

Abstract
Both lipoprotein-associated phospholipase A2 (Lp-PLA2) activity, a biomarker of inflammation, and concentration of its primary associated lipoprotein, LDL, are correlated with adverse coronary outcomes. We previously reported a quantitative trait locus (QTL) corresponding to HSA2p24.3–p23.2 with pleiotropic effects on Lp-PLA2 activity and LDL-cholesterol (LDL-C) concentration in baboons fed a basal diet. Here, our goal was to locate pleiotropic QTLs influencing both traits in the same baboons fed a high-cholesterol, high-fat (HCHF) diet, and to assess whether shared genetic effects on these traits differ between diets. We assayed Lp-PLA2 activity and LDL-C concentration in 683 baboons fed the HCHF diet. We used a bivariate maximum likelihood-based variance components approach in whole-genome linkage screens to locate a QTL [logarithm of odds (LOD) = 3.13, genome-wide P = 0.019] corresponding to HSA19q12–q13.2 with pleiotropic effects on Lp-PLA2 activity and LDL-C levels in the HCHF diet. We additionally found significant evidence of genetic variance in response to diet for Lp-PLA2 activity (P = 0.0017) and for LDL-C concentration (P = 0.00001), revealing a contribution of genotype-by-diet interaction to covariation in these two traits. We conclude that the pleiotropic QTLs detected at 2p24.3–p23.2 and 19q12–q13.2 on the basal and HCHF diets, respectively, exert diet-specific effects on covariation in Lp-PLA2 activity and LDL-C concentration.
Keywords: lipoprotein-associated phospholipase A2, genotype-by-diet interaction, atherosclerosis, pleiotropy, baboon
Both lipoprotein-associated phospholipase A2 (Lp-PLA2) activity, a biomarker of inflammation, and LDL-cholesterol (LDL-C) concentration are associated with early onset atherosclerosis, endothelial dysfunction, and future cardiac events (1, 2). Additionally, Lp-PLA2 activity and LDL-C concentration are positively correlated (3, 4), an observation consistent with the knowledge that the major portion of Lp-PLA2 in the circulation is bound to LDL. The positive relationship between Lp-PLA2 activity and LDL-C concentration and their association with cardiovascular disease probably reflects the enzymatic action of Lp-PLA2 on LDL particles, which produces end products with pro-inflammatory activity that may account for many of the atherogenic properties of oxidized LDL (5, 6).
Although studies addressing the effect of genes on Lp-PLA2 activity and LDL-C concentration are limited, evidence supports a genetic contribution to covariation (i.e., pleiotropy) in both traits. Polymorphism in PLA2G7 (coding for Lp-PLA2) has substantial effects on variation in LDL-C concentration in healthy controls, and on Lp-PLA2 activity in healthy controls and patients with cardiovascular disease (CVD) (7, 8). Additionally, we recently reported a genetic correlation between Lp-PLA2 activity and LDL-C concentration in baboons fed a basal diet, and presented evidence for a quantitative trait locus (QTL) on chromosome 2p that influences covariation in these traits (9).
Because Lp-PLA2 activity is both genetically correlated and physically associated with LDL, and both traits predict risk of CVD, knowledge of genes with pleiotropic effects on the response of these traits to an atherogenic [i.e., high-cholesterol, high-fat (HCHF)] diet would be of considerable value. However, to our knowledge, no studies have searched for genes regulating the response of Lp-PLA2 activity to dietary challenge. In contrast to Lp-PLA2, substantial research has addressed the location of genes that influence the response of LDL-C concentration to diets enriched in cholesterol and fat, in both animal models and humans (10, as reviewed in Refs. 11–13). To date, the majority of evidence for genes that influence the response of LDL-C levels to dietary fat and cholesterol supports the involvement of polymorphisms in LDLR, which codes for the LDL receptor, or in APOE and the APOAI-APOCIII-APOAIV gene cluster, which code for lipid transport proteins. Despite these important advances, we still have not identified many genes that regulate the response of LDL and associated Lp-PLA2 activity to an HCHF diet.
