

Abstract
Structural and functional diversity among the aminoacyl-tRNA synthetases prevent infiltration of the genetic code by noncognate amino acids. To explore whether these same features distinguish the synthetases as potential sources of resistance against antibiotic amino acid analogues, we investigated bacterial growth inhibition by S-(2-aminoethyl)-L-cysteine (AEC). Wild-type lysyl-tRNA synthetase (LysRS) and a series of active site variants were screened for their ability to restore growth of an Escherichia coli LysRS null strain at increasing concentrations of AEC. While wild-type E. coli growth is completely inhibited at 5 μM AEC, two LysRS variants, Y280F and F426W, provided substantial resistance and allowed E. coli to grow in the presence of up to 1 mM AEC. Elevated resistance did not reflect changes in the kinetics of amino acid activation or tRNALys aminoacylation, which showed at best 4–6-fold improvements, but instead correlated with the binding affinity for AEC, which was decreased ~50-fold in the LysRS variants. In addition to changes in LysRS, AEC resistance has also been attributed to mutations in the L box riboswitch, which regulates expression of the lysC gene, encoding aspartokinase. The Y280F and F426W LysRS mutants contained wild-type L box riboswitches that responded normally to AEC in vitro, indicating that LysRS is the primary cellular target of this antibiotic. These findings suggest that the AEC resistance conferred by L box mutations is an indirect effect resulting from derepression of lysC expression and increased cellular pools of lysine, which results in more effective competition with AEC for binding to LysRS.
The fidelity of protein synthesis largely depends on the accuracy of two processes, the interaction of mRNA codons with cognate tRNA anticodons on the ribosome and the attachment of amino acids to their cognate tRNAs by the aminoacyl-tRNA synthetases (aaRS). aaRSs maintain error rates lower than 1 in 5000 during aminoacyl-tRNA (aa-tRNA) synthesis through a combination of exquisite substrate specificity and elimination of errors by intrinsic proofreading mechanisms (1). The central role of aaRSs in defining the genetic code places a strong selective pressure on these enzymes to prevent mistakes during cognate aa-tRNA formation (2–4). Cognate tRNA selection requires the identification of a unique combination of nucleotides at particular positions, which together offer sufficiently diverse recognition elements to allow their specific selection by the corresponding aaRSs (5). Distinguishing between structurally related amino acids and other small molecules is more problematic, and errors in substrate selection are unavoidable. To get around this problem, certain aaRSs have acquired appended or inserted domains that proofread noncognate amino acids, thereby preventing the synthesis of incorrectly matched aa-tRNAs (6).
Other strategies that ensure the specificity of amino acid discrimination during translation include screening by elongation factors (7, 8), proofreading by free-standing domains (9, 10), and duplication of aaRSs (11). Duplication of synthetase activities is widespread and occurs mostly within the same aaRS family, as seen, for example, in the acquisition of antibiotic resistance alleles by pathogenic clinical isolates (12, 13). Non-orthologous duplication of synthetase activities is seen less frequently, as in the case of the two structurally unrelated forms of lysyl-tRNA synthetase, LysRS1 and LysRS2. Although their overall architectures share no common features, the structures of LysRS1 and LysRS2 in complexes with lysine reveal that recognition of the R-group of L-lysine relies on similar residues arranged in different active site architectures (14). These differences in the mechanism of recognition impact noncognate substrate discrimination, as reflected by the in vivo resistance to growth inhibition imparted by LysRS1 and LysRS2 individually against S-(2-aminoethyl)-L-cysteine (AEC) and γ-aminobutyric acid, respectively (15–18). Differential resistance to naturally occurring noncognate amino acids revealed how LysRS1 or LysRS2 acting alone can exclude lysine analogues and maintain specificity during translation of lysine codons.
The finding that LysRS offers a potential mechanism of resistance to lysine analogues is consistent with previous studies showing that other aaRSs also confer resistance to antibiotics (19). An alternative mechanism of resistance to AEC linked to changes in the regulation of genes encoding components of the lysine biosynthetic pathway has been described in bacteria and yeast (20–22). Both Escherichia coli and Bacillus subtilis encode lysine-binding L box riboswitches upstream of the corresponding lysine biosynthesis genes (23, 24), and mutations in these regulatory leader regions provide AEC resistance by increasing aspartokinase production (25, 26). Based upon these and other studies, it has been proposed that the L box riboswitch rather than LysRS may in fact be the primary cellular target for AEC (27). Here we show that E. coli LysRS2 active site variants confer antibiotic resistance despite the presence of a wild-type L box sensitive to AEC, indicating that LysRS2 is the cellular target for this antibiotic.
RESULTS AND DISCUSSION
In Vivo Activity of LysRS2 Variants
E. coli LysRS2 variants (encoded by lysS) were analyzed in vivo using the E. coli strain PALΔSΔUTR in which both the genomic lysS and lysU genes, which encode two isoforms of LysRS2, have been disrupted. The LysRS variants all contain changes to the lysine binding pocket in the active site (Figure 1). Growth of E. coli PALΔSΔUTR is maintained at permissive temperatures by a copy of lysU carried in the plasmid pMAK705, which contains a temperature-sensitive origin of replication (28). Growth at nonpermissive temperatures is dependent on the presence of a second stable plasmid, in this case pXLysSK1, which is compatible with pMAK705 and encodes wild-type lysS or a variant capable of supporting growth. The empty vector pXLysSK1-EcoRI did not restore growth following replica-plating and counterselection at a nonpermissive temperature, indicating efficient loss of pMAK705. Plasmids encoding the LysRS2 variants E240Q and E278D, both of which result in severe loss of in vitro aminoacylation activity (17), were unable to restore growth of PALΔSΔUTR after the loss of pMAK705. All other LysRS variants tested were able to support growth at nonpermissive temperatures on Luria–Bertani (LB) or M9 minimal medium (MM) (Table 1). These derivatives of E. coli PALΔSΔUTR in which growth is dependent on various LysRS2 active site variants were tested for sensitivity to AEC in vivo.
