (for IL-10) than the levels of pro-inflammatory cytokines. The results of RPA were further validated by testing of cytokines by ELISA. Furthermore, pretreatment with R465 caused an increased expression of pro-inflammatory cytokines in response to the arthritogenic epitope B177 in Mtb-immunized LEW rats compared to cytokine secretion by B177 in rats pre-treated with PBS/IFA and immunized with Mtb. Thus, a pro-inflammatory type of cytokine response to the pathogenic determinant of Bhsp65 was associated with decreased severity of AA. Our results regarding the protective effect of IFN-γ are supported by those of others showing that the administration of recombinant IFN-γ led to reduced severity of AA and improved recovery from the disease [43], and that the injection of anti-IFN-γ antibodies before induction of AA [44] as well as to recipient rats prior to adoptive transfer of arthritogenic LNC [23] aggravated the severity of arthritis. Taken together, these two complementary approaches demonstrate the protective effect of IFN-γ against AA. In addition, other investigators have also reported that Ag-induced T cells that are capable of suppressing AA secrete relatively higher levels of IFN-γ compared to that of IL-10 [31].
Furthermore, it has been shown in another model of arthritis that IFN-γ−/− mice exhibited increased severity of collagen-induced arthritis (CIA) [14], and that IFN-γ regulates susceptibility to CIA [22]. The results of the disease-protective effect of IFN-γ in two different models of arthritis (AA in this study and CIA [22]) involving two different target antigens as well as two rodent species corroborate the significance of this physiologically relevant observation. Also reported are the failure of IFN-γR−/− mice to recover from EAE [7, 20] and the role of IL-12/IFN-γ–mediated protective mechanisms in animal models of diabetes [15], uveitis [12] myocarditis [13], and cardiac allograft [45]. These studies coupled with ours show that IFN-γ can regulate autoimmune pathology involving pro-inflammatory cytokines.
Besides IFN-γ and TNF-α other cytokines such as IL-17 and IL-23 have been shown to be important in inflammation and autoimmune diseases such as RA [46] and CIA [7, 47, 48]. IL-17 was found in RA synovium, and it has been shown to synergize with IL-1 to induce IL-6 production by synovial fibroblasts from RA patients [46]. A study in AA showed that IL-17 was expressed transiently in the LN during early part of the disease (Inc phase) [24]. In our study, we have shown that B177-reactive T cells express IL-17, and that the suppression of this IL-17 production is associated with decreased severity of AA following R465-pretreatment of LEW rats. This result is indirectly supported by the finding that IFN-γ may negatively regulate the differentiation of naïve CD4+ T cells into IL-17-producing T cells [19]. Finally, the AA-protective effects of R465 treatment might also involve antigen-induced CD4+CD25+ T cells [49–51] and disease-regulating antibodies [33, 42]. Besides peptide-based approaches, the regulatory attribute of IFN-γ in arthritis also can be further explored for therapeutic purposes by inducing appropriate modulation of cytokine signaling [52].
Our results show that high levels of IFN-γ have a regulatory role in AA. However, this cytokine is also known to play a role in the induction and maintenance of inflammation [8, 9]. Therefore, to reconcile these contrasting roles of IFN-γ, we propose a model in which a particular threshold of this cytokine is required for the initiation of inflammation, but the secretion of the same cytokine beyond a critical level triggers regulatory mechanisms that suppress the ongoing inflammation. Furthermore, the modulation of IL-17 production can help explain the seemingly unexpected results of some of the earlier studies in animal models of autoimmunity in which the interpretations regarding the outcome of altered IFN-γ level/activity were based primarily on the classical Th1-Th2 paradigm [7, 18, 19]. In this regard, the Th17 hypothesis has emerged as a new paradigm in fully understanding the pathogenesis and immune regulation of autoimmunity [7].
