

Abstract
Implicit solvent based methods play an increasingly important role in molecular modeling of biomolecular structure and dynamics. Recent methodological developments have mainly focused on extension of the generalized Born (GB) formalism for variable dielectric environments and accurate treatment of nonpolar solvation. Extensive efforts in parameterization of GB models and implicit solvent force fields have enabled ab initio simulation of protein folding to native or near-native structures. Another exciting area that has benefitted from the advances in implicit solvent models is the development of constant pH molecular dynamics methods, which have recently been applied to calculations of protein pKa values and studies of pH-dependent peptide and protein folding.
Keywords: aggregation, continuum electrostatics, force field, generalized Born, hydrophobic interactions, length-scale, misfolding, molecular dynamics, protein folding, solvent accessible surface area, van der Waals
Introduction
Proper description of the solvent environment is critical in simulations of biological molecules and assemblies. While the accuracy of general purpose molecular mechanics force fields has consistently improved over the years, substantial limitations remain when it comes to exploring large conformational reorganization of biomolecules, such as in protein folding. Many of these limitations are related to the need for an accurate and efficient description of solvent. Explicit inclusion of water molecules (and/or membrane lipid molecules) arguably provides the highest level of detail, but, at the same time, significantly increases the system size and thus the associated computational cost. Alternatively, one may explicitly include only the solute degrees of freedom, and assume that the mean influence of solvent can be captured by the free energy cost of solvating the solute in a given configuration. Such an implicit treatment of solvent not only substantially reduces the system size, but also allows faster sampling of the solute conformation due to the absence of solvent friction. Recent methodological developments in implicit solvent models have allowed accurate calculation of the solvation free energy with moderate computational cost, leading to an explosion of applications, ranging from protein folding/misfolding to drug design. The rapid calculation of the solvation free energy (i.e., without the necessity to average over the solvent degrees of freedom) has also greatly facilitated the development of other novel simulation methods, in particular, constant pH molecular dynamics techniques, which have opened a door to the theoretical study of many important pH-dependent conformational processes. There have been a number of excellent reviews on different aspects of implicit solvent models as well as the constant pH molecular dynamics methods in the last few years [1, 2, 3, 4]. Here, we will briefly review the main approximations underlying the implicit solvent models, and focus on the important advances in development and applications that were published during the period from early 2004 to mid 2007. Since the physical and mathematical details of constant pH molecular dynamics methods have been discussed by Mongan and Case in a recent review [4], we will focus on the methodological advances and application studies that emerged after that review. We will conclude this review with a discussion of possible areas for improvement and future directions of research.
General Principles of Implicit Solvent Theories
In general, an implicit treatment of solvent aims to capture the mean influence of solvent via direct estimation of the solvation free energy, defined as the reversible work required to transfer the solute in a fixed configuration from vacuum to solution. In fully empirical approaches, the solvation free energy is estimated directly from certain geometric properties of the solute, such as the atomic solvent-exposed surface area or solvent-exclusion volume, using empirical free energy scales [5, 6]. A more rigorous approach is to decompose the total solvation free energy into nonpolar and electrostatic contributions. This allows both contributions to be related to appropriate continuum models of water, and is generally more accurate than the fully empirical approaches. Continuum electrostatics is the most well-established theory for the description of electrostatic solvation, where the solute is considered as a cavity embedded in a high dielectric medium. The corresponding electrostatic solvation free energy can be calculated rigorously by solving the Poisson-Boltzmann (PB) equation [2], or approximately using the generalized Born (GB) theory [7]. GB offers higher computational efficiency and readily allows the analytical evaluation of forces. Therefore, it is often preferred over PB methods for molecular dynamics (MD) simulations. Furthermore, with continual improvement over the last few years, many GB models are now capable of achieving the same level of accuracy as PB [1].
In contrast to the advances in the development of GB theory, methods for accurate treatment of nonpolar solvation are lacking. The nonpolar solvation free energy is either largely ignored, or estimated from the solvent-accessible surface area (SA). With the significant improvement in the electrostatic solvation models, limitations of simple SA models for biomolecule simulation are becoming increasingly evident [8, 9]. Furthermore, the dramatic reduction in the system size with elimination of the solvent molecules comes with some inevitable consequences. For example, implicit solvent may fail to capture short-range effects where detailed interplay of a few water molecules is important. Nonetheless, the GB/SA combination is now recognized as a prime choice for implicit treatment of solvent in biomolecular simulations [1]. Various implementations are available in major molecular modeling software packages, and have found applications to a wide range of biological problems.
Recent Methodological Developments
Modeling variable dielectric environments
Standard GB models are designed and parameterized for the typical situation with a solute dielectric constant of εp=1 and a solvent dielectric constant of εw ∼80. In these models, the effective Born radii only depend on the molecular geometry, but not on the solute and solvent dielectric constants. This can be problematic, for example, if one wants to study the free energy of transfer of a solute between two solvents with different dielectric constants. Feig et al. proposed an empirical extension that includes explicit dependence of the effective Born radii on the dielectric environment and demonstrated improved agreement with the reference “exact” PB solutions [10]. The theory was later extended to model biological membranes as multiple layers of infinite dielectric slabs [11]. Starting from Kirkwood's exact solution for the case of a random charge distribution in a sphere, Onufriev and coworkers derived an approximate analytical solution that does not require summing infinite series, and later extended it to handle arbitrary molecular shape [12, 13]. More recently, Zhou and coworkers proposed heavily parameterized GB models that directly scale the total solvation free energy using empirical functions derived from fitting to the PB results, in addition to several other modifications [14]. While the errors of the total solvation free energy of proteins relative to the PB results are effectively reduced, it is not clear how well these models reproduce the exact effective Born radii for both buried and exposed atoms.