Figure 1.
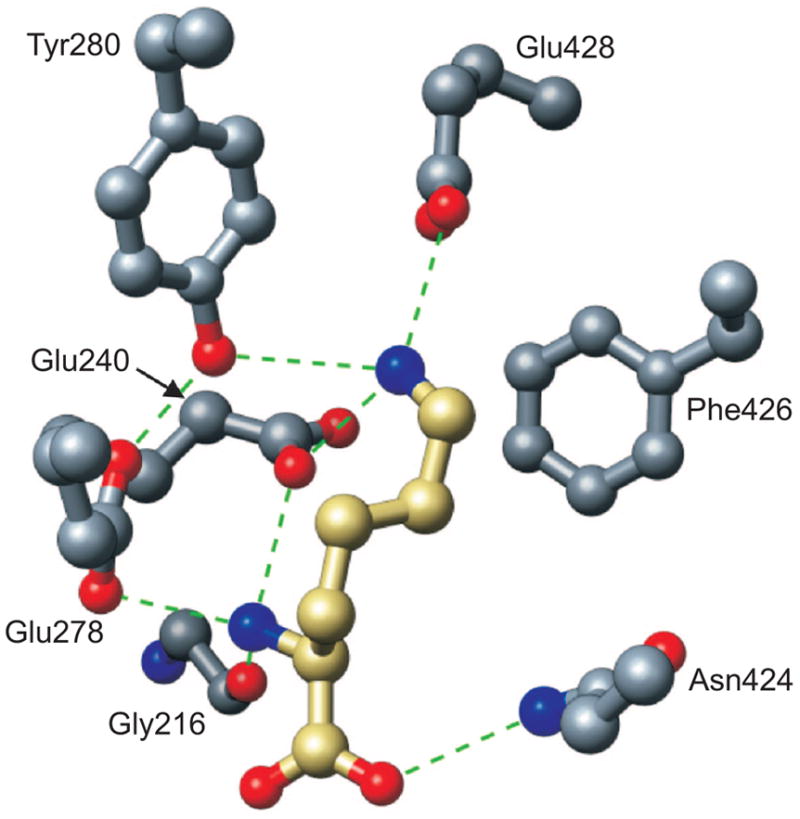
Model of L-lysine recognition by the active site of E. coli LysRS (40). H-bonds are shown as dashed lines. The carbon backbone for the substrate lysine is gold, while the carbon backbone of the LysRS2 active site residues is gray. Oxygen and nitrogen are colored red and blue, respectively, for enzyme and substrate.
TABLE 1.
Doubling times in liquid media of E. coli PALΔSΔUTR derivatives containing pXLysSK1 vectors encoding LysRS2 variants
| LysRS2 | LB (min)a | LB (R)b | MM (min)a | MM (R)b | kcat/KM lysine (R)c |
|---|---|---|---|---|---|
| Wild type | 43 ± 2 | 1.0 | 100 ± 5 | 1.0 | 1.0 |
| G216A | 91 ± 4 | 2.1 | 120 ± 6 | 1.2 | 33 |
| E240D | 83 ± 4 | 1.9 | 100 ± 6 | 1 | 33 |
| E240Q | d | d | d | d | 330 |
| E278D | d | d | d | d | 1100 |
| E278Q | 37 ± 3 | 0.86 | 110 ± 5 | 1.2 | 50 |
| Y280F | 90 ± 5 | 2.0 | 93 ± 4 | 0.9 | 500 |
| Y280S | 91 ± 4 | 2.1 | 120 ± 5 | 1.2 | 50 |
| N424D | 76 ± 3 | 1.7 | 130 ± 5 | 1.3 | 2.5 |
| N424Q | 83 ± 4 | 1.9 | 150 ± 4 | 1.5 | 50 |
| F426H | 58 ± 3 | 1.3 | 190 ± 6 | 1.9 | 50 |
| F426W | 110 ± 5 | 2.5 | 120 ± 5 | 1.2 | 20 |
| E428D | 90 ± 5 | 2.0 | 120 ± 4 | 1.2 | 14 |
| E428Q | 55 ± 3 | 1.2 | 150 ± 5 | 1.5 | 50 |
Values are the means of at least three independent experiments with standard deviations indicated.
Relative increase or decrease in growth rate compared with wild-type.
Relative decrease compared with wild-type for in vitro tRNA lysine aminoacylation (17).
Not determined.
LysRS2 Variants and in Vivo AEC Resistance
LysRS2 variants able to sustain growth of E. coli were tested for their ability to grow on MM supplemented with 5 μM AEC, conditions that inhibit growth of the parental strain. The pattern of growth inhibition was consistent with previous in vitro studies, in that those LysRS2 variants with kinetics for AEC inhibition similar to wild-type (E240D, E278Q, N424D/Q, and E428Q) were unable to sustain growth in the presence of AEC. LysRS2 variants G216A, Y280F, Y280S, F426H, F426W, and E428D, all of which sustained growth at 5 μM AEC (Figure 2), were subsequently tested for growth in MM at elevated AEC concentrations. LysRS2 Y280F and F426W variant strains grew at 100 and 500 μM AEC, respectively, but none of the other LysRS2 variants could sustain growth above 5 μM AEC (Figure 3; data not shown). LysRS2 Y280F imparted dose-dependent AEC tolerance (i.e., poorer growth at higher concentrations), whereas F426W resulted in similar growth in the presence of AEC concentrations ranging from 5 to 500 μM. The absence of dose dependence for F426W suggested that factors in addition to LysRS2, such as indirect effects on lysine metabolism, may contribute to resistance, and this variant was not investigated further in vivo.
Figure 2.
