

Abstract
Mutations in superoxide dismutase 1 (SOD1; EC 1.15.1.1) are responsible for a proportion of familial amyotrophic lateral sclerosis (ALS) through acquisition of an as-yet-unidentified toxic property or properties. Two proposed possibilities are that toxicity may arise from imperfectly folded mutant SOD1 catalyzing the nitration of tyrosines [Beckman, J. S., Carson, M., Smith, C. D. & Koppenol, W. H. (1993) Nature (London) 364, 584] through use of peroxynitrite or from peroxidation arising from elevated production of hydroxyl radicals through use of hydrogen peroxide as a substrate [Wiedau-Pazos, M., Goto, J. J., Rabizadeh, S., Gralla, E. D., Roe, J. A., Valentine, J. S. & Bredesen, D. E. (1996) Science 271, 515–518]. To test these possibilities, levels of nitrotyrosine and markers for hydroxyl radical formation were measured in two lines of transgenic mice that develop progressive motor neuron disease from expressing human familial ALS-linked SOD1 mutation G37R. Relative to normal mice or mice expressing high levels of wild-type human SOD1, 3-nitrotyrosine levels were elevated by 2- to 3-fold in spinal cords coincident with the earliest pathological abnormalities and remained elevated in spinal cord throughout progression of disease. However, no increases in protein-bound nitrotyrosine were found during any stage of SOD1-mutant-mediated disease in mice or at end stage of sporadic or SOD1-mediated familial human ALS. When salicylate trapping of hydroxyl radicals and measurement of levels of malondialdehyde were used, there was no evidence throughout disease progression in mice for enhanced production of hydroxyl radicals or lipid peroxidation, respectively. The presence of elevated nitrotyrosine levels beginning at the earliest stages of cellular pathology and continuing throughout progression of disease demonstrates that tyrosine nitration is one in vivo aberrant property of this ALS-linked SOD1 mutant.
Keywords: nitration, peroxidation
Point mutations in Cu/Zn superoxide dismutase 1 (SOD1; EC 1.15.1.1) cause approximately 20% of cases of familial amyotrophic lateral sclerosis (ALS) (1–5), a progressive neurodegenerative disorder characterized by degeneration of upper and lower motor neurons (2, 6). SOD1, a cytosolic enzyme abundant in neural tissue, plays an important role in regulation of oxidative stress and protection against oxygen radical-induced cellular damage (7). Despite an early focus on a loss of activity as a potential mechanism underlying human disease (8), subsequent in vitro studies demonstrated that familial ALS mutations vary in their effects on enzymatic activity and for some mutations, such as the substitution of arginine for glycine at position 37 (G37R), the mutant enzyme retains full specific activity (9). A series of transgenic mouse models expressing familial ALS-linked human SOD1 mutations [G93A (10), G37R (11), and G85R (12)], as well as one transgenic line expressing a mutation in mouse SOD1 [G86R (13)], have clearly demonstrated that disease in mice is not due to loss of SOD1 activity, but rather arises from a toxic property or properties of the mutant enzymes. This is further supported by the absence of motor neuron disease in mice homozygous for deletion of the SOD1 gene (14). Further, despite over 42 different point mutations known in human disease (3–5), there are no null mutations that preclude the synthesis of full-length, or nearly full-length, SOD1 polypeptides.
Central questions in the pathogenesis of SOD1-linked familial ALS are (i) the identity of the toxic property or properties of the mutant subunits; and (ii) the basis for the selective vulnerability of motor neurons. There are currently two hypotheses for the aberrant properties of the mutant enzymes. First, Beckman and colleagues (15) proposed that the mutants may be less perfectly folded, allowing more efficient use of peroxynitrite as an enzyme substrate. Formed by the rapid, spontaneous reaction of superoxide with nitric oxide, peroxynitrite may be converted by the catalytic copper of the mutant enzymes into a highly reactive nitronium intermediate which readily reacts with tyrosine residues of proteins (15, 16). While nitric oxide is not thought normally to be present in motor neurons, nitric oxide synthase is induced after injury (17, 18). A second hypothesis, proposed by Bredesen and colleagues (19), focuses on the ability of mutant SOD1s to use hydrogen peroxide as a substrate to generate highly toxic hydroxyl radicals that can damage a variety of cellular targets [e.g., mitochondrial membranes by lipid peroxidation and glutamate transporters by oxidation (20)]. At least two of the familial ALS-linked mutant SOD1 subunits can increase the rate of hydroxyl radical formation in vitro, probably due to increased availability of the active Cu2+ site to H2O2 (19).
In light of these earlier efforts, we report here the use of biochemical and immunologic methods to test whether enhanced tyrosine nitration and/or peroxidation occurs in vivo in transgenic mice that develop progressive motor neuron disease mediated by expression of human familial ALS-linked mutation G37R SOD1 (11).
EXPERIMENTAL PROCEDURES
Measurement of Free 3-Nitrotyrosine.
Spinal cords and brain stems from control mice, transgenic mice overexpressing wild-type human SOD1, and two transgenic lines (lines 42 and 106) expressing human mutant G37R SOD1 were rapidly removed at 6, 12, and 20 weeks (near end-stage disease) of age. Six to 12 animals were used for each age group. Lumbar and cervical spinal cords as well as brain stems were placed in 0.25 ml of chilled 0.1 M perchloric acid. The samples were sonicated and centrifuged, and 3-nitrotyrosine levels in the supernatant were quantified by HPLC with a 16-electrode chemical detector (21). 3-Nitrotyrosine data were expressed as ratios of 3-nitrotyrosine to tyrosine.