Optimization of GB implicit solvent force fields
Consistent with the view that the GB framework has reached a mature stage (for standard aqueous simulations), there have been extensive efforts directed towards optimizing the GB/SA implicit solvent force fields in recent years. We emphasize the important difference between the “numerical” accuracy and “physical” accuracy of the GB models. The former property refers to the ability of a GB theory to numerically reproduce the corresponding “exact” PB solution, given the same solvent boundary and other physical parameters (e.g., the intrinsic atomic radii); the latter refers to the ability in reproducing actual (often experimental) physical properties, such as the solvation free energy, native structure and thermodynamic stability. Achieving high physical accuracy does not only require a GB model to be numerically accurate, but also depends on a range of physical parameters including the intrinsic atomic radii, definition of the solvent boundary (if adjustable), and very importantly, the underlying protein force field. Also note that the optimized physical parameters (e.g., intrinsic radii and torsion potentials) should be directly transferable to the corresponding PB models. Chen et al. recently reported an extensive optimization of the GBSW/SA implicit solvent model in CHARMM [15]. The resulting force field appears to improve the balance between solvation and intramolecular interactions and correctly predicts the secondary structure preferences and stabilities for a range of helical and beta peptides. The same force field successfully folded both beta-hairpin trpzip2 and mini-protein Trp-cage to about 1.0 Åbackbone RMSD, as illustrated in Figure 1. A GB/SA model in Amber was also carefully optimized to improve the balance of secondary structure preferences [16], following several previous modifications [17, 18]. These optimizations represent important advances toward the accurate modeling of protein conformational equilibrium. It is also worth noting that they all directly benefitted from extensive folding and unfolding simulations of model peptides, which has only been possible with implicit solvent.
Figure 1.

Representative folded structures of trpzip2 beta hairpin (top) and Trp-cage mini-protein (bottom), in comparison with the corresponding NMR structures. Both folded structures correspond to the average structures of the largest ensembles at 270 K sampled during the last 10 nanoseconds of replica-exchange folding simulations [15]. RMSD values shown were computed using all backbone heavy atoms.
With continual improvement in the accuracy of modeling electrostatic solvation, nonpolar solvation is becoming a critical bottleneck. Levy and coworkers have demonstrated that it is important to decompose the nonpolar solvation free energy further into a repulsive cavity term and an attractive solute-solvent dispersion interaction term [8]. While the cavity term scales well with the surface area, the dispersion term depends strongly on the atomic composition of the solute and only approximately tracks the surface area. The decomposition allows proper description of solvent screening of the solute-solute dispersion interactions. Levy and coworkers further proposed a continuum van der Waals solvent model [8], and later implemented an empirical approximation for quick estimation of the dispersion term [19]. The importance of an explicit solute-solvent dispersion term has been confirmed in a study where the nonpolar mean solvation forces from explicit solvent simulations were examined [20]. Following the theoretical work of Chandler and coworkers [21], Chen and Brooks illustrated several important consequences of neglecting the length-scale dependence of hydrophobic solvation in current SA-based nonpolar models [9]. Specifically, a SA model with conformational independent surface tension coefficient overstabilizes pair-wise nonpolar interactions and predicts anti-cooperativity instead of cooperativity in three-body hydrophobic association. The importance of length-scale dependence of hydrophobic solvation in protein conformational equilibrium was also demonstrated by microsecond time-scale MD simulations of a β-hairpin in explicit and implicit solvents [22]. McCammon and coworkers argued that electrostatic and nonpolar solvation are coupled and that one needs to solve an optimization problem to derive the most appropriate solvent boundary for both components [23]. However, it appears that current implicit solvent models have not reached the level of accuracy to meaningfully accout for such secondary effects in biomolecular simulations.
Other developments of implicit solvent models
The original GB formalism has also been extended and modified for specialized applications, including a residue pair-wise GB for efficient rotamer placement in computational protein design [24] and an approximate GB potential suitable for Monte Carlo simulations [25]. Recently, Haberthur and Caflisch described a new analytical GB model named FACTS that is analogous to the pairwise descreening approximations in nature but utilizes both the solute volume and symmetry to improve the accuracy of the effective Born radii [26]. Lazaridis introduced the Gouy-Chapman theory for modeling charged membranes, and this can be used as a supplementary term to existing implicit membrane models [6]. Advances also continue to be made in PB methodologies. Boundary element methods were developed as an alternative to the popular finite difference method for computing both electrostatic potential and forces [27]. Schnieders et al. developed a polarizable multiple PB model that compares well with explicit AMOEBA water [28]. Substantial efforts continue to be made in testing and benchmarking various implicit solvent models [17, 29, 30, 31]. It is also worth noting that ongoing discussions have been directed toward the optimal choice of solvent-solute boundary, a critical parameter in implicit solvent models [32, 33].