The goal of the current study was to locate QTLs influencing covariation in Lp-PLA2 activity and LDL-C concentration in an HCHF diet, and to determine whether genetic effects on these traits differ between basal and HCHF diets (genotype-by-diet interaction) in pedigreed baboons, a valuable model for studying the genetics of risk factors for human atherosclerosis.
MATERIALS AND METHODS
Animals and dietary protocol
We obtained data for this study from a sample of 683 pedigreed baboons (Papio hamadryas) comprising 442 females and 241 males maintained outdoors in social groups at the Southwest National Primate Research Center (SNPRC), located at the Southwest Foundation for Biomedical Research (SFBR) in San Antonio, Texas. The age of baboons in this sample ranged from 2–29 years, corresponding approximately to a human developmental age range of 6–87 years. Animal care personnel and staff veterinarians provided routine and emergency health care to all animals in accordance with the Guide for the Care and Use of Laboratory Animals. The SFBR facility is certified by the Association for Assessment and Accreditation of Laboratory Animal Care International, and all procedures were approved by the Institutional Animal Care and Use Committee.
Baboons sampled in this study were maintained on a commercial monkey diet (basal diet, SWF Primate Diet; Harlan Teklad, Madison, WI), composed of 0.02 mg/g cholesterol and 7% fat from plant oils. The same baboons were then fed an HCHF diet for 7 weeks, composed of 6.37 mg/g cholesterol and 41% fat from lard [a detailed description of both basal and HCHF diets is given in (14)]. Animals had ad libitum access to either basal or HCHF diets, and blood samples were drawn from all baboons before and after the 7-week dietary challenge period.
For the purposes of this study, these baboons were organized into 11 distinct pedigrees, yielding a diverse array of relative pair classes: 650 parent–offspring; 577 sibling; 66 grandparent–grandchild; 81 avuncular; 6,270 half-sibling; 1,668 half-avuncular; 3 first cousin; 40 half-first cousin; 5 half-first cousin, once removed; 21 half-sibling and first cousin; 520 half-sibling and half-first cousin; 7 half-sibling and half-avuncular; and 31 double half-avuncular. These 11 baboon pedigrees may be viewed at the SNPRC website: http://www.snprc.org/baboon/map/BPPpedigrees/bpppeds.htm.
Phenotyping
Lp-PLA2 activity and LDL-C concentration were assayed in serum samples, obtained as part of an ongoing study of the effects of diet and genotype on variation in atherosclerosis risk factors. Blood samples were taken from the femoral vein of overnight-fasted baboons sedated with ketamine. Serum was separated from whole blood by low-speed centrifugation and stored in individual, single-use aliquots at −80°C, protected from oxidation and desiccation (15).
Serum Lp-PLA2 enzyme activity was measured at 30°C using a kit provided by Cayman Chemical Co. (Ann Arbor, MI). Hydrolysis of the substrate, 2-thio platelet-activating factor, produced a free thiol that was quantified using 5,5′-dithio-bis-(2-nitrobenzoic acid). The reaction was monitored at 405 nm using a BioTek (Winooski, VT) ELx808 microplate reader running in kinetic data acquisition mode. Rates were calculated from at least 15 min of readings in the linear phase and converted to nmol/min/ml plasma using an extinction coefficient value of 13.6/mM-cm. The between-assay coefficient of variation, based on a control sample run on each plate, was 4.3%.
Cholesterol concentrations were measured enzymatically (16) with a reagent supplied by Boehringer Mannheim Diagnostics (Indianapolis, IN) and using a Ciba-Corning (Norwood, MA) Express Plus clinical chemistry analyzer. Cholesterol carried by HDL was estimated following precipitation of apolipoprotein B (apoB)-containing lipoproteins with heparin-Mn2+ as described (17), and LDL-C was calculated as the difference between total and HDL-C [in baboons, the major proportion of apoB-associated cholesterol is found on particles in the LDL size/density range (18)]. Between-assay coefficients of variation for these determinations were 2.2% and 4.6% for total and HDL-C, respectively.