Preparation of Spinal Cord, Sciatic Nerve, and Neurofilament-Enriched Extracts.
Whole spinal cords and sciatic nerves were homogenized in buffer containing 50 mM Tris⋅HCl at pH 7.00, 150 mM NaCl, 5 mM EDTA, 1% SDS, 1% Nonidet P-40, and 1 mM each of leupeptin, pepstatin, and chymostatin, boiled for 10 min, and clarified by centrifugation at 16,000 × g. To prepare intermediate filament-enriched cytoskeletons, whole spinal cords from control and transgenic mice were homogenized in a 1:1 mixture of buffer A (100 mM sodium phosphate, pH 7.15/150 mM NaCl/2 mM EDTA/2% Triton X-100) and buffer B (1.8 M sucrose/0.001% sodium azide) containing 2 mM phenylmethanesulfonyl fluoride. Homogenates were clarified by centrifugation at 16,000 × g for 20–30 min, and the neurofilament enriched pellets were reconstituted in 50 mM sodium phosphate, pH 7.00/2 mM EDTA/1% SDS. Similarily, intermediate filament-enriched cytoskeletons were prepared from spinal cords from two patients with familial ALS caused by SOD1 mutation A4V, four patients with sporadic ALS, and three control individuals (all but the familial samples were kindly provided by Jeff Rothstein).
Ten micrograms of partially purified neurofilament protein and 50 μg of whole spinal cord and sciatic nerve extracts were loaded on 7.5% polyacrylamide gels, electrophoresed, and transferred to nitrocellulose filters (22). Immunoblots were blocked in 2% nonfat dry milk in PBS and allowed to react with monoclonal antibodies to 3-nitrotyrosine (Upstate Biotechnology, Lake Placid, NY; and antibodies 1A6 and 7A2 kindly provided by Joe Beckman, University of Alabama) at 4°C, overnight. Horseradish peroxidase-conjugated mouse IgG was used as a secondary antibody, and antibody binding was visualized by enhanced chemiluminescence. An in vitro nitrated sample of neurofilaments (see below) was analyzed in parallel. Primary antibody specificity was determined by inhibition of binding in the presence of 10 mM 3-nitro-l-tyrosine (Sigma). Neurofilament preparations were immunoblotted with rabbit polyclonal antibodies recognizing glial fibrillary acidic protein (GFAP; Dako), the carboxyl termini of neurofilament light (NF-L) and heavy (NF-H) subunits (23), and monoclonal antibodies to medium (NF-M) subunits (Boehringer Mannheim). Bound primary antibody was detected with 125I-labeled protein A.
Signals generated by chemiluminescence were quantified by laser densitometry (LKB) using a dilution series of in vitro nitrated neurofilaments as quantitation standards. 125I signals were quantified with a PhosphorImager (Molecular Dynamics).
In Vitro Nitration of Neurofilaments.
Forty micrograms of partially purified neurofilaments was nitrated with newly synthesized peroxynitrate (24) at a final concentration of 10 mM in 100 μl of 0.1 M potassium phosphate, pH 7.4, for 10 min at room temperature.
Measurement of 2,3- and 2,5-Dihydroxybenzoic Acid (DHBA).
The salicylate hydroxyl trapping method (25–27) was used for measuring levels of OH− radicals in control mice, transgenic mice overexpressing human SOD1, and two transgenic lines of mice (42 and 106) expressing human mutant G37R SOD1. Measurements were made from animals aged 6 weeks (at the time of the earliest observable pathology), 12 weeks (still prior to clinical disease), and 20 weeks (near end-stage of disease). Six to 12 animals were used for each age group. Salicylate (100 mg/kg) was administered intraperitoneally and 60 min later the animals were sacrificed. Spinal cords were rapidly removed and dissected on ice into cervical and lumbar regions, and the brain stem was removed. All tissues were placed in 0.25 ml of chilled 0.1 M perchloric acid. The samples were sonicated and centrifuged, and levels of salicylate and its metabolites 2,3- and 2,5-DHBA in the supernatant were quantified by HPLC as described above. The data were expressed as the ratio of 2,3- and 2,5-DHBA to salicylate to normalize for potential animal-to-animal differences in the concentrations of injected salicylate.
RESULTS
Elevation of Free Nitrotyrosine Levels Early in Disease and Throughout Disease Progression.
Levels of free nitrotyrosine were measured in extracts of spinal cords from the following: normal mice, transgenic mice expressing the G37R mutant at levels 5 (line 106) or 12 (line 42) times the amount of endogenous mouse SOD1, and transgenic mice (line 76) expressing wild-type human SOD1 to levels 11 times mouse SOD1. The two G37R-expressing lines of mice develop progressive motor neuron disease beginning at 4 months (for line 42) or 6–9 months (for line 106) of age (11). Although histological examination with hemotoxylin and eosin suggested that abnormalities in motor neurons first appear at ≈11 weeks of age for both lines 42 and 106 (11), immunocytochemistry with antibodies to neurofilaments identified vaculolar abnormalities in the route exit zones of spinal cords as early as 5 weeks, and these were markedly increased at 6 weeks of age (Fig. 1, arrowheads).