Implicit Solvent Studies of Biomolecular Structure and Dynamics
Along with the methodological advances, implicit solvent models have enjoyed success in a range of areas, such as protein design [34], structure refinement [35, 36, 37, 38], protein-protein, protein-nucleic acid and protein-ligand interactions [39, 40, 41, 42]. One of the most exciting applications of efficient implicit solvent models is arguably to understand protein folding and misfolding at atomic detail. Recent studies have focused on fast folding of small peptides and mini-proteins [15, 16]. Folding simulations of larger and more complex proteins are emerging [18, 43, 44, 45, 46]. In addition to predicting native structures, recent studies also emphasize obtaining thermodynamics and kinetics of folding that are consistent with experiment. These efforts represent an encouraging step toward realistic modeling of biomolecular conformational equilibria. Nonetheless, it has yet to be demonstrated that a single implicit solvent force field can be consistently applied to fold a range of sequences with non-trivial folds (i.e., at least three regular secondary structure elements arranged in a non-trivial tertiary structure). This is an indication that substantial challenges (and thus opportunities) remain in deriving a balanced and transferable implicit solvent force fields for the reliable simulation of protein conformational equilibrium and ab initio folding. However, recognizing that ab initio simulation of protein folding is one of the most challenging and stringent tests of the force field and sampling technique, one should not be overly discouraged by the limited success in folding simulations conducted to date.
Implicit solvent models have also been applied to study other thermodynamic and kinetic properties of proteins. For example, MD simulations with a GB implicit solvent model were recently used to demonstrate the electrostatic basis for the stability of thermophilic proteins [47]. Hills and Brooks investigated the kinetics of the self association of model amyloidogenic hexapeptides and suggested that hydrophobic cooperativity is sufficient to allow for nucleation-dependent fibril formation [48].
Constant pH Molecular Dynamics
A realistic representation of aqueous environment for modeling biological processes requires the consideration of solution conditions such as salt concentration and acidity (or pH). A traditional way to include solution pH effects in molecular simulations of proteins is to assign and fix protonation states for ionizable side chains. In the past five years, significant progress has been made in the development of methods that enable molecular dynamics to be performed in equilibrium with an infinite proton bath (constant pH) [4]. These methods, often referred to as constant pH molecular dynamics, explicitly allow for protein dielectric response to the correlated event of charging and conformational dynamics, and are mainly distinguished in the way by which the protonation states are modeled.
Methods based on discrete protonation states
In a discrete protonation states method, also called stochastic titration MD, sampling of conformational space by MD is combined with sampling of discrete protonation states through Monte Carlo (MC) moves. Within this scheme, different approaches have been developed, which mainly differ in the solvent representation for the MD and MC steps but also in the protocol for updating the protonation states as well as the method for calculating the deprotonation free energy for the MC step. In explicit solvent MD simulations, Bürgi et al. employed short-time free energy simulation in explicit solvent to estimate the free energy of deprotonation [49] for the trial MC move, while Baptista et al. [50, 63] made use of the PB electrostatics calculation. Discrete protonation states methods using implicit solvent models for both MD and MC steps have significantly lower computational cost. Dlugosz and Antosiewicz combined PB calculation for protonation states sampling with an implicit solvent model, Analytical Continuum Electrostatics, for conformational sampling [51]. Mongan et al. developed a protocol that employs a GB implicit solvent model for both MC and MD sampling [52].
One potential pitfall in discrete states methods is associated with the discontinuous energy and force as a result of the abrupt switch in charge states. In explicit solvent simulations, unfavorable interactions with the solvent may arise, thereby lowering the acceptance ratio for MC moves. This problem can be alleviated by allowing solvent to equilibrate at a fixed solute conformation after the change in protonation states, as demonstrated by Baptista and coworker in their mixed solvent scheme [50, 63]. In implicit solvent simulations, this problem is largely circumvented due to the instantaneous adjustment of solvent to the new protonation states [53]. Another strategy that may ease the problem is to change the protonation state of one site at a time, although this may slow the convergence for strongly coupled titrations [52, 4]. Most recently, Stern proposed an explicit solvent based method, in which MC trial moves consist of short molecular dynamics runs with a time-dependent Hamiltonian that interpolates between the old and new protonation states [53]. This method has the advantage that the trial MC moves are not instantaneous, thereby increasing the probability for acceptance in the MC steps. Also, it may offer a reduction in computational expense as compared to the method based on free energy calculations [49]. Besides the pitfall due to discontinuous energies, a discrete protonation states simulation is significantly more expensive than a standard MD simulation, because of the extra computational time spent on the energy evaluations in the MC trial moves.
The accuracy of constant pH molecular dynamics methods and their feasibility for studying pH-dependent conformational phenomena of proteins can be assessed by pKa calculations. Several methods based on discrete protonation states have been tested in pKa calculations for hen egg lysozyme [49, 52, 54]. The most recent results by Machuqueiro and Baptista [54] using the mixed solvent scheme are especially encouraging. The overall root-mean-square deviation between the computed and experimental pKa values is below 1 pK unit.
Methods based on continuous protonation states
In the continuous protonation states approach, continuous titration coordinates are propagated simultaneously with the conformational degrees of freedom in explicit or implicit solvent molecular dynamics simulations. Börjesson and Hünenberger developed a so-called acidostat method for explicit solvent MD simulations in which the extent of deprotonation is relaxed towards the equilibrium value via a first-order coupling scheme in analogy to Berendsen's thermostat [55]. However, the theoretical basis for the equilibrium condition seems somewhat unclear [56].