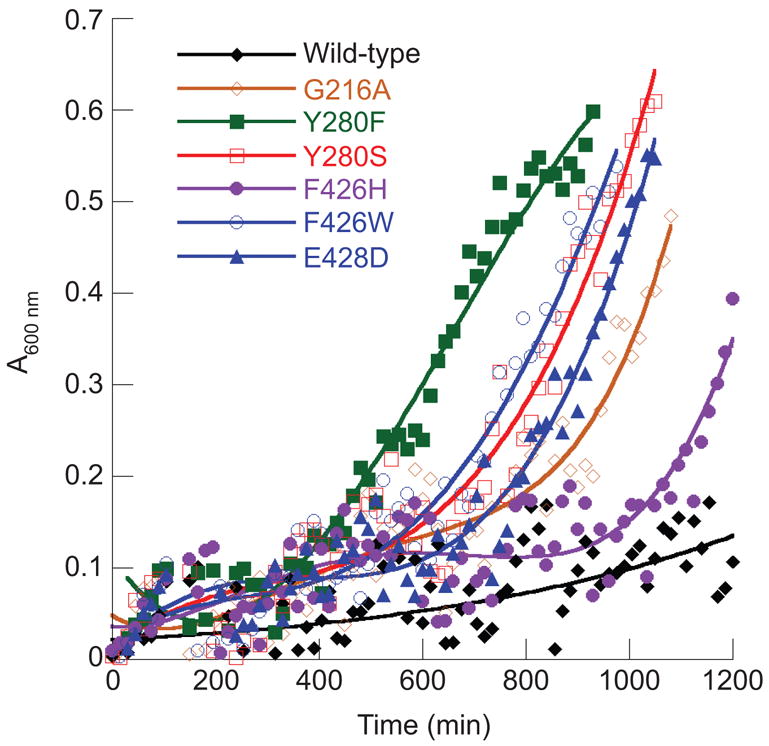
Growth of E.coli PALΔSΔUTR derivatives on MM supplemented with 5 μM AEC. Cells contained the vector pXLysSK1 encoding either wild-type LysRS or active site variants as indicated. Each time point represents an average reading derived from three independent cultures grown in 96 well microtiter plates.
Figure 3.
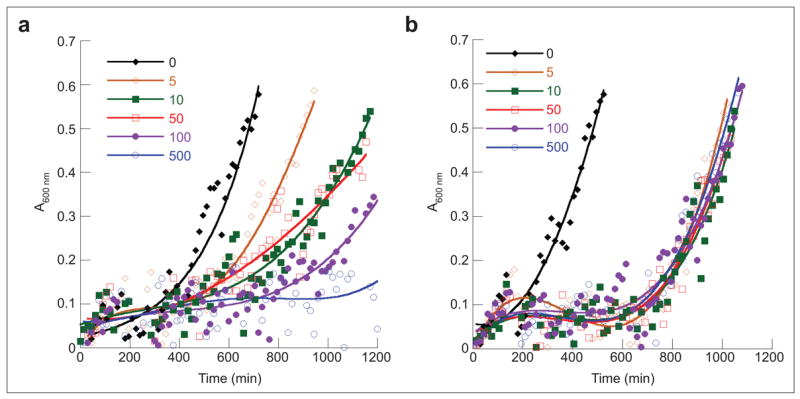
Growth of E. coli PALΔSΔUTR derivatives in the presence of AEC. Cells containing the vector pXLysSK1 encoding LysRS2 Y280F (a) or F426W (b) were grown on MM supplemented with different concentrations of AEC (μM) as indicated. Each time point represents an average reading derived from three independent cultures grown in 96 well microtiter plates.
The Y280F LysRS2 variant offered substantially higher resistance to AEC in vivo than indicated by in vitro steady-state kinetic studies, which had predicted resistance comparable to wild-type (17). This variant was then used as a starting point to search for additional mutations in LysRS2 that, together with Y280F, could provide similar levels of resistance to AEC as previously associated with L box mutants (1–10 mM) (21, 29).
LysRS2 Variants with Enhanced AEC Resistance
Y280F LysRS2-encoding plasmids were subjected to chemical mutagenesis using hydroxylamine, and the resulting mutants were replica plated to MM supplemented with 1 mM AEC. After 5 rounds of plasmid purification, transformation, and screening, one isolate (Y280F4) was obtained that grew on 5 mM AEC after re-transformation of fresh PALΔSΔUTR cells. Sequencing of the lysS gene from the corresponding plasmid did not reveal any additional mutations in either the LysRS2 encoding region or the promoter. The absence of mutations in the Y280F4-encoding gene led us to investigate whether increased abundance of LysRS2 protein accounted for AEC resistance, analogous to the elevated levels of tryptophanyl-tRNA synthetase observed during growth in the presence of the antibiotic indolmycin (30). E. coli PALΔSΔUTR transformants producing wild-type LysRS2 or the Y280F or Y280F4 variants were grown in LB or MM with or without AEC (5 μM), cells were harvested toward the end of log phase (A600 ≈ 0.8), and LysRS2 was quantified by immunoblotting. The most notable difference between Y280F and Y280F4 was observed during growth on MM+AEC (Figure 4) where LysRS2 levels were higher for Y280F4, suggesting that elevated AEC resistance of this variant is due to increased abundance of the LysRS2 protein. The absence of changes in lysS indicates that these increases in LysRS2 result from other undefined mechanisms, for example, mutations in the origin of replication, which increase plasmid copy number, or other mutations that enhance mRNA abundance.
Figure 4.