Figure 1.
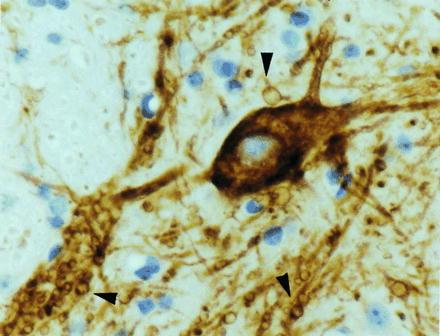
Early pathology in transgenic mice expressing SOD1 mutant G37R. Immunocytochemistry on a spinal cord section of a 6-week-old, G37R (line 42) transgenic mouse using antibodies against a phosphorylated determinant on neurofilament proteins (SMI 32). Numerous small vacuoles are evident in processes (arrowheads) in the spinal cord. (×800.)
Temporally coincident with the first pathology in motor neuron processes of G37R (line 42), levels of free 3-nitrotyrosine were elevated 2-fold in extracts of lumbar spinal cords of 6-week-old transgenic animals (Fig. 2A, line 42), compared with similar measurements from spinal cords from nontransgenic control mice. Since no increase in nitrotyrosine levels was found in mice accumulating wild-type human SOD1 to a similar level [Fig. 2A, line hSOD(76)], the elevation in nitrotyrosine content must reflect a property unique to the G37R mutant SOD1. Moreover, 3-nitrotyrosine was present at lesser elevations at 6 weeks in the cervical spinal cord and brain stem of these G37R (line 42) transgenic mice (Fig. 2A). In the other G37R SOD1 mutant (line 106), levels of free 3-nitrotyrosine at 6 weeks of age were also increased, this time by 25% in the lumbar spinal cord, 80% in the cervical spinal cord, and 20% in the brain stem (Fig. 3A).
Figure 2.

Elevated levels of 3-nitrotyrosine early and throughout disease progression in transgenic mice expressing G37R SOD1 (line 42). Ratios of 3-nitrotyrosine to tyrosine were measured in cervical and lumbar spinal cord and brain stem of control mice, transgenic mice overexpressing wild-type human SOD1 (hSOD76), and transgenic mice expressing human mutant G37R SOD1 (line 42). (A) At 6 weeks of age [n = 12, 10, 11 (lumbar spinal cord); n = 13, 11, 11 (cervical spinal cord); n = 13, 6, 6 (brain stem)]. (B) At 20 weeks of age [n = 7, 3, 5 (lumbar spinal cord); n = 7, 3, 5 (cervical spinal cord); n = 7, 3, 5 (brainstem)]. Values are expressed as ratios (3-nitrotyrosine/tyrosine) × 1000.
Figure 3.

Elevated levels of 3-nitrotyrosine early in disease in transgenic mice expressing G37R SOD1 (line 106). Ratio of 3-nitrotyrosine to tyrosine in cervical and lumbar spinal cord and brain stem of control mice and transgenic mice expressing human mutant G37R SOD1 (line 42). (A) At 6 weeks of age [n = 6 (lumbar and cervical spinal cord, brain stem)]. (B) At 12 weeks of age [n = 6 (lumbar and cervical spinal cord, brain stem)]. Values are expressed as ratios (3-nitrotyrosine/tyrosine) × 1000.
With age the levels of 3-nitrotyrosine in the spinal cords in both lines of SOD1 mutant mice remained elevated: for line 106, by 12 weeks of age nitrotyrosine levels increased to 300% of control in cervical spinal cord (Fig. 3B), while levels in lumbar spinal cord and brain stem remained ≈25% above control (Fig. 3B). By 20 weeks of age (Fig. 2B), when G37R (line 42) mice have developed severe hind limb paralysis and muscle wasting, levels of 3-nitrotyrosine were elevated 25% in cervical and lumbar spinal cord. Given the severe motor neuron loss at this stage of disease, the reduction of 3-nitrotyrosine levels compared with earlier in disease progression may arise from loss of the most affected neurons. In the brain stem, which shows early pathology similar to that in the spinal cord, there was a 2-fold elevation in nitrotyrosine levels at 20 weeks of age.
Levels of Protein-Bound 3-Nitrotyrosine in Spinal Cords of Transgenic Mice Expressing G37R Mutant SOD1.
In light of the elevated levels of free 3-nitrotyrosine in spinal cords of transgenic mice expressing G37R, an obvious question is whether this corresponds to elevated levels of nitrated proteins and whether some proteins are selectively targeted for nitration. To address this, immunoblots of spinal cord and sciatic nerve extracts were analyzed for protein-bound nitrotyrosine, using a monoclonal antibody previously reported to react with nitrotyrosine (28). In addition, in view of the aberrant neurofilament accumulation as prominent pathology features of both sporadic (29) and familial (30) disease mediated by SOD1 mutant A4V (31), neurofilament-enriched cytoskeletal preparations were examined in parallel.