Lee, Salsbury and Brooks developed a method based on λ dynamics [57] and GB implicit solvent simulations, in which fictitious λ particles are used to propagate the titration degrees of freedom [58]. This method is later referred to as the CPHMD method. The end-point states of λ represent the deprotonated and protonated states. To deal with simultaneous titration at two competing sites, such as the δ and ε nitrogens in the histidine side chain, Khandogin and Brooks developed a two-dimensional λ dynamics technique, in which a second variable, x, representing the interconversion between proton tautomers, is introduced to allow for the description of the protonation process at competing sites on an equal footing [59]. The forces on λ and x coordinates are attenuated by electrostatic and van der Waals interactions involving atoms in titrating side chains. The two-dimensional λ dynamics is also applied to the titration of carboxyl groups, because exchange of the titrating oxygens is slow on a MD timescale. Realizing that fluctuations in protonation states depend largely on local conformational rearrangement, Khandogin and Brooks improved the convergence of protonation state sampling by making use of the replica-exchange (REX) enhanced conformational sampling protocol [60]. The accuracy of pKa prediction using CPHMD simulations is intimately linked to the accuracy of the underlying solvent model. Overstabilization of salt bridges in the GB model has led to the systematic underestimation of pKa's for carboxylates that interact with positively charged groups [59]. Significant improvement was observed [60] by employing an optimized set of GB input atomic radii [15]. One remaining problem is related to the underestimation of desolvation energies of buried groups as well as buried charge-charge interactions, which may be largely attributed to the employed dielectric boundary model that neglects solvent excluded volumes. These effects manifest themselves as pronounced deviations from experiment in the pKa calculation for buried residues [60]. Finally, Khandogin and Brooks demonstrated that, by employing a simple Debye-Hückel screening function, salt effects on the titration property of proteins can be largely accounted for [60].
REX-CPHMD simulations using the tautomer model, an improved GB parameterization and accounting for salt effects, are able to offer quantitative prediction of pKa's for proteins. In a benchmark study using 10 proteins that exhibit very large pKa shifts, the root-mean-square deviation from experiment ranges from 0.6 to 1.0 pK units [60]. The quantitative accuracy in predicting protein ionization equilibria and the additional advantage of being only marginally slower than a standard GB simulation have enabled REX-CPHMD simulations to be applied to unravel detailed mechanisms of many important pH-coupled conformational processes that were previously inaccessible to theoretical studies. We will outline some of these applications in the next section.
Applications to pH-dependent conformational phenomena
The ultimate goal for the development of constant pH molecular dynamics methods is to study pH-coupled conformational transitions, such as the proton-gradient driven ATP synthesis, pH induced ion-exchange process in the integral membrane, and pH-dependent protein misfolding/aggregation. Although the ability for modeling these processes is ultimately limited by the bottleneck in conformational sampling and the accuracy of force field and solvent model, emerging applications using constant pH molecular dynamics have demonstrated great promise.
An important question related to the necessity for the development of constant pH simulation methodologies is the significance of solvent-solute proton exchange for the structure and dynamics of polypeptides under pH conditions such that both protonated and deprotonated states are occupied. Dlugosz and Antosiewicz investigated this question using an implicit solvent stochastic titration method [61]. They showed that the distributions of Asp-Lys distance can not be reproduced by a linear combination of those resulting from simulations with fixed protonation states.
The pH-dependent helicity of decalysine has been studied by Börjesson and Hünenberger using the acidostat method in explicit solvent [62], and by Machuqueiro and Baptista using the stochastic titration method with an mixed solvent scheme [63]. In the latter study, the effect of ionic strength was included in the PB calculation for MC moves and through the reaction field parameters for the explicit solvent molecular dynamics. The calculated helix profile as a function of pH was compared with experiment.
The origin of the pH-dependent helicity of proteins has been a subject of study in the experimental community for the past two decades. Khandogin and Brooks investigated pH-dependent folding of the C-peptide from ribonuclease A, using REX-CPHMD folding simulations [64]. The study revealed not only a bell-shaped pH profile for the total helix content, in agreement with experiment, but also a pH-dependent conformational equilibrium consisting of unfolded and partially folded states of various helical lengths. Furthermore, specific electrostatic interactions responsible for the helix-coil transition were identified that are consistent with, and complementary to the existing experimental data.
Khandogin and Brooks went on to elucidate the folding behavior of two β amyloid peptides (Aβ) from Alzheimers disease, Aβ(1-28) and Aβ(10-42) [65]. Unlike the C-peptide and many globular proteins, which are maximally folded at an intermediate pH that minimizes the total charge, Aβ is helical with an inverse bell-shaped pH profile in aqueous TFE solution but it is largely unfolded in water [65]. The study of Khandogin and Brooks reconciled this discrepancy by showing that Aβ is mainly coil-like but contains several short, nascent helical segments that have elevated helix propensity under low and high pH conditions. Moreover, this study revealed pH-dependent solvent exposure in the central hydrophobic cluster (residues 17-21), which forms the minimum fibril forming sequence, and the pH-dependent β-turn formation in residues 23-26. Taken together, these data predicted that, at pH 6, Aβ adopts conformational states that are most prone for the formation of β-sheet based aggregates (Figure 2). Finally, this study suggested that, although minimum charge-charge repulsion at the isoelectric point provides an initial driving force for aggregation, the pH modulation of the folding landscape of Aβ results in an enhanced intermolecular hydrophobic association, which stabilizes β-sheet based oligomers.
Figure 2.