Acknowledgments
This work was supported by grants (AI-047790 and AI-059623) from the National Institute of Health, Bethesda, MD, and the Arthritis Foundation, Atlanta, GA. We thank Martin Flajnik, Peter Calabresi, John Sacci, Dean Mann, and Stefanie Vogel for their critique and suggestions. We also thank Swamy Kumar Polumuri for help in the optimization of the real time PCR assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berner B, Akca D, Jung T, Muller GA, Reuss-Borst MA. Analysis of Th1 and Th2 cytokines expressing CD4+ and CD8+ T cells in rheumatoid arthritis by flow cytometry. J Rheumatol. 2000;27:1128–1135. [PubMed] [Google Scholar]
- 2.Park SH, Min DJ, Cho ML, Kim WU, Youn J, Park W, Cho CS, Kim HY. Shift toward T helper 1 cytokines by type II collagen-reactive T cells in patients with rheumatoid arthritis. Arthritis Rheum. 2001;44:561–569. doi: 10.1002/1529-0131(200103)44:3<561::AID-ANR104>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 3.Krakauer M, Sorensen PS, Sellebjerg F. CD4(+) memory T cells with high CD26 surface expression are enriched for Th1 markers and correlate with clinical severity of multiple sclerosis. J Neuroimmunol. 2006;181:157–164. doi: 10.1016/j.jneuroim.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Dhib-Jalbut S. Pathogenesis of myelin/oligodendrocyte damage in multiple sclerosis. Neurology. 2007;68:S13–21. doi: 10.1212/01.wnl.0000275228.13012.7b. discussion S43–54. [DOI] [PubMed] [Google Scholar]
- 5.Karlsson MG, Lawesson SS, Ludvigsson J. Th1-like dominance in high-risk first-degree relatives of type I diabetic patients. Diabetologia. 2000;43:742–749. doi: 10.1007/s001250051372. [DOI] [PubMed] [Google Scholar]
- 6.Wilson SB, Kent SC, Patton KT, Orban T, Jackson RA, Exley M, Porcelli S, Schatz DA, Atkinson MA, Balk SP, Strominger JL, Hafler DA. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature. 1998;391:177–181. doi: 10.1038/34419. [DOI] [PubMed] [Google Scholar]
- 7.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 8.Romagnani S. Regulation of the T cell response. Clin Exp Allergy. 2006;36:1357–1366. doi: 10.1111/j.1365-2222.2006.02606.x. [DOI] [PubMed] [Google Scholar]
- 9.O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2:37–45. doi: 10.1038/nri702. [DOI] [PubMed] [Google Scholar]
- 10.Adorini L. Cytokine-based immunointervention in the treatment of autoimmune diseases. Clin Exp Immunol. 2003;132:185–192. doi: 10.1046/j.1365-2249.2003.02144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gor DO, Rose NR, Greenspan NS. TH1-TH2: a procrustean paradigm. Nat Immunol. 2003;4:503–505. doi: 10.1038/ni0603-503. [DOI] [PubMed] [Google Scholar]
- 12.Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, Caspi RR. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis. J Exp Med. 1999;189:219–230. doi: 10.1084/jem.189.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afanasyeva M, Wang Y, Kaya Z, Stafford EA, Dohmen KM, Sadighi Akha AA, Rose NR. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 2001;104:3145–3151. doi: 10.1161/hc5001.100629. [DOI] [PubMed] [Google Scholar]
- 14.Guedez YB, Whittington KB, Clayton JL, Joosten LA, van de Loo FA, van den Berg WB, Rosloniec EF. Genetic ablation of interferon-gamma up-regulates interleukin-1beta expression and enables the elicitation of collagen-induced arthritis in a nonsusceptible mouse strain. Arthritis Rheum. 2001;44:2413–2424. doi: 10.1002/1529-0131(200110)44:10<2413::aid-art406>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 15.Trembleau S, Penna G, Gregori S, Giarratana N, Adorini L. IL-12 administration accelerates autoimmune diabetes in both wild-type and IFN-gamma-deficient nonobese diabetic mice, revealing pathogenic and protective effects of IL-12-induced IFN-gamma. J Immunol. 2003;170:5491–5501. doi: 10.4049/jimmunol.170.11.5491. [DOI] [PubMed] [Google Scholar]