Baboon genotyping and whole-genome linkage map
Statistical genetic analyses of these two traits took advantage of a baboon whole-genome linkage map based on genotype data at nearly 300 microsatellite marker loci (mean intermarker interval = 8.9 cM) from 984 pedigreed baboons in these same 11 extended pedigrees. The physical locations in the human genome for nearly all marker loci in the baboon map are known, thus facilitating the identification of orthologous chromosomal regions in the two species. Construction of the current baboon linkage map is described in detail elsewhere (19), and additional information can be found at the SNPRC website: http://www.snprc.org/baboon/genome/index.html.
Statistical genetic methods
All statistical genetic analyses were conducted using a maximum likelihood-based variance decomposition approach implemented in the computer package SOLAR (Sequential Oligogenic Linkage Analysis Routines) (20). We used this approach to partition the phenotypic variance in each trait (σP2) into components corresponding to additive genetic effects (σG2), estimated as a function of relatedness among pedigreed baboons, and environmental effects (σE2). We define heritability (h2) as the proportion of residual phenotypic variance unexplained by covariates that can be attributed to additive genetic effects (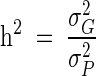 ).
).
Accounting for random and measured environmental contributions to the phenotypic variance can improve power to detect genetic effects. After regressing out nominally significant mean effects of age, sex, age2, age×sex, and age2×sex, we applied an inverse Gaussian transformation to the residuals to correct for departures from multivariate normality that might inflate evidence for linkage (21). This transformation results in standardized traits with means and standard deviations approaching 0 and 1, respectively. All polygenic and linkage analyses were conducted using these normalized residual data.
The bivariate polygenic model
To determine the extent to which phenotypic variation in serum Lp-PLA2 activity and LDL-C concentration may be affected by shared genes and shared nongenetic factors, we conducted bivariate analyses in which both traits are considered simultaneously. In addition to the heritability for each trait, the bivariate polygenic model also estimates the additive genetic and environmental correlations between both traits. The genetic correlation (ρG) is an estimate of pleiotropy between the two traits, and ρG2 thus estimates the portion of the additive genetic variance in each trait due to shared genetic effects. The random environmental correlation (ρE) is an estimate of the shared effects of nonadditive genetic factors and unmeasured environmental variables. Using these estimates, we calculated the phenotypic correlation between the trait pair as 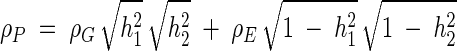 (22). We assessed the significance of ρG and ρE by means of likelihood ratio tests comparing the likelihoods of models in which the correlation was estimated to those in which it was constrained to zero (rejection of ρG = 0 indicates pleiotropy) or to 1 (failure to reject ∣ρG∣ = 1 indicates complete pleiotropy).
(22). We assessed the significance of ρG and ρE by means of likelihood ratio tests comparing the likelihoods of models in which the correlation was estimated to those in which it was constrained to zero (rejection of ρG = 0 indicates pleiotropy) or to 1 (failure to reject ∣ρG∣ = 1 indicates complete pleiotropy).
The bivariate linkage model
To search for pleiotropic QTLs affecting phenotypic variation in both Lp-PLA2 activity and LDL-C concentration, we conducted bivariate multipoint linkage analyses using an extension to the bivariate polygenic model in which we modeled the phenotypic covariance among relatives as the sum, for each trait, of the additive genetic covariance attributable to a specified marker locus, the additive genetic covariance due to the effects of other loci, and the covariance due to unmeasured environmental factors. This extension allows estimation of the proportion of the residual variance in each of i traits that is attributable to the effects of a QTL as hQi2 = σQi2/σPi2, where σQi2 and σPi2, respectively, are the QTL-specific additive genetic variance and the residual phenotypic variance for trait i. The hypothesis of linkage is supported when σQi2 is significantly greater than zero (20). In addition to the same parameters as are estimated in the bivariate polygenic model, the additive genetic correlation between the traits due to the effects of the QTL (ρQ) is also estimated in the bivariate linkage model.