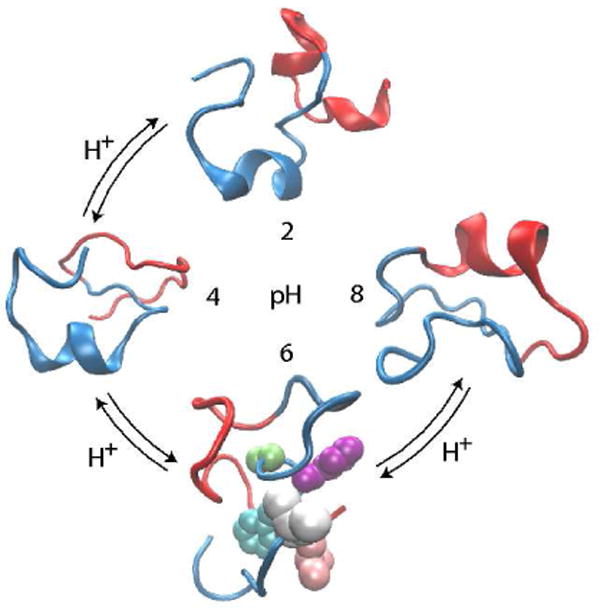
Representative conformations of the β amyloid peptide (10-42) under different pH conditions. The conformations were obtained as centroids of the most populated clusters from the replica-exchange CPHMD folding simulations [65]. The N-terminal residues 10-28 are shown in blue; the C-terminal residues 29-42 are shown in red. In the most aggregation-prone state (pH 6), the side chains of the central hydrophobic cluster Leu-17, Val-18, Phe-19, Phe-20 and Ala-21 are shown as van der Waals spheres in pink, grey, cyan, purple and green, respectively.
Khandogin and Brooks also explored the formation of folding intermediate states involving ionizable side chains [66]. REX-CPHMD simulations initiated from an X-ray crystal and a minimized average solution NMR structure of the villin headpiece domain gave rise to two conformational states that display distinct titration behavior for a histidine residue (H41), similar to the measurements for the native and intermediate states, respectively. Supported by the dynamical, structural and titration properties, the simulation data suggested that the state derived from the solution NMR structure resembles a putative intermediate that has a largely unfolded N-terminal subdomain. Moreover, the formation of the putative intermediate was found to be the result of the loss of a hydrogen-bonded network centered at H41. Thus, this work put forth a proposal that the hydrogen-bonded network and not the protonation state of H41 is a prerequisite for folding in the villin headpiece domain.
Conclusions
Recent methodological advances have continued to push the level of accuracy and complexity of continuum electrostatics based solvation theories. In particular, GB/SA theory has become a prime choice for biomolecular simulation due to the favorable balance in accuracy and efficiency. GB/SA based atomistic simulations have been applied to investigate various conformational properties of biological assemblies, which can not be directly studied using explicit solvent based methods. It is particularly encouraging that GB/SA based ab initio folding simulations combined with advanced conformational sampling protocols are now able to reproduce native or near-native structures for peptides and mini-proteins, and capture thermodynamics and kinetics of folding at a quantitative level. Nonetheless, substantial challenges and opportunities remain, particularly in the treatment of nonpolar solvation. As implicit solvent models are maturing for biomolecular simulations, one major goal is to achieve a force field consistent with the implicit solvent model and vice versa. The initial successes in extensive co-optimization of the GB/SA models and the protein force field, albeit limited, are very encouraging.
The advancement in implicit solvent models has also facilitated the development of constant pH molecular dynamics methods. Of particular interest are the stochastic titration method that combines PB calculations for protonation states sampling and explicit solvent molecular dynamics for conformational sampling and the CPHMD method based on λ dynamics and GB/SA models. The accuracy of the CPHMD method will benefit significantly from further improvement of the underlying GB models and a more rigorous treatment of salt screening effects. The CPHMD method can be combined with other implicit solvent models such as PB. However, since PB requires numerical solutions, the potential of mean force for the protonation of model compound is no longer analytical and has to be approximated by a continuous function. In principle, the CPHMD formalism can be extended to explicit solvent simulations, although, with the current CPU and sampling capability, convergence properties are expected to be poor. Another effect that may play a significant role is electronic polarization. In this regard, one avenue is to employ a polarizable force field.
Emerging applications of constant pH molecular dynamics have mainly focused on first principles pKa predictions and pH-coupled conformational dynamics. The accuracy and speed of the REX-CPHMD simulation have enabled novel insights into the pH-modulated peptide folding, formation of intermediates, and the aggregation behavior of amyloidogenic peptides. Ongoing studies (JK and CLB, unpublished data) indicate that the REX-CPHMD technique can be applied to investigate many other pH-dependent conformational phenomena, such as the pH-dependent folding at the membrane interface, electrostatic interactions in the unfolded states, and backbone registry shifts in amyloid fibrils. The detailed insights gained from these and other studies greatly complement and extend the information accessible by experimental techniques, making implicit solvent based molecular simulation a powerful tool for exploring unanswered questions related to mechanisms that govern a wide variety of biological processes.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM48807, GM57513 and RR12255).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feig M, Brooks CL., III Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol. 2004;14:217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Baker NA. Improving implicit solvent simulations: a poisson-centric view. Curr Opin Struct Biol. 2005;15:137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Koehl P. Electrostatics calculations: latest methodological advances. Curr Opin Struct Biol. 2006;16:142–151. doi: 10.1016/j.sbi.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Mongan John, Case David A. Biomolecular simulations at constant pH. Curr Opin Struct Biol. 2005;15:157–163. doi: 10.1016/j.sbi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara P, Apostolakis J, Caflisch A. Evaluation of a fast implicit solvent model for molecular dynamics simulations. Proteins. 2002;46:24–33. doi: 10.1002/prot.10001. [DOI] [PubMed] [Google Scholar]
- 6.Lazaridis T. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins. 2005;58:518–527. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- 7.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 8.Levy RM, Zhang LY, Gallicchio E, Felts AK. On the nonpolar hydration free energy of proteins: Surface area and continuum solvent models for the solute-solvent interaction energy. J Am Chem Soc. 2003;125:9523–9530. doi: 10.1021/ja029833a. [DOI] [PubMed] [Google Scholar]; ** This paper demonstrated the importance of explicit inclusion of the solute-solvent dispersion for accurate estimation of nonpolar solvation free energy and proposed a continuum vdW model that can be used to estimate the dispersion term.