- 16.Campbell IK, O’Donnell K, Lawlor KE, Wicks IP. Severe inflammatory arthritis and lymphadenopathy in the absence of TNF. J Clin Invest. 2001;107:1519–1527. doi: 10.1172/JCI12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 19.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol. 1999;163:5278–5286. [PubMed] [Google Scholar]
- 21.Zhang J. Yin and yang interplay of IFN-gamma in inflammation and autoimmune disease. J Clin Invest. 2007;117:871–873. doi: 10.1172/JCI31860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56:1145–1151. doi: 10.1002/art.22453. [DOI] [PubMed] [Google Scholar]
- 23.Brasted M, Spargo LD, Mayrhofer G, Cleland LG. Blockade of IFN-gamma does not affect the arthritogenicity of T cells generated during the induction of adjuvant arthritis but exacerbates the polyarthritis produced by adoptive transfer of arthritogenic effector cells. Immunol Cell Biol. 2005;83:189–195. doi: 10.1111/j.1440-1711.2005.01313.x. [DOI] [PubMed] [Google Scholar]
- 24.Bush KA, Walker JS, Lee CS, Kirkham BW. Cytokine expression and synovial pathology in the initiation and spontaneous resolution phases of adjuvant arthritis: interleukin-17 expression is upregulated in early disease. Clin Exp Immunol. 2001;123:487–495. doi: 10.1046/j.1365-2249.2001.01469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holoshitz J, Naparstek Y, Ben-Nun A, Cohen IR. Lines of T lymphocytes induce or vaccinate against autoimmune arthritis. Science. 1983;219:56–58. doi: 10.1126/science.6336851. [DOI] [PubMed] [Google Scholar]
- 26.Pearson CM. Development of arthritis, periarthritis and periostitis in rats given adjuvants. Proc Soc Exp Biol Med. 1956;91:95–101. doi: 10.3181/00379727-91-22179. [DOI] [PubMed] [Google Scholar]
- 27.Taurog JD, Sandberg GP, Mahowald ML. The cellular basis of adjuvant arthritis. II. Characterization of the cells mediating passive transfer. Cell Immunol. 1983;80:198–204. doi: 10.1016/0008-8749(83)90106-5. [DOI] [PubMed] [Google Scholar]
- 28.van Eden W, Thole JE, van der Zee R, Noordzij A, van Embden JD, Hensen EJ, Cohen IR. Cloning of the mycobacterial epitope recognized by T lymphocytes in adjuvant arthritis. Nature. 1988;331:171–173. doi: 10.1038/331171a0. [DOI] [PubMed] [Google Scholar]
- 29.Moudgil KD, Chang TT, Eradat H, Chen AM, Gupta RS, Brahn E, Sercarz EE. Diversification of T cell responses to carboxy-terminal determinants within the 65-kD heat-shock protein is involved in regulation of autoimmune arthritis. J Exp Med. 1997;185:1307–1316. doi: 10.1084/jem.185.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moudgil KD. Diversification of response to hsp65 during the course of autoimmune arthritis is regulatory rather than pathogenic. Immunol Rev. 1998;164:175–184. doi: 10.1111/j.1600-065x.1998.tb01219.x. [DOI] [PubMed] [Google Scholar]
- 31.Quintana FJ, Carmi P, Mor F, Cohen IR. Inhibition of adjuvant arthritis by a DNA vaccine encoding human heat shock protein 60. J Immunol. 2002;169:3422–3428. doi: 10.4049/jimmunol.169.6.3422. [DOI] [PubMed] [Google Scholar]
- 32.Anderton SM, van der Zee R, Prakken B, Noordzij A, van Eden W. Activation of T cells recognizing self 60-kD heat shock protein can protect against experimental arthritis. J Exp Med. 1995;181:943–952. doi: 10.1084/jem.181.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulmansky R, Cohen CJ, Szafer F, Moallem E, Fridlender ZG, Kashi Y, Naparstek Y. Resistance to adjuvant arthritis is due to protective antibodies against heat shock protein surface epitopes and the induction of IL-10 secretion. J Immunol. 2002;168:6463–6469. doi: 10.4049/jimmunol.168.12.6463. [DOI] [PubMed] [Google Scholar]
- 34.Durai M, Kim HR, Moudgil KD. The regulatory C-terminal determinants within mycobacterial heat shock protein 65 are cryptic and cross-reactive with the dominant self homologs: implications for the pathogenesis of autoimmune arthritis. J Immunol. 2004;173:181–188. doi: 10.4049/jimmunol.173.1.181. [DOI] [PubMed] [Google Scholar]