Specificity of the anti-nitrotyrosine monoclonal antibody (1A6) was initially demonstrated by intense reactivity with a preparation of partially purified intermediate filaments derived from spinal cord primarily containing NF-L (68 kDa), NF-M (160 kDa), NF-H (200 kDa), and GFAP (48 kDa) subunits (Fig. 4A, lane 1) and nitrated in vitro with peroxynitrite. The antibody yielded very weak signals with NF-L and GFAP subunits in the unmodified preparation (Fig. 4A, lane 2), and this background reactivity was not reduced by addition of 10 mM 3-nitro-l-tyrosine (Fig. 4A, lane 3). One hundred nanograms of the nitrated sample (1/100 the amount of protein in the unmodified sample) yielded intense immunoreactivity (Fig. 4A, lane 4) that was completely blocked by simultaneous addition of 10 mM 3-nitro-l-tyrosine (Fig. 4A, lane 5), confirming ability of this antibody to bind selectively to nitrated proteins.
Figure 4.

Immunoblot analysis for protein-bound nitrotyrosine in mice expressing SOD1 mutant G37R. (A) Partially purified intermediate filaments extracted from whole spinal cord and nitrated in vitro were electrophoresed on 7.5% polyacrylamide gels. Lane 1, unmodified filament preparation stained with Coomassie blue; lanes 2 and 4, the unmodified (lane 2) and nitrated (lane 4) filament preparation immunoblotted with a monoclonal antibody (1A6) that recognizes nitrotyrosine; lanes 3 and 5, immunoblots as in lanes 2 and 4 except with antibody binding in the presence of 10 mM 3-nitro-l-tyrosine. (B) Intermediate filament-enriched, cytoskeletal extracts from control mice (lanes 1, 4, and 7), mice expressing human SOD1 (lanes 2, 4, and 8), and mice expressing G37R SOD1 (lanes 3, 6, and 9) at 6 weeks (lanes 1–3), 12 weeks (lanes 4–6), and 20 weeks (lanes 7–9) immunoblotted using rabbit polyclonal antibodies that recognized NF-L and NF-H and a monoclonal antibody that recognizes NF-M. (C and D) (Top) Coomassie blue-stained gel of total spinal cord extracts (C) and neurofilament-enriched, cytoskeletal extracts (D) from 6-, 12-, or 20-week-old control and transgenic mice overexpressing human SOD1 or G37R SOD1. (Middle) Immunoblot of the same extracts along with an in vitro nitrated neurofilament extract (lane 10) probed for protein-bound 3-nitrotyrosine. (Bottom) Immunoblot of the same spinal cord extracts probed for 3-nitrotyrosine in the presence of 10 mM 3-nitro-l-tyrosine.
Whole spinal cord extracts (Fig. 4C) or partially purified neurofilament preparations (Fig. 4D) from control mice (Fig. 4 C and D, lanes 1, 4, and 7), transgenic mice overexpressing human SOD1 (Fig. 4 C and D, lanes 2, 5, and 8), and transgenic mice expressing mutant G37R (line 42) SOD1 (Fig. 4 C and D, lanes 3, 6, and 9) at 6 weeks (Fig. 4 C and D, lanes 1–3), 12 weeks (Fig. 4 C and D, lanes 4–6), and 20 weeks (Fig. 4 C and D, lanes 7–9) were electrophoresed on 7.5% polyacrylamide gels alongside the intermediate filament preparation nitrated in vitro (Fig. 4 C and D, lane 10). Staining with Coomassie blue (Fig. 4 C and D Top), as well as immunoblotting for the major neurofilament subunits NF-L, NF-M, and NF-H (Fig. 4B) demonstrated comparable loadings of proteins from each sample. Immunoblotting with the monoclonal nitrotyrosine antibody 1A6 (Fig. 4 C and D Middle) revealed two prominent bands (only after 20-min exposures) corresponding to apparent molecular masses of 68 kDa and 48 kDa in both the total extract (Fig. 4C) and neurofilament-enriched samples (Fig. 4D). However, antibody binding to neither of these could be blocked by incubation with 10 mM 3-nitro-l-tyrosine (Fig. 4 C and D Bottom), whereas immunoreactivity with the in vitro nitrated intermediate filament preparation was completely blocked (Fig. 4 C and D Bottom, lane 10). Moreover, not only was there no increase in apparent nitration during progression of disease in G37R animals (compare lanes 3, 6, and 9 of Fig. 4 C and D), there were also no obvious differences in apparent nitration in whole extracts or neurofilament-enriched preparations from control mice (Fig. 4 C and D, lanes 1, 4, and 7), transgenic mice overexpressing human SOD1 (Fig. 4 C and D, lanes 2, 5, and 8), and transgenic mice expressing G37R SOD1 (Fig. 4 C and D, lanes 3, 6, and 9) of any age. Identical results were observed for sciatic nerve extracts (data not shown). For transgenic mice expressing human SOD1 mutant G37R these results indicate that if protein-bound nitration is mediated by this SOD1 mutant, the levels of accumulated nitration throughout disease are below the detection limits of this method and antibody.
Absence of Elevated Protein-Bound Nitrotyrosine at the End Stage of Sporadic or SOD1 Mutant-Mediated Familial ALS.