- 9.Chen J, Brooks CL., III Critical importance of length-scale dependence in implicit modeling of hydrophobic interactions. J Am Chem Soc. 2007;129:2444–2445. doi: 10.1021/ja068383+. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The authors demonstrated several important implications of the length-scale dependence of hydrophobic interactions in implicit treatment of nonpolar solvation and proposed a simple empirical model to incorporate such property in the framework of the traditional SA models.
- 10.Feig M, Im W, Brooks CL., III Implicit solvation based on generalized Born theory in different dielectric environments. J Chem Phys. 2004;120:903–911. doi: 10.1063/1.1631258. [DOI] [PubMed] [Google Scholar]; * A simple empirical extension of the original GBMV model to allow for explicit dependence of the Born radii on the dielectric environments.
- 11.Tanizaki S, Feig M. A new generalized born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes. J Chem Phys. 2005;122:124706. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 12.Sigalov G, Scheffel P, Onufriev A. Incorporating variable dielectric environments into the generalized born model. J Chem Phys. 2005;122:094511. doi: 10.1063/1.1857811. [DOI] [PubMed] [Google Scholar]
- 13.Sigalov G, Fenley A, Onufriev A. Analytical electrostatics for biomolecules: Beyond the generalized born approximation. J Chem Phys. 2006;124:124902. doi: 10.1063/1.2177251. [DOI] [PubMed] [Google Scholar]; * An efficient analytical extension of the original GB approximation to reproduce the dependence of the Born radii on the solute and solvent dielectrics.
- 14.Tjong H, Zhou HX. GBr6: A parameterization-free, accurate, analytical generalized born method. J Phys Chem B. 2007;111:3055–3061. doi: 10.1021/jp066284c. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Im W, Brooks CL., III Balancing solvation and intramolecular interactions: Towards a consistent generalized Born force field. J Am Chem Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** An extensive co-optimization of the GB input atomic radii and peptide backbone torsion potential, guided by pair-wise interactions between amino acid polar groups and conformational equilibria of model peptides. The optimized GBSW/SA force field not only reproduces the experimental conformational equilibria for a range of helical and β peptides, but also correctly folds designed peptides TrpZip2 and Trp-Cage.
- 16.Jang S, Kim E, Pak Y. Direct folding simulation of alpha-helices and beta-hairpins based on a single all-atom force field with an implicit solvation model. Proteins. 2007;66:53–60. doi: 10.1002/prot.21173. [DOI] [PubMed] [Google Scholar]; * An optimized Amber-based GB/SA protein force field that demonstrates more balanced secondary structure preference.
- 17.Geney R, Layten M, Gomperts R, Hornak V, Simmerling C. Investigation of salt bridge stability in a generalized Born solvent model. J Chem Theory Comput. 2006;2:115–127. doi: 10.1021/ct050183l. [DOI] [PubMed] [Google Scholar]
- 18.Jang S, Kim E, Pak Y. Free energy surfaces of miniproteins with αββα motif: Replica exchange molecular dynamics simulation with an implicit solvation model. Proteins. 2006;62:663–671. doi: 10.1002/prot.20771. [DOI] [PubMed] [Google Scholar]
- 19.Gallicchio E, Levy RM. AGBNP: An analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. J Comput Chem. 2004;25:479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]; * The first GB model with a full nonpolar solvation term that is suitable for molecular dynamics simulations.
- 20.Wagoner Jason A, Baker Nathan A. Assessing implicit models for nonpolar mean solvation forces: The importance of dispersion and volume terms. Proc Natl Acad Sci USA. 2006;103:8331–8336. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 22.Daidone I, Ulmschneider MB, Di Nola A, Amadei A, Smith JC. Dehydration-driven solvent exposure of hydrophobic surfaces as a driving force in peptide folding. Proc Natl Acad Sci USA. 2007;104:15230–15235. doi: 10.1073/pnas.0701401104. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Two microsecond time-scale simulations of a β-hairpin were used to demonstrate the importance of the length-scale dependence of hydrophobic solvation.
- 23.Dzubiella J, Swanson JMJ, McCammon JA. Coupling hydrophobicity, dispersion, and electrostatics in continuum solvent models. Phys Rev Lett. 2006;96:087802. doi: 10.1103/PhysRevLett.96.087802. [DOI] [PubMed] [Google Scholar]
- 24.Archontis G, Simonson T. A residue-pairwise generalized Born scheme suitable for protein design calculations. J Phys Chem B. 2005;109:22667–22673. doi: 10.1021/jp055282+. [DOI] [PubMed] [Google Scholar]
- 25.Michel J, Taylor RD, Essex JW. Efficient generalized Born models for Monte Carlo simulations. J Chem Theory Comput. 2006;2:732–739. doi: 10.1021/ct600069r. [DOI] [PubMed] [Google Scholar]
- 26.Haberthur U, Caflisch A. FACTS: fast analytical continuum treatment of solvation. J Comput Chem. 2007 doi: 10.1002/jcc.20832. in press. [DOI] [PubMed] [Google Scholar]; * An efficient analytical GB model that yields accurate effective Born radii for both buried and surface atoms.