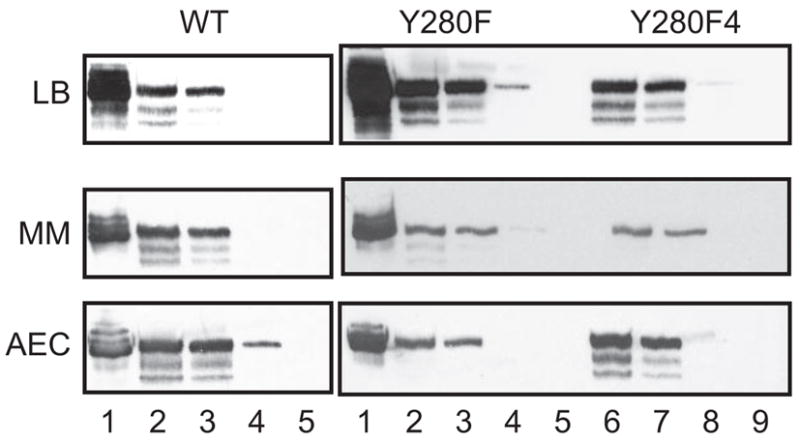
LysRS in E. coli PALΔSΔUTR derivatives. E. coli PALΔSΔUTR cells producing wild-type (WT), Y280F, and Y280F4 LysRS were grown in different media as indicated. Total cell protein from late log phase (A600 = 0.8) cells grown on LB, MM supplemented with 2 mM lysine (MM), or MM supplemented with 5 μM AEC (AEC) were separated by SDS–PAGE, transferred to membranes, and detected by using anti-LysRS2 polyclonal antibodies. Total protein concentrations loaded were 20 μg (lanes 2 and 6), 10 μg (lanes 3 and 7), 1 μg (lanes 4 and 8), and 0.1 μg (lanes 5 and 9). Purified LysRS (10 ng, lane 1) was loaded as control.
AEC Binding by the L Box Riboswitch
It has previously been shown that mutations in the leader region upstream of the E. coli lysC gene cause derepression of aspartokinase production, resulting in increased intracellular lysine and resistance to AEC (21). To investigate whether mutations in the lysC leader might contribute to the AEC resistance of the E. coli PALΔSΔUTR derivatives producing the LysRS2 Y280F and F426W variants, the sequences of the corresponding chromosomal regions were determined. The lysC leader sequences from each of these strains were found to be identical to that of the original parental strain, E. coli K-12 MG1655.
The B. subtilis lysC leader RNA regulates gene expression through a series of lysine-dependent structural rearrangements that include formation of a helix, designated helix 1, which results in the folding of the RNA into the helix of an intrinsic terminator. This mechanism results in attenuation of transcription both in vivo and in vitro when lysine levels are high (23, 24). It was suggested (23) that the lysC leader RNA in E. coli, which was previously shown to be involved in lysC regulation (25), can sense the concentration of lysine and form a repressor helix that sequesters the Shine–Dalgarno sequence and inhibits translation initiation. Phylogenetic and mutational analyses of L box RNAs predict that positions 202–214 of the E. coli lysC leader RNA participate in formation of helix 1 in the presence of lysine (23, 31). To determine whether helix 1 forms in E. coli lysC, binding of an oligonucleotide complimentary to positions 202–214 within the 3′ side of helix 1 was monitored by cleavage by RNase H, which specifically cleaves RNA/DNA hybrids. Transcription in the absence of lysine resulted in protection of 20% of the RNA from cleavage by RNase H; addition of lysine resulted in a 3-fold increase in protection from cleavage, supporting the model that lysine binding causes stabilization of helix 1 (Figure 5, panel a, lanes 1 and 7). Transcription in the presence of AEC resulted in a similar 60% protection from cleavage (Figure 5, panel b, lane 14). The RNase H cleavage assay was performed using concentrations of lysine or AEC ranging from 100 μM to 6.4 mM (Figure 5, panel a) to determine the concentration resulting in half-maximal protection. Protection from RNase H cleavage increased as the lysine concentration was increased, resulting in an apparent KD of ~360 μM; the apparent KD for AEC was ~830 μM (Figure 5, panel c). RNase H cleavage of the B. subtilis leader showed a similar 2-fold decrease in affinity for AEC as compared with lysine (data not shown). These results are in good agreement with other studies of lysC leader RNAs, which indicate a decreased affinity for AEC (23, 24) and support the hypothesis that the L box RNAs in the LysRS2 variants, which are identical to wild-type, exhibit normal response to AEC in vivo.
Figure 5.

RNase H cleavage of the E. coli lysC leader RNA. Labeled RNAs generated in the presence of increasing concentrations of lysine (a) or AEC (b) were incubated with a DNA oligonucleotide complimentary to the 3′ side of helix 1 to allow hybridization, which was followed by digestion of DNA/RNA hybrids with RNase H. FL indicates full-length transcripts; C indicates RNase H cleavage products. Lysine or AEC concentrations (mM) used in each transcription are shown above each lane. c) Quantitation of protection in response to lysine (■) or AEC (▲).
AEC Binding by LysRS2 Variants
In vitro steady-state kinetic parameters indicated little difference in the apparent specificity for AEC between wild-type LysRS2 and the Y280F and F426W variants (17). These data are in contrast to the in vivo studies described above, which showed dramatic increases in AEC resistance as a result of single amino acid replacements of Tyr280 and Phe426. In an attempt to reconcile the apparent discrepancy between the in vivo and in vitro data, dissociation constants (KD) for lysine and AEC binding to wild-type and variant LysRS2 were determined (Table 2). In agreement with previous steady-state data, lysine and AEC were found to bind wild-type LysRS2 with comparable affinities. LysRS2 F426W bound AEC 23-fold less tightly than wild-type, contradicting previous steady-state data that indicated an apparent binding affinity comparable to wild-type. The Y280F variant showed a 45-fold increase in the KD for AEC compared to wild-type, in contrast to the steady-state data, which had suggested a decrease in apparent affinity of only 5-fold. The reduced affinity for AEC of the Y280F and F426W variants is consistent with increased resistance in vivo and also suggests that antibiotic binding might be rate-limiting for these variants. Substrate binding was assumed not to be a rate-limiting step in the classical Michaelis–Menten steady-state kinetic analyses employed previously; in the event that substrate binding becomes rate-limiting, as may be the case here for AEC, the derived steady-state parameters would be invalid (32), explaining their lack of correlation with the in vivo data.