To determine whether protein-bound nitration could be detected in human ALS samples, whole spinal cord extracts or neurofilament-enriched extracts from five sporadic ALS patients, two familial ALS patients (both with SOD1 mutation A4V), and four age-matched non-ALS controls were immunoblotted as before with antibodies against nitrotyrosine (analysis of 8 of these 11 samples is shown in Fig. 5). In whole spinal cord extracts and using either monoclonal antibody 1A6 or 7A2, this revealed one nitrated polypeptide of the size predicted for NF-L and another of about 50 kDa, the size of GFAP. Binding of both antibodies was partially blocked by competition with 3-nitrotyrosine (Fig. 5A Middle). Apparent nitrotyrosine signals (Fig. 5A Top) were generally proportional to the overall polypeptide pattern as seen in Coomassie blue staining (Fig. 5A Bottom), but there was no preferential increase in either the sporadic or familial ALS samples compared with the controls, with the highest signal arising in a control sample, possibly from an unusually large amount of human serum albumin in this sample.
Figure 5.

Immunoblot analysis for protein-bound nitrotyrosine in familial and sporadic ALS patients. Soluble fractions (A) or neurofilament-enriched fractions (B) from sporadic ALS patients (lanes 1–3), control individuals (lanes 4–6), and familial ALS patients with SOD1 mutation A4V (lanes 7 and 8) were immunoblotted with nitrotyrosine antibody 1A6 (Top). (Middle) Parallel immunoblots with the same antibody but in the presence of 10 mM 3-nitro-l-tyrosine. (Bottom in A) The same whole spinal cord extracts stained with Coomassie blue. (Bottom in B) The blot of the neurofilament fractions in B Top was stripped and reprobed with an antibody to NF-L. Lanes 9, an in vitro nitrated neurofilament sample. The A4V samples in lanes 7 and 8 were from 63- and 42-year-old patients 6 or 17 hr post mortem. The SALS samples in lanes 1, 2, and 3 are from 81-, 62-, and 61-year-old patients 7, 14, and 13 hr post mortem.
Examination of the neurofilament-enriched spinal cord fractions revealed several species reactive with nitrotyrosine antibodies (Fig. 5B Top), and the reactions were at least partially blocked by competition with free nitrotyrosine (Fig. 5B Middle). Among these was NF-L, which showed modest apparent differences in nitration among the various samples. However, reprobing the immunoblot with antibodies to NF-L (Fig. 5B Bottom) demonstrated that these differences simply paralleled the NF-L content of each sample, rather than representing selective differences in NF-L nitration level. Laser densitometry of the nitration signals (along with a dilution series to provide appropriate quantitation standards), coupled with phosphorimaging to quantify the NF-L levels, confirmed this qualitative impression: NF-L nitration was not elevated in either the sporadic or the A4V-mediated familial ALS patients. Further, a polypeptide of size appropriate for GFAP was intensely reactive in some samples, but not others. Whatever the source(s) of this variation, there was no correlation with sporadic or familial ALS. Neither was there a correlation with the intensities of any of the additional polypeptides apparently reactive with the nitrotyrosine antibodies (Fig. 5B Top).
Nitrated Proteins Cannot Be Detected Immunocytochemically in Motor Neurons of Transgenic Mice Expressing G37R SOD1.
Although elevated levels of protein-bound nitrotyrosine could not be found in bulk tissue extracts from mice expressing SOD1 mutation G37R or from familial or sporadic ALS patients, it remained possible that significant local increases in nitrated proteins (e.g., a subcellular domain of motor neurons) were present, but obscured by the more abundant, unaffected tissues. To determine whether localized accumulations of nitrotyrosine were present, cross sections of lumbar and cervical spinal cord from control and G37R (line 42) transgenic mice at 8, 12, and 20 weeks of age were probed with the 3-nitrotyrosine antibody. Parallel sections were immunostained with an antibody to choline acetyltransferase (ChAT) to unambiguously mark the presence of motor neurons. Although strong ChAT immunoreactivity indicated the presence of surviving motor neurons in the ventral horn, 3-nitrotyrosine immunoreactivity was not demonstrated in motor neurons or in other cell types in the early or late stages of disease (data not shown).
Salicylate Hydroxyl Trapping Does Not Reveal a Significant Increase in Hydroxyl Radicals in Mice with Motor Neuron Disease Mediated by SOD1 Mutant G37R.
To test whether the G37R mutant SOD1 increases hydroxyl radicals by acting as a peroxidase (19), levels of hydroxyl radicals were measured in G37R transgenic mice by using a salicylate hydroxyl-trapping assay. In this method, which has previously been used to demonstrate the involvement of free radicals in excitotoxicity in vivo (32), salicylate is administered intraperitoneally and tissues are collected 60 min later. Salicylate reacts with hydroxyl radicals, resulting in the formation of 2,3- and 2,5-DHBA. Comparison of 2,3- and 2,5-DHBA levels in a cohort of 12 6-week-old G37R (line 42) mice with age-matched control littermates revealed no increase in either salicylate metabolite in upper or lower spinal cords or in brain stems (Fig. 6A Upper). Even at 12 weeks, well after the appearance of initial pathology, neither DHBA product was elevated in G37R mice, with the possible exception of a very modest increase of 2,3-DHBA in the lower spinal cord (Fig. 6A Lower). Moreover, similar analysis in G37R (line 106) mice confirmed the absence of higher levels of DHBA products compared with control mice, at both 6- and 12-week age points (Fig. 6B). Curiously, expression of wild-type human SOD1 in mice did result in significant increases in 2,5-DHBA, particularly in the upper spinal cord, and to a lesser extent there were increases in 2,3-DHBA in brain stem (Fig. 6A).