- 27.Lu BZ, Cheng XL, Huang JF, McCammon JA. Order N algorithm for computation of electrostatic interactions in biomolecular systems. Proc Natl Acad Sci USA. 2006;103:19314–19319. doi: 10.1073/pnas.0605166103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnieders MJ, Baker NA, Ren PY, Ponder JW. Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum. J Chem Phys. 2007;126:124114. doi: 10.1063/1.2714528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XF, Hassan SA, Mehler EL. Long dynamics simulations of proteins using atomistic force fields and a continuum representation of solvent effects: Calculation of structural and dynamic properties. Proteins. 2005;60:464–484. doi: 10.1002/prot.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan H, Mark AE, Zhu J, Honig B. Comparative study of generalized born models: Protein dynamics. Proc Natl Acad Sci USA. 2005;102:6760–6764. doi: 10.1073/pnas.0408857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan CH, Yang LJ, Luo R. How well does Poisson-Boltzmann implicit solvent agree with explicit solvent? a quantitative analysis. J Phys Chem B. 2006;110:18680–18687. doi: 10.1021/jp063479b. [DOI] [PubMed] [Google Scholar]
- 32.Yu ZY, Jacobson MP, Friesner RA. What role do surfaces play in GB models? a new-generation of surface-generalized born model based on a novel gaussian surface for biomolecules. J Comput Chem. 2006;27:72–89. doi: 10.1002/jcc.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mongan J, Simmerling C, McCammon JA, Case DA, Onufriev A. Generalized Born model with a simple, robust molecular volume correction. J Chem Theory Comput. 2007;3:156–169. doi: 10.1021/ct600085e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes A, Alexandrov A, Bathelt C, Archontis G, Simonson T. Computational sidechain placement and protein mutagenesis with implicit solvent models. Proteins. 2007;67:853–867. doi: 10.1002/prot.21379. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Im W, Brooks CL., III Refinement of NMR structures using implicit solvent and advanced sampling techniques. J Am Chem Soc. 2004;126:16038–16047. doi: 10.1021/ja047624f. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Won HS, Im W, Dyson HJ, Brooks CL. Generation of native-like models from limited NMR data, modern force fields anda dvanced conformational sampling. J Biomol NMR. 2005;31:59–64. doi: 10.1007/s10858-004-6056-z. [DOI] [PubMed] [Google Scholar]
- 37.Lee MS, Olson MA. Assessment of detection and refinement strategies for de novo protein structures using force field and statistical potentials. J Chem Theory Comput. 2007;3:312–324. doi: 10.1021/ct600195f. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Brooks CL., III Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins. 2007;67:922–930. doi: 10.1002/prot.21345. [DOI] [PubMed] [Google Scholar]
- 39.Chocholousova J, Feig M. Implicit solvent simulations of DNA and DNA-protein complexes: Agreement with explicit solvent vs experiment. J Phys Chem B. 2006;110:17240–17251. doi: 10.1021/jp0627675. [DOI] [PubMed] [Google Scholar]
- 40.Liu HY, Zou XQ. Electrostatics of ligand binding: Parametrization of the generalized Born model and comparison with the Poisson-Boltzmann approach. J Phys Chem B. 2006;110:9304–9313. doi: 10.1021/jp060334w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bu LT, Im W, Brooks CL., III Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys J. 2007;92:854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]; *A simple protocol for predicting transmembrane helix packing interface based on the GBSW implicit membrane model.
- 42.Sun Y, Dominy BN, Latour RA. Comparison of solvation-effect methods for the simulation of peptide interactions with a hydrophobic surface. J Comput Chem. 2007;28:1883–1892. doi: 10.1002/jcc.20716. [DOI] [PubMed] [Google Scholar]
- 43.Roe DR, Hornak V, Simmerling C. Folding cooperativity in a three-stranded beta-sheet model. J Mol Biol. 2005;352:370–381. doi: 10.1016/j.jmb.2005.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jagielska A, Scheraga HA. Influence of temperature, friction, and random forces on folding of the B-domain of staphylococcal protein A: All-atom molecular dynamics in implicit solvent. J Comput Chem. 2007;28:1068–1082. doi: 10.1002/jcc.20631. [DOI] [PubMed] [Google Scholar]
- 45.Lei H, Wu C, Liu H, Duan Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc Natl Acad Sci USA. 2007;104:4925–4930. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Ab initio folding simulations of a small protein HP35 that reach fully folded conformations as close as 0.46 Å from the crystal structure.
- 46.Lei HX, Duan Y. Ab initio folding of albumin binding domain from all-atom molecular dynamics simulation. J Phys Chem B. 2007;111:5458–5463. doi: 10.1021/jp0704867. [DOI] [PubMed] [Google Scholar]
- 47.Dominy BN, Minoux H, Brooks CL., III An electrostatic basis for the stability of thermophilic proteins. Proteins. 2004;57:128–141. doi: 10.1002/prot.20190. [DOI] [PubMed] [Google Scholar]; * An interesting study where MD simulations in a GB implicit solvent reveal the interplay of electrostatic interactions and dielectric response for the enhanced stability of thermophilic proteins.