TABLE 2.
Fluorescence titration of LysRS2 variants with lysine and AECa
| LysRS
|
|||
|---|---|---|---|
| wild-type | Y280F | F426W | |
| lysine KD (μM) | 1.6 ± 0.6 | 57 ± 0.4 | 37 ± 0.2 |
| AEC KD (μM) | 1.7 ± 0.5 | 77 ± 1 | 39 ± 0.4 |
Values are the means of at least four independent experiments with standard deviations indicated.
Conclusions
The inability of wild-type LysRS2 to discriminate AEC from lysine results in toxicity via the co-translational insertion of AEC into proteins in place of lysine (33). This mechanism of antibiotic action was supported by the finding that nonorthologous replacement of B. subtilis LysRS2 with the AEC-resistant Borrelia burgdorferi LysRS1 conferred resistance in vivo (15). However, because resistance in this case was dependent on a heterologous protein, this approach did not directly identify LysRS2 as the natural target of the antibiotic. More recently another study in B. subtilis concluded instead that the L box riboswitch rather than LysRS is the cellular target of AEC (27), since resistance alleles consistently map to the 5′ leader region of the lysC gene (25, 26). Such mutations lead to increased aspartokinase protein levels and elevated intracellular lysine concentrations, thereby allowing growth in the presence of 1–10 mM AEC (21, 29). If lysC were indeed the primary target of AEC, then the introduction of resistant variants of LysRS would not be expected to alleviate AEC toxicity. Our data show that LysRS2 variants can impart resistance to up to 5 mM AEC, despite the presence of a wild-type lysC gene with a functional L box leader region. Together with previous studies, our findings clearly show that LysRS is the primary cellular target for growth inhibition by AEC while the L box of lysC is the primary site for acquiring resistance. This conclusion is consistent with previous studies selecting for AEC resistant strains, which consistently isolate L box but not LysRS mutants (21, 25, 29). This clear separation of the bacterial target and resistance sites suggests that the future design of effective lysine analog antibiotics will depend on targeting both LysRS and the L box.
The LysRS1 and LysRS2 proteins are unrelated in structure but perform the identical function, synthesis of Lys-tRNALys (34). Biochemical studies in vitro have shown that LysRS2 is inherently more sensitive than LysRS1 to inhibition by AEC (15–18). Surprisingly, our present data show that LysRS2 can readily acquire AEC resistance comparable to that of LysRS1 through single amino acid substitutions in the active site. Among those tested, the Y280F and F426W variants provided the most significant improvements to AEC resistance in vivo. The AEC resistance of LysRS2 Y280F likely stems from the disruption of an H-bond interaction network with the ε-NH2 group of the amino acid substrate lysine and Glu240 in the back of the active site (Figure 6). The F426W replacement is expected to be marginally less disruptive in that direct H-bonding interactions are not affected, but instead the bulkier tryptophan side chain would be expected to hinder binding near the amino acid substrate backbone (Figure 6). Previous steady-state analyses failed to reveal the substantial losses in AEC affinity resulting from both the Y280F and F426W substitutions, which likely lower kon to the point that amino acid binding becomes rate-limiting. The absence of a substantial increase in AEC tolerance among the other LysRS2 variants, several of which display comparable steady-state kinetic parameters to Y280F and F426W, suggests that this mechanistic change is the predominant factor determining resistance. This is supported by the finding that mutants selected for further increases in AEC resistance did not contain protein-coding changes but instead showed modest increases in LysRS2 levels. These mutants and the corresponding parental strains all grew twice as slowly as wild-type, indicating that LysRS2-dependent AEC resistance adversely impacts growth. This correlation between growth and AEC toxicity is in contrast to LysRS1, which confers AEC-resistance without any reduction in growth rate (35). Taken together, these data suggest that the LysRS1 and LysRS2 active sites have similar capacities for AEC resistance, but only LysRS1 can attain resistance without critically compromising its ability to efficiently synthesize Lys-tRNALys.
Figure 6.
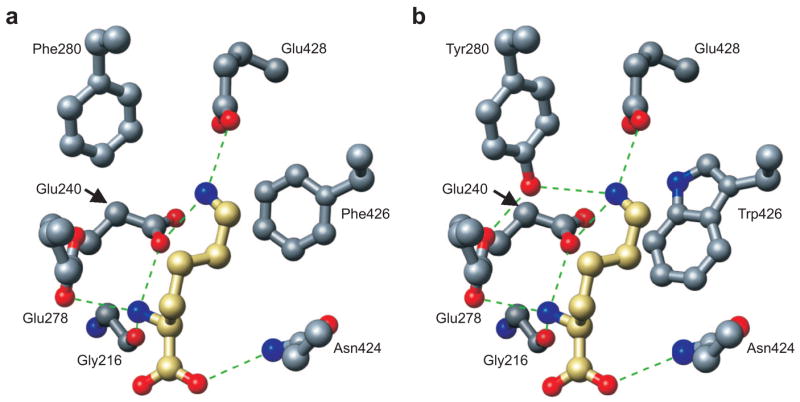
Model of L-lysine recognition by LysRS variants Y280F and F426W. Modeling of lysine binding at the active site of E. coli LysRS (40) for the Y280F (a) and F426W (b) variants. Residues were modified appropriately, and the structures were energy minimized using Swiss-Pdb Viewer v 3.7. The resulting models were visualized using PyMOL (41). H-bonds are shown as dashed lines. The carbon backbone for the substrate lysine is gold, while the carbon backbone of the LysRS2 active site residues is gray. Oxygen and nitrogen are colored red and blue, respectively, for enzyme and substrate.