Figure 6.

Salicylate hydroxyl-trapping method reveals no increase in hydroxyl radicals in transgenic mice expressing G37R SOD1. (A) Ratios of 2,5-DHBA (Left) and 2,3-DHBA (Right) to salicylate × 1000 in cervical and lumbar spinal cord and brain stem of control mice, transgenic mice overexpressing human SOD1 [hSOD(76)], and transgenic mice expressing human mutant G37R SOD1 line 42 at 6 weeks of age [n = 7, 6, 5 (lumbar and cervical spinal cord and brain stem)] (Upper) and 12 weeks of age [n = 12, 7, 11 (lumbar spinal cord); n = 12, 6, 11 (cervical spinal cord and brain stem)] (Lower). (B) Levels of 2,5- and 2,3-DHBA relative to salicylate × 1000 in control mice and transgenic mice expressing G37R (line 106) at 6 weeks of age [n = 6 (lumbar cord, cervical cord, and brain stem)] (Upper) and 12 weeks of age [n = 6 (lumbar cord, cervical cord, and brain stem)] (Lower).
A biologically relevant target for hydroxyl radical damage is lipid peroxidation arising from free-radical-mediated damage to polyunsaturated fatty acids of membranes, whose decomposition produces aldehydes such as malondialdehyde (MDA) (33). We used the thiobarbituric acid (TBA) test, in which levels of MDA are measured after reaction with TBA (34). With one exception in which a G37R mouse yielded a 2-fold increase in MDA-TBA, in six additional G37R mice no differences were seen compared with age-matched littermate control mice. Moreover, immunocytochemistry with antibodies recognizing MDA (data not shown) could not detect MDA on cross sections of spinal cords from G37R or control mice at 8 or 20 weeks of age.
DISCUSSION
We have demonstrated here that levels of free 3-nitrotyrosine are elevated in the tissues at risk in G37R SOD1 mutant-mediated disease, beginning at the early preclinical phases and continuing throughout progression of clinical signs. This offers definitive in vivo evidence that the G37R mutant does elevate tyrosine nitration. Mechanistically, the increase in nitration seems most likely to arise from an increased efficiency of the SOD1 mutant in using peroxynitrite as a substrate, although an alternative that the G37R mutant provokes an increase in nitric oxide production (and hence in peroxynitrite) is also possible. Whichever is correct, at a minimum, this evidence establishes that in vulnerable tissues tyrosine nitration is one aberrant property of this SOD1 mutant. The overall evidence thus implicates protein nitration in initiation and spread of motor neuron disease.
As to the protein targets for nitration, neurofilaments are attractive candidates for SOD1 mutant-mediated damage in that they are both very abundant and very long lived [requiring transit times of a year or more for travel from the cell body to the synapses in the sciatic nerve (35)]. Not only are aberrant accumulations of neurofilaments in both perikarya and axons prominent pathologic features of many instances of sporadic (29, 36, 37) and SOD1 mutant-mediated familial (30, 38) ALS, increases in filament number or organization by expressing high levels of wild-type mouse NF-L (39) or human NF-H (40, 41) in transgenic mice are direct causes of motor neuronopathies. Even more ALS-like disease characterized by progressive degeneration and death of motor neurons with large caliber (neurofilament-rich) axons can be caused by expression of a mutant NF-L subunit to levels appropriate for a dominantly inherited disease (42). While no primary mutation in NF-L, NF-M, or NF-H genes has been found in examination of more than 100 familial ALS patients known not to have mutations in SOD1 (43), these findings strongly support the view that SOD1-mutant-mediated damage to neurofilament subunits could lead to disorganized filaments that would preferentially affect the largest-caliber motor and sensory axons known to be at risk in human ALS (44). Indeed, the major neurofilament subunit, NF-L, contains 20 tyrosines (45), including 9 in the 96-amino acid head domain required for filament assembly (46). However, immunoblotting of either whole extracts or cytoskeletal fractions strongly enriched in neurofilaments did not reveal measurable levels of nitrotyrosine on neurofilament subunits, indicating that in the SOD1-mutant-expressing mice, neurofilaments are not targets for damage via tyrosine nitration, or levels of nitration are below limits detectable using the available antibodies.
Because we can detect signals from as little as 2 ng of our nitrated standards and assuming that in vitro nitration using saturating amounts of peroxynitrite yielded modification of 50% of available tyrosines on our NF-L standard (20 mol of tyrosine per 68 kDa of protein), our sensitivity for detecting nitration on a 68-kDa protein of tyrosine composition similar to that of NF-L would require an abundance of 0.16% at an efficiency of nitration of 3% of available tyrosines (the percentage of nitrotyrosine relative to tyrosine in affected tissues). The failure to identify even abundant proteins, such as the neurofilaments or GFAP, as nitration products in G37R-mediated disease, especially in the neurofilament-enriched cytoskeletal fraction, strongly suggests that the stoichiometry of nitration must be well below 1%. Of course, it should be obvious that if one or more rare proteins were selectively targeted for nitration, these could easily be below our detection limit. In any event, what is clear is that despite chronic, significant increases in tyrosine nitration detected biochemically, at no point in disease does the level of protein-bound nitrotyrosine ever rise to the level necessary for detection by a bulk measure, nor is there a sufficiently local increase that we could detect it with immunocytochemistry. While the absence of evidence for protein-bound nitrotyrosine could imply that this is not the, or even an, aberrant in vivo property responsible for disease mediated by this SOD1 mutant, the fact that increased tyrosine nitration is found in affected tissues offers more persuasive evidence that nitration is at least one aberrant property relevant to G37R toxicity. The absence of elevated bound nitrotyrosine in the sporadic and SOD1 mutant A4V-mediated familial ALS human spinal cords, as compared with control spinal cords, demonstrates that bulk nitration is probably not involved in human disease, although localized increases cannot be discounted, as two previous reports have claimed modest elevations in nitrotyrosine immunoreactivity in Betz cells or anterior horn cells of some ALS patients (47, 48). It must be noted, however, that the absence of analysis of SOD1-mutant-mediated samples in the first of these reports (47) coupled with the absence of non-ALS controls or evidence of specificity of staining in the second (48), limits the compellingness of the immunocytochemical evidence.