Our linkage analyses incorporate identity-by-descent (IBD) allele sharing estimated from genotype data at the microsatellite markers in the baboon linkage map. For marker-locus specific linkage analyses (e.g., markers within candidate genes), we estimated IBD probabilities for the pedigrees using a pair-wise maximum likelihood-based procedure (23). To facilitate our multipoint, whole-genome QTL searches we estimated multipoint probabilities of IBD among relatives throughout the baboon linkage map using Markov Chain Monte Carlo routines implemented in the computer package Loki (24). We tested linkage hypotheses at 1 cM intervals along each chromosome using likelihood ratio tests, and converted the resulting likelihood ratio statistic to the logarithm of odds (LOD) score of classic linkage analysis (25). Bivariate LOD scores are adjusted to be equivalent to univariate LOD scores in terms of degrees of freedom (26).
To control for the genome-wide false-positive rate, we calculated genome-wide P values for each LOD score using a modification of a method suggested by Feingold, Brown, and Siegmund (27) that takes into account pedigree complexity and the finite marker density of the linkage map. Accordingly, our threshold for significant evidence of linkage (corresponding to genome-wide α = 0.05) was LOD = 2.69, and that for suggestive evidence of linkage was LOD = 1.46.
Co-incident linkage versus pleiotropy at the QTL
In a bivariate linkage analysis, QTLs may be found that appear to influence phenotypic variation in both traits. To distinguish the event where two traits may each be independently influenced by closely linked genes (“co-incident linkage”) from QTL pleiotropy, we conducted a likelihood ratio test of the hypothesis ρQ = 0. In accordance with Almasy, Dyer, and Blangero (28), failure to reject this hypothesis supports the coincident linkage of two QTLs over pleiotropic effects at the same locus.
Detecting genotype-by-diet interaction
Genotype-by-diet interaction occurs when there is a significant additive genetic component to the variance in response to dietary environment (29). The additive genetic variance in a phenotypic response to diet is a function of both the trait's additive genetic variance in the two diets and the additive genetic correlation between measures of that trait in the two diets. Accordingly, to assess the contribution of genotype-by-diet interaction to Lp-PLA2 activity and LDL-C concentration, we tested the null hypotheses of equal additive genetic variance (i.e., σG12 = σG22) and complete additive genetic correlation (i.e., ρG = 1) for each trait between the two diets (30). The rejection of either null hypothesis is indicative of genotype-by-diet interaction.
To test the null hypothesis of equal additive genetic variance between the basal and the HCHF diets for each trait, we employed a Wald test, using estimates of the additive genetic variances computed directly as the product of the heritability and the phenotypic variance (i.e., the square of the phenotypic standard deviation), and using standard formulae to calculate exact sampling variances and covariances of parameter estimates for the additive genetic variances (31–33). To test the null hypothesis of complete additive genetic correlation between the two diets for each trait, we performed tests of significant deviation from ρG = 1 between the basal and the HCHF diets using likelihood ratio tests wherein the likelihood of the null hypothesis constraining the additive genetic correlation to 1 (i.e., complete association) is compared with the likelihood of the estimated additive genetic correlation.
RESULTS
Sample sizes and distribution of Lp-PLA2 activity and LDL-C concentration measured in basal and HCHF diets
Sample sizes and summary statistics describing the distribution of raw data for Lp-PLA2 activity and LDL-C concentration measured in both the basal and HCHF diets are provided in Table 1. All summary statistics for Lp-PLA2 activity and LDL-C concentration increased from the basal to the HCHF diet. Mean Lp-PLA2 activity increased ∼26%, whereas mean LDL-C concentration increased 101% from the basal to the HCHF diet. As a proportion of the mean, the variance increased ∼127% and ∼156% from the basal to the HCHF diet, for Lp-PLA2 activity and LDL-C concentration, respectively. These initial findings suggested a possible contribution