METHODS
In Vivo Analysis of LysRS Variants
The pXLysSK1 vector (28) containing the E. coli lysS gene under both its endogenous promoter and the T7 RNAP promoter was used as a template for generation of LysRS variants. Sets of two complementary primers, 27 nucleotides each, were designed to introduce each LysRS2 mutation. Mutations were confirmed by complete sequencing of each gene. The empty vector, with no lysS gene, was prepared by digestion of plasmid pXLysSK1 with EcoRI and ligation of the resulting empty vector fragment with T4 DNA ligase. The E. coli strain PALΔSΔUTR (F−(lac-pro) gyrA rpoB metB argE(Am) ara supF ΔlysS::kan ΔlysU srl-300::Tn10 recA56 (pMAK705 lysU) (36) was transformed with 100 ng of plasmid pXLysSK1 containing either a LysRS variant, wild-type LysRS as a positive control, or no insert as a negative control. Transformants were selected on LB plates supplemented with 100 μg mL−1 ampicillin at 43 °C, a prohibitive temperature for pMAK705 lysU replication. Single colonies were counted and then replica-plated and grown on LB supplemented with 100 μg mL−1 ampicillin at 43 °C for two subsequent rounds. After the second replica-plating, plates were replicated both on LB supplemented with 100 μg mL−1 ampicillin and LB supplemented with 100 μg mL−1 ampicillin and 30 μg mL−1 chloramphenicol (to check for continued retention of pMAK705 lysU) and grown at 30 °C for 24 h. Colonies able to grow on both plates were excluded from further analysis. Purified isolates were then replicated to MM plates supplemented with 100 μg mL−1 ampicillin and grown at 37 °C. Colonies were selected from LB plates grown at 30 °C and inoculated into liquid LB supplemented with 100 μg mL−1 of ampicillin and grown to saturation at 37 °C, washed, and diluted into LB or MM supplemented with 2 mM lysine or 5 μM AEC and incubated in a microplate reader for growth rate determination at 37 °C. Each growth curve was determined in triplicate and averaged. Colonies able to grow on MM supplemented with 5 μM AEC were subjected to growth in increasing concentrations of AEC up to 500 μM. Plasmids encoding LysRS variants able to promote growth on media containing more than 5 μM AEC were isolated, sequenced and reintroduced into the PALΔSΔUTR strain, and the resulting isolates were retested for growth in the presence of AEC.
Construction and Screening of LysRS Variant Libraries
Plasmid pXK-YF, coding for the LysRS Y280F variant, was isolated and used to transform E. coli DH5α for large scale plasmid DNA isolation. Ten micrograms of plasmid DNA was incubated with 500 μL of 1 M hydroxylamine solution, prepared just prior to the reaction, for 20 h at 37 °C. After incubation, DNA was recovered using a plasmid DNA preparation kit (Qiagen). A total of 1 μg of recovered plasmid DNA was used to transform E. coli CJ236, an ung−strain unable to correct the chemical modification of U caused by hydroxylamine (37, 38) and approximately 50,000 transformants were then pooled. Cells were grown to saturation, re-inoculated in 500 mL of LB, and grown for 16 h, and plasmid DNA was then isolated. The resulting plasmid preparation was used to retransform the PALΔSΔUTR strain, and approximately 50,000 transformants were obtained. Colonies were replica-plated onto LB containing 100 μg mL−1 of ampicillin, and after a second replica-plating at 43 °C, colonies were replicated to MM supplemented with 1 mM AEC and grown at 37 °C. Colonies able to grow at this concentration of AEC were isolated and grown to saturation in liquid LB, washed, and diluted on MM supplemented with increasing concentrations of AEC (up to 10 mM), and incubated in a microplate reader in triplicate. Mutants able to grow at concentrations higher than 2 mM AEC were isolated and the entire lysS gene was sequenced to identify additional mutations. The isolated plasmids were then used to transform E. coli PALΔSΔUTR and screened a second time to confirm their phenotypes.
Detection of LysRS Variants by Immunoblotting
E. coli PALΔSΔUTR containing wild-type LysRS, or the Y280F or Y280F4 variants, was grown in LB or MM supplemented with 2 mM lysine or 500 μM AEC at 37 °C, and 100 mL samples were taken at A600= 1.0 (late log phase). Cells were centrifuged at 6000 × g at 4 °C for 15 min and suspended in 5 mL of sample buffer (100 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 1 mM EDTA, and protease inhibitor cocktail [Hoffmann-La Roche, Inc.]). Cells were then passed twice through a French Press cell and centrifuged at 30,000 × g for 30 min at 4 °C. Cell extracts were concentrated in dialysis bags covered with PEG 20,000 MW overnight at 4 °C. Protein concentrations were determined by the Bradford method (BioRad, Inc.). Cell extracts with varying amounts of protein were separated on 10% SDS/PAGE gels and then blotted onto nitrocellulose membranes. LysRS was detected using primary rabbit polyclonal antibodies (against LysRS from E. coli) and horseradish peroxidase conjugated secondary antibodies followed by visualization using standard chemiluminescence protocols.
RNase H Cleavage
The DNA template for transcription was generated by PCR of E. coli PALΔSΔUTR chromosomal DNA using a 5′ primer containing the T7 RNAP promoter fused to position +18 of the E. coli lysC transcript. Position +232 was selected as the end point of the lysC fragment and 20 random nucleotides were added downstream to allow resolution of cleaved RNA from the full-length transcript. The resulting fragment was confirmed by sequencing and used as template DNA for T7 RNAP transcription using a T7-MEGAshortscript transcription kit (Ambion). RNAs were synthesized in the presence of [α-32P]-UTP (GE Healthcare; 800 Ci/mmol [30 TBq/mmol]) for 30 min at 37 °C in the presence or absence of lysine or AEC. After transcription, a DNA oligonucleotide (47 μM) complimentary to positions 202–214 of the E. coli lysC RNA leader was added, followed by incubation for 5 min. RNase H (0.45 U μL−1; Ambion) was then added and the incubation was continued for 10 min at 37 °C. The reactions were stopped by the addition of phenol. The resulting cleavage products were subjected to electrophoresis on 6% denaturing polyacrylamide gel, visualized by PhosphorImager analysis, and quantified using ImageQuant 5.2. The percentage of RNA protected from cleavage was calculated as the ratio of full length transcript relative to the total amount of RNA for each reaction.