- 35.Durai M, Gupta RS, Moudgil KD. The T cells specific for the carboxyl-terminal determinants of self (rat) heat-shock protein 65 escape tolerance induction and are involved in regulation of autoimmune arthritis. J Immunol. 2004;172:2795–2802. doi: 10.4049/jimmunol.172.5.2795. [DOI] [PubMed] [Google Scholar]
- 36.Firestein GS. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol. 2005;11:S39–44. doi: 10.1097/01.rhu.0000166673.34461.33. [DOI] [PubMed] [Google Scholar]
- 37.Carter RH. B cells in health and disease. Mayo Clin Proc. 2006;81:377–384. doi: 10.4065/81.3.377. [DOI] [PubMed] [Google Scholar]
- 38.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5:399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 39.Mia MY, Durai M, Kim HR, Moudgil KD. Heat shock protein 65-reactive T cells are involved in the pathogenesis of non-antigenic dimethyl dioctadecyl ammonium bromide-induced arthritis. J Immunol. 2005;175:219–227. doi: 10.4049/jimmunol.175.1.219. [DOI] [PubMed] [Google Scholar]
- 40.Bouziane M, Miao F, Bates SE, Somsouk L, Sang BC, Denissenko M, O’Connor TR. Promoter structure and cell cycle dependent expression of the human methylpurine-DNA glycosylase gene. Mutat Res. 2000;461:15–29. doi: 10.1016/s0921-8777(00)00036-7. [DOI] [PubMed] [Google Scholar]
- 41.Youssef JA, Bouziane M, Badr MZ. Age-dependent effects of nongenotoxic hepatocarcinogens on liver apoptosis in vivo. Mech Ageing Dev. 2003;124:333–340. doi: 10.1016/s0047-6374(02)00189-6. [DOI] [PubMed] [Google Scholar]
- 42.Kim HR, Kim EY, Cerny J, Moudgil KD. Antibody responses to mycobacterial and self heat shock protein 65 in autoimmune arthritis: epitope specificity and implication in pathogenesis. J Immunol. 2006;177:6634–6641. doi: 10.4049/jimmunol.177.10.6634. [DOI] [PubMed] [Google Scholar]
- 43.Nakajima H, Takamori H, Hiyama Y, Tsukada W. The effect of treatment with recombinant gamma-interferon on adjuvant-induced arthritis in rats. Agents Actions. 1991;34:63–65. doi: 10.1007/BF01993239. [DOI] [PubMed] [Google Scholar]
- 44.Wiesenberg I, Van der Meide PH, Schellekens H, Alkan S. Suppression and augmentation of rat adjuvant arthritis with monoclonal anti-interferon-gamma antibody. Clin Exp Immunol. 1989;78:245–249. [PMC free article] [PubMed] [Google Scholar]
- 45.Guillonneau C, Hill M, Hubert FX, Chiffoleau E, Herve C, Li XL, Heslan M, Usal C, Tesson L, Menoret S, Saoudi A, Le Mauff B, Josien R, Cuturi MC, Anegon I. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-gamma, and indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:1096–1106. doi: 10.1172/JCI28801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–414. [PubMed] [Google Scholar]
- 47.Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, Schwarzenberger P, van de Loo FA, van den Berg WB. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- 48.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A. 2003;100:5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.La Cava A, Ebling FM, Hahn BH. Ig-reactive CD4+CD25+ T cells from tolerized (New Zealand Black x New Zealand White)F1 mice suppress in vitro production of antibodies to DNA. J Immunol. 2004;173:3542–3548. doi: 10.4049/jimmunol.173.5.3542. [DOI] [PubMed] [Google Scholar]
- 50.Prakken BJ, Samodal R, Le TD, Giannoni F, Yung GP, Scavulli J, Amox D, Roord S, de Kleer I, Bonnin D, Lanza P, Berry C, Massa M, Billetta R, Albani S. Epitope-specific immunotherapy induces immune deviation of proinflammatory T cells in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2004;101:4228–4233. doi: 10.1073/pnas.0400061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Kyttaris VC, Juang YT, Tsokos GC. Immune cells and cytokines in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2005;17:518–522. doi: 10.1097/01.bor.0000170479.01451.ab. [DOI] [PubMed] [Google Scholar]