- 48.Hills RD, Brooks CL., III Hydrophobic cooperativity as a mechanism for amyloid nucleation. J Mol Biol. 2007;368:894–901. doi: 10.1016/j.jmb.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bürgi Roland, Kollman Peter A, van Gunsteren Wilfred F. Simulating proteins at constant pH: an approach combining molecular dynamics and Monte Carlo simulation. Proteins. 2002;47:469–480. doi: 10.1002/prot.10046. [DOI] [PubMed] [Google Scholar]
- 50.Baptista António M, Teixeira Vitor H, Soares Cláudio M. Constant-pH molecular dynamics using stochastic titration. J Chem Phys. 2002;117:4184–4200. [Google Scholar]; ** A mixed solvent scheme that combines the strengths of the PB electrostatics method for protonation sampling and explicit solvent molecular dynamics for conformational sampling. An empirically adjustable dielectric constant for solute is needed for the PB calculations.
- 51.Dlugosz Maciej, Antosiewicz Jan M. Constant-pH molecular dynamics simulations: a test case of succinic acid. Chem Phys. 2004;302:161–170. [Google Scholar]
- 52.Mongan John, Case David A, McCammon J Andrew. Constant pH molecular dynamics in generalized Born implicit solvent. J Comput Chem. 2004;25:2038–2048. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- 53.Stern Harry A. Molecular simulation with variable protonation states at constant pH. J Chem Phys. 2007;126:164112. doi: 10.1063/1.2731781. [DOI] [PubMed] [Google Scholar]
- 54.Machuqueiro Miguel, Baptista António M. Acid range titration of HEWL using a constant-pH molecular dynamics method. Proteins. 2007 doi: 10.1002/prot.21923. in press. [DOI] [PubMed] [Google Scholar]
- 55.Börjesson Ulf, Hünenberger Philippe H. Explicit-solvent molecular dynamics simulation at constant pH: Methodology and application to small amines. J Chem Phys. 2001;114(22):9706–9719. [Google Scholar]
- 56.Baptista António M. Explicit-solvent molecular dynamics simulation at constant pH: Methodology and application to small amines. J Chem Phys. 2002;116:7766–7768. Comment on. [Google Scholar]
- 57.Kong Xianjun, Brooks Charles L., III λ-dynamics: A new approach to free energy calculations. J Chem Phys. 1996;105:2414–2423. [Google Scholar]
- 58.Lee Michael S, Salsbury Freddie R, Jr, Brooks Charles L., III Constant-pH molecular dynamics using continuous titration coordinates. Proteins. 2004;56:738–752. doi: 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- 59.Khandogin Jana, Brooks Charles L., III Constant pH molecular dynamics with proton tautomerism. Biophys J. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** A two-dimensional λ-dynamics based method and algorithms were developed to allow for coupled double site titration in CPHMD simulations. Significant improvement in the accuracy of pKa prediction for histidine and carboxylate groups was achieved.
- 60.Khandogin Jana, Brooks Charles L., III Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry. 2006;45:9363–9373. doi: 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]; * pKa calculations for a stringent test set of 10 proteins using REX-CPHMD simulations with an improved GB parameterization and a Debye-Huckel screening function. The root-mean-square deviation from experiment was below 1 pK unit for all proteins.
- 61.Dlugosz M, Antosiewicz JM. Effects of solute-solvent proton exchange on polypeptide chain dynamics: a constant-pH molecular dynamics study. J Phys Chem B. 2005;109:13777–13784. doi: 10.1021/jp0505779. [DOI] [PubMed] [Google Scholar]; * An interesting study trying to address the question as to whether and how structural and dynamical properties derived from constant pH molecular dynamics simulations differ from those using fixed charge states.
- 62.Börjesson Ulf, Hünenberger Philippe H. pH-dependent stability of a decalysine α-helix studied by explicit-solvent molecular dynamics simulations at constant pH. J Phys Chem B. 2004;108:13551–13559. [Google Scholar]
- 63.Machuqueiro Miguel, Baptista António M. Constant-pH molecular dynamics with ionic strength effects: protonation-conformation coupling in decalysine. J Phys Chem B. 2006;110:2927–2933. doi: 10.1021/jp056456q. [DOI] [PubMed] [Google Scholar]
- 64.Khandogin Jana, Chen Jianhan, Brooks Charles L., III Exploring atomistic details of pH-dependent peptide folding. Proc Natl Acad Sci USA. 2006;103:18546–18550. doi: 10.1073/pnas.0605216103. [DOI] [PMC free article] [PubMed] [Google Scholar]; * An application of the REX-CPHMD method to the pH-dependent folding of the C-peptide. A pH-dependent conformational equilibrium was shown, and specific electrostatic interactions responsible for helix-coil transitions were identified.
- 65.Khandogin Jana, Brooks Charles L., III Linking folding with aggregation in Alzheimer's β amyloid peptides. Proc Natl Acad Sci USA. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** An intriguing study of pH-dependent conformational properties of the largely unfolded β amyloid peptides. The authors revealed a hidden link between pH-modulated folding and aggregation propensity.
- 66.Khandogin Jana, Raleigh Daniel P, Brooks Charles L., III Folding intermediate in the villin headpiece domain arises from disruption of a N-terminal hydrogen-bonded network. J Am Chem Soc. 2007;129:3056–3057. doi: 10.1021/ja0688880. [DOI] [PMC free article] [PubMed] [Google Scholar]