KD Determination
Fluorescence spectra were measured at 20 °C in a Hitachi F3010 spectrofluorometer in 100 mM Tris-HCl, pH 8.0, containing 16 mM MgCl2. The excitation wavelength was 295 nm, and emission was recorded at 340 nm. The excitation and emission bandpasses were 5 and 10 nm, respectively. The protein concentration was 2 μM. The absorbance of the solution was less than 0.1 cm−1 at the excitation wavelength. The fluorescence intensity was corrected for dilution due to addition of ligand solution. The values of the dissociation constants for enzyme–ligand complexes were determined as described previously (39). The data were fitted to a single site binding equation, using KYPLOT (Koichi Yoshioka, 1997–2000 version 2.0 beta 13). At least four experiments were done under the same conditions, and average values and standard deviations were used to construct the plots.
Characterization of the lysC Leader Region
Regions of 477 bp including the lysC leader sequence were amplified by PCR from genomic DNA of the parental E. coli strain PALΔSΔUTR and AEC-resistant transformants of the same strain producing LysRS Y280F and F426W. PCR primers were complementary to sequences 425 bp upstream (5′-GTGTAAACCCTTAGCGCAG-3′) and 49 bp downstream (5′-CAAAATCAGCTACGCTGG-3′) of the ATG translation start site. The PCR products were purified by gel extraction (QIAGEN, QIAEX II) and then directly sequenced.
Acknowledgments
We would like to thank S. Blanquet for providing strains and plasmids, F. Grundy for technical advice, and J. Ling and H. Roy for critical reading of the manuscript. This work was supported by Grants GM65183 (M.I.) and GM63615 (T.H.) from the National Institutes of Health, and a NIH predoctoral fellowship F31GM076894 (S.W.).
References
- 1.Ibba M, Söll D. Science. Vol. 286. 1999. Quality control mechanisms during translation; pp. 1893–1897. [DOI] [PubMed] [Google Scholar]
- 2.Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D’Hooghe M, Van Gerwen V, Callaerts P, Van Den BL, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, Tim-merman V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 3.Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 4.Nangle LA, Motta CM, Schimmel P. Global effects of mistranslation from an editing defect in mammalian cells. Chem Biol. 2006;13:1091–1100. doi: 10.1016/j.chembiol.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Giegé R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hendrickson TL, Schimmel P. In: Translation Mechanisms. Lapointe J, Brakier-Gingras L, editors. Kluwer Academic/Plenum Publishers; New York: 2003. pp. 34–64. [Google Scholar]
- 7.LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science. 2001;294:165–168. doi: 10.1126/science.1064242. [DOI] [PubMed] [Google Scholar]
- 8.Dale T, Uhlenbeck OC. Amino acid specificity in translation. Trends Biochem Sci. 2005;30:659–665. doi: 10.1016/j.tibs.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Ahel I, Korencic D, Ibba M, Söll D. Trans-editing of mischarged tRNAs. Proc Natl Acad Sci USA. 2003;100:15422–15427. doi: 10.1073/pnas.2136934100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An S, Musier-Forsyth K. Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J Biol Chem. 2004;279:42359–42362. doi: 10.1074/jbc.C400304200. [DOI] [PubMed] [Google Scholar]
- 11.Brown JR, Zhang J, Hodgson JE. A bacterial antibiotic resistance gene with eukaryotic origins. Curr Biol. 1998;8:R365–R367. doi: 10.1016/s0960-9822(98)70238-6. [DOI] [PubMed] [Google Scholar]
- 12.Brown JR, Gentry D, Becker JA, Ingraham K, Holmes DJ, Stanhope MJ. Horizontal transfer of drug-resistant aminoacyl-transfer-RNA synthetases of anthrax and Gram-positive pathogens. EMBO Rep. 2003;4:1–7. doi: 10.1038/sj.embor.embor881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yanagisawa T, Kawakami M. How does Pseudomonas fluorescens avoid suicide from its antibiotic pseudomonic acid? J Biol Chem. 2003;278:25887–25894. doi: 10.1074/jbc.M302633200. [DOI] [PubMed] [Google Scholar]
- 14.Terada T, Nureki O, Ishitani R, Ambrogelly A, Ibba M, Söll D, Yokoyama S. Functional convergence of two lysyl-tRNA synthetases with unrelated topologies. Nat Struct Biol. 2002;9:257–262. doi: 10.1038/nsb777. [DOI] [PubMed] [Google Scholar]
- 15.Jester B, Levengood J, Roy H, Ibba M, Devine K. Non-orthologous replacement of lysyl-tRNA synthetase prevents addition of lysine analogs to the genetic code. Proc Natl Acad Sci USA. 2003;100:14351–14356. doi: 10.1073/pnas.2036253100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levengood J, Ataide SF, Roy H, Ibba M. Divergence in noncognate amino acid recognition between class I and class II lysyl-tRNA synthetases. J Biol Chem. 2004;279:17707–17714. doi: 10.1074/jbc.M313665200. [DOI] [PubMed] [Google Scholar]
- 17.Ataide SF, Ibba M. Discrimination of cognate and noncognate substrates at the active site of class II lysyl-tRNA synthetase. Biochemistry. 2004;43:11836–11841. doi: 10.1021/bi0490542. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Praetorius-Ibba M, Ataide SF, Roy H, Ibba M. Discrimination of cognate and noncognate substrates at the active site of class I lysyl-tRNA synthetase. Biochemistry. 2006;45:3646–3652. doi: 10.1021/bi0523005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ataide SF, Ibba M. Small molecules - big players in the evolution of protein synthesis. ACS Chem Biol. 2006;1:285–297. doi: 10.1021/cb600200k. [DOI] [PubMed] [Google Scholar]