Finally, although in vitro at least two mutations (A4V and G93A) can increase the rate of hydroxyl radical formation (19), probably due to the increased availability of the active Cu2+ to H2O2, examination in disease mediated by G37R for increase in hydroxyl radicals using salicylate trapping or search for the expected peroxidation product malondialdehyde yielded no in vivo support for H2O2 as an aberrant in vivo substrate for this SOD1 mutant. In contrast, increases in wild-type human SOD1, but not the G37R mutant, yielded reproducible increases in the products of hydroxyl radical action on salicylate (Fig. 6A). This would be consistent with increased SOD1 activity elevating the rate of H2O2 production and with the wild-type SOD1, but not G37R, promoting hydroxyl radical formation through the use of H2O2 as a substrate.
Acknowledgments
We give heartfelt thanks to Dr. Joe Beckman for providing antibodies to nitrotyrosine, to Dr. Michael Strong for generously sharing with us his unpublished immunoblotting protocol for identifying protein nitration, and to Dr. Jeff Rothstein for the sporadic ALS samples. We thank Scott Anderson for maintaining the mouse colony. This work has been supported by grants from the National Institutes of Health (NS 27036) and the ALS Association to D.W.C.; L.I.B is supported by a postdoctoral fellowship from the Muscular Dystrophy Association. Salary support for D.W.C. is provided by the Ludwig Institute for Cancer Research.
ABBREVIATIONS
- ALS
amyotrophic lateral sclerosis
- SOD1
superoxide dismutase 1
- GFAP
glial fibrillary acidic protein
- NF-L
NF-M, and NF-H subunits, neurofilament light, medium, and heavy subunits
- DHBA
dihydroxybenzoic acid
References
- 1.Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan J P, Deng H X, Brown R. Nature (London) 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Brown R H., Jr Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 3.Radunovic A, Leigh P N. Neurol Neurosurg Psych. 1996;61:565–567. doi: 10.1136/jnnp.61.6.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddique T, Nijhawan D, Hentati A. Neurology. 1996;47:S27–S34. doi: 10.1212/wnl.47.4_suppl_2.27s. ; discussion S34–S35. [DOI] [PubMed] [Google Scholar]
- 5.Cudkowicz M E, McKenna-Yasek D, Sapp P E, Chin W, Geller B, Hayden D L, Schoenfeld D A, Hosler B A, Horvitz H R, Brown R H. Ann Neurol. 1997;41:210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- 6.Banker B Q. In: Myology. Engle A G, Banker B Q, editors. New York: McGraw Hill; 1986. , Part II, pp. 2031–2066. [Google Scholar]
- 7.Fridovich I. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 8.Bowling A C, Schulz J B, Brown R H, Jr, Beal M F. J Neurochem. 1993;61:2322–2325. doi: 10.1111/j.1471-4159.1993.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 9.Borchelt D R, Lee M K, Slunt H S, Guarnieri M, Xu Z S, Wong P C, Brown R H, Jr, Price D L, Sisodia S S, Cleveland D W. Proc Natl Acad Sci USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurney M E, Pu H, Chiu A Y, Dal Canto M C, Polchow C Y, Alexander D D, Caliendo J, Hentati A, Kwon Y W, Deng H X. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 11.Wong P C, Pardo C A, Borchelt D R, Lee M K, Copeland N G, Jenkins N A, Sisodia S S, Cleveland D W, Price D L. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 12.Bruijn L I, Becher M W, Lee M K, Anderson K L, Jenkins N A, Copeland N G, Sisodia S S, Rothstein J D, Borchelt D R, Price D L, Cleveland D W. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 13.Ripps M E, Huntley G W, Hof P R, Morrison J H, Gordon J W. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reaume A B, Elliott J L, Hoffman E K, Kowall N W, Ferrante R J, Siwek D F, Wilcox H M, Flood D G, Beal M F, Brown R H, Scott R W, Snider W D. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 15.Beckman J S, Carson M, Smith C D, Koppenol W H. Nature (London) 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 16.Beckman J S. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 17.Verge V M K, Xu Z, Xu X, Wiesenfeld-Hallin Z, Hökfelt T. Proc Natl Acad Sci USA. 1992;89:11617–11621. doi: 10.1073/pnas.89.23.11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Li Y, Liu H, Wu W. Neurosci Lett. 1995;194:109–112. doi: 10.1016/0304-3940(95)11741-e. [DOI] [PubMed] [Google Scholar]
- 19.Wiedau-Pazos M, Goto J J, Rabizadeh S, Gralla E D, Roe J A, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 20.Pogun S, Dawson V, Kuhar M J. Synapse. 1994;18:21–26. doi: 10.1002/syn.890180104. [DOI] [PubMed] [Google Scholar]
- 21.Beal M F, Matson W R, Swartz K J, Gamache P H, Bird E D. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 22.Lopata M A, Cleveland D W. J Cell Biol. 1987;105:1707–1720. doi: 10.1083/jcb.105.4.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z, Cork L C, Griffin J W, Cleveland D W. J Cell Sci Suppl. 1993;17:101–108. doi: 10.1242/jcs.1993.supplement_17.15. [DOI] [PubMed] [Google Scholar]
- 24.Beckman J S, Beckman T W, Chen J, Marshall P A, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao W, Carney J M, Duchon A, Floyd R A, Chevion M. Neurosci Lett. 1988;88:233–238. doi: 10.1016/0304-3940(88)90132-2. [DOI] [PubMed] [Google Scholar]
- 26.Floyd R A, Watson J J, Wong P K. J Biochem Biophys Methods. 1984;10:221–225. doi: 10.1016/0165-022x(84)90042-3. [DOI] [PubMed] [Google Scholar]
- 27.Hall E D, Andrus P K, Yonkers P A. J Neurochem. 1993;60:588–594. doi: 10.1111/j.1471-4159.1993.tb03189.x. [DOI] [PubMed] [Google Scholar]
- 28.Beckman J S, Ye Y Z, Anderson P G, Chen J, Accavotti M A, Tarpey M M, White C R. Biol Chem Hoppe-Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 29.Hirano A, Donnenfeld H, Sasaki S, Nakano I. J Neuropathol Exp Neurol. 1984;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Hirano A, Nakano I, Kurland L T, Mulder D W, Holley P W, Saccomanno G. J Neuropathol Exp Neurol. 1984;43:471–480. doi: 10.1097/00005072-198409000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Shibata N, Hirano A, Kobayashi M, Siddique T, Deng H-X, Hung W-Y, Kato T, Asayama K. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Schulz J B, Henshaw D R, Siwek D, Jenkins B G, Ferrante R J, Cipolloni P B, Kowall N W, Rosen B R, Beal M F. J Neurochem. 1995;64:2239–2247. doi: 10.1046/j.1471-4159.1995.64052239.x. [DOI] [PubMed] [Google Scholar]
- 33.Janero D R. Free Radical Biol Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- 34.Halliwell B, Kaur H, Ingleman-Sundberg M. Free Radical Biol Med. 1991;10:439–441. doi: 10.1016/0891-5849(91)90052-5. [DOI] [PubMed] [Google Scholar]
- 35.Cleveland D W. Cell. 1996;84:663–666. doi: 10.1016/s0092-8674(00)81044-2. [DOI] [PubMed] [Google Scholar]
- 36.Carpenter S. Neurology. 1968;18:841–851. doi: 10.1212/wnl.18.9.841. [DOI] [PubMed] [Google Scholar]
- 37.Hirano A. In: Cytopathology of Amyotrophic Lateral Sclerosis. Rowland L P, editor. Vol. 56. New York: Raven; 1991. pp. 91–101. [PubMed] [Google Scholar]
- 38.Rouleau G A, Clark A W, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien J-P, Figlewicz D. Ann Neurol. 1996;39:128–131. doi: 10.1002/ana.410390119. [DOI] [PubMed] [Google Scholar]
- 39.Xu Z, Cork L C, Griffin J W, Cleveland D W. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 40.Cote F, Collard J F, Julien J P. Cell. 1993;73:35–46. doi: 10.1016/0092-8674(93)90158-m. [DOI] [PubMed] [Google Scholar]
- 41.Collard J F, Cote F, Julien J P. Nature (London) 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 42.Lee M K, Marszalek J R, Cleveland D W. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 43.Vechio J V, Bruijn L I, Xu Z-S, Brown R H, Jr, Cleveland D W. Ann Neurol. 1996;40:603–610. doi: 10.1002/ana.410400410. [DOI] [PubMed] [Google Scholar]
- 44.Kawamura Y, Dyck P J, Shimono M, Okazaki H, Tateishi J, Doi H. J Neuropathol Exp Neurol. 1981;40:667–675. doi: 10.1097/00005072-198111000-00008. [DOI] [PubMed] [Google Scholar]
- 45.Julien J P, Grosveld F, Yazdanbaksh K, Flavell D, Meijer D, Mushynski W. Biochim Biophys Acta. 1987;909:10–20. doi: 10.1016/0167-4781(87)90041-8. [DOI] [PubMed] [Google Scholar]
- 46.Gill S R, Wong P C, Monteiro M J, Cleveland D W. J Cell Biol. 1990;111:2005–2019. doi: 10.1083/jcb.111.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abe K, Pan L, Watanabe M, Kato T, Itoyama Y. Neurosci Lett. 1995;199:152–154. doi: 10.1016/0304-3940(95)12039-7. [DOI] [PubMed] [Google Scholar]
- 48.Chou S M, Wang H S, Komai K. J Neuroanat. 1996;10:249–258. doi: 10.1016/0891-0618(96)00137-8. [DOI] [PubMed] [Google Scholar]