- 20.Zwolshen JH, Bhattacharjee JK. Genetic and biochemical properties of thialysine-resistant mutants of Saccharomyces cerevisiae. J Gen Microbiol. 1981;122:281–287. doi: 10.1099/00221287-122-2-281. [DOI] [PubMed] [Google Scholar]
- 21.Di Girolamo M, Busiello V, Di Girolamo A, Foppoli C, De Marco C. Aspartokinase III repression in a thialysine-resistant mutant of E. coli. Biochem Int. 1988;17:545–554. [PubMed] [Google Scholar]
- 22.Kalinowski J, Bachmann B, Thierbach G, Puhler A. Aspartokinase genes lysC alpha and lysC beta overlap and are adjacent to the aspartate beta-semialdehyde dehydrogenase gene asd in Corynebacterium glutamicum. Mol Gen Genet. 1990;224:317–324. doi: 10.1007/BF00262424. [DOI] [PubMed] [Google Scholar]
- 23.Grundy FJ, Lehman SC, Henkin TM. The L box regulon: lysine sensing by leader RNAs of bacterial lysine biosynthesis genes. Proc Natl Acad Sci USA. 2003;100:12057–12062. doi: 10.1073/pnas.2133705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 2003;17:2688–2697. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Y, Chen NY, Paulus H. Identification of aecA mutations in Bacillus subtilis as nucleotide substitutions in the untranslated leader region of the aspartokinase II operon. J Gen Microbiol. 1991;137:1135–1143. doi: 10.1099/00221287-137-5-1135. [DOI] [PubMed] [Google Scholar]
- 26.Patte JC, Akrim M, Mejean V. The leader sequence of the Escherichia coli lysC gene is involved in the regulation of LysC synthesis. FEMS Microbiol Lett. 1998;169:165–170. doi: 10.1111/j.1574-6968.1998.tb13313.x. [DOI] [PubMed] [Google Scholar]
- 27.Blount KF, Wang JX, Lim J, Sudarsan N, Breaker RR. Antibacterial lysine analogs that target lysine riboswitches. Nat Chem Biol. 2007;3:44–49. doi: 10.1038/nchembio842. [DOI] [PubMed] [Google Scholar]
- 28.Leveque F, Plateau P, Dessen P, Blanquet S. Homology of lysS and lysU, the two Escherichia coli genes encoding distinct lysyl-tRNA synthetase species. Nucleic Acids Res. 1990;18:305–312. doi: 10.1093/nar/18.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vold B, Szulmajster J, Carbone A. Regulation of dihydrodipicolinate synthase and aspartate kinase in Bacillus subtilis. J Bacteriol. 1975;121:970–974. doi: 10.1128/jb.121.3.970-974.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitabatake M, Ali K, Demain A, Sakamoto K, Yokoyama S, Söll D. Indolmycin resistance of Streptomyces coelicolor A3(2) by induced expression of one of its two tryptophanyl-tRNA synthetases. J Biol Chem. 2002;277:23882–23887. doi: 10.1074/jbc.M202639200. [DOI] [PubMed] [Google Scholar]
- 31.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch? Nucleic Acids Res. 2003;31:6748–6757. doi: 10.1093/nar/gkg900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fersht AR. Structure and Mechanism in Protein Science. W.H. Freeman; New York: 1999. [Google Scholar]
- 33.Christner P, Yankowski RL, Benditt M, Jimenez SA. Alteration in the conformational stability of collagen caused by the incorporation of the lysine analogue S-2-aminoethylcysteine. Biochim Biophys Acta. 1996;1294:37–47. doi: 10.1016/0167-4838(95)00261-8. [DOI] [PubMed] [Google Scholar]
- 34.Ibba M, Morgan S, Carnow AW, Pridmore DR, Vothknecht UC, Gardner W, Ling W, Woese CR, Soll D. A euryarcheal lysyl-tRNA sythetase: resemblance to class I synthetases. Science. 1997;278:1119–1122. doi: 10.1126/science.278.5340.1119. [DOI] [PubMed] [Google Scholar]
- 35.Ataide SF, Jester BC, Devine KM, Ibba M. Stationary-phase expression and aminoacylation of a transfer-RNA-like small RNA. EMBO Rep. 2005;6:742–747. doi: 10.1038/sj.embor.7400474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Commans S, Blanquet S, Plateau P. A single substitution in the motif 1 of Escherichia coli lysyl-tRNA synthetase induces cooperativity toward amino acid binding. Biochemistry. 1995;34:8180–8189. doi: 10.1021/bi00025a025. [DOI] [PubMed] [Google Scholar]
- 37.Joyce CM, Grindley ND. Method for determining whether a gene of Escherichia coli is essential: application to the polA gene. J Bacteriol. 1984;158:636–643. doi: 10.1128/jb.158.2.636-643.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 39.Takita T, Ohkubo Y, Shima H, Muto T, Shimizu N, Sukata T, Ito H, Saito Y, Inouye K, Hiromi K, Tonomura B. Lysyl-tRNA synthetase from Bacillus stearothermophilus. Purification, and fluorometric and kinetic analysis of the binding of substrates, L-lysine and ATP. J Biochem (Tokyo) 1996;119:680–689. doi: 10.1093/oxfordjournals.jbchem.a021296. [DOI] [PubMed] [Google Scholar]
- 40.Onesti S, Desogus G, Brevet A, Chen J, Plateau P, Blanquet S, Brick P. Structural studies of lysyl-tRNA synthetase: conformational changes induced by substrate binding. Biochemistry. 2000;39:12853–12861. doi: 10.1021/bi001487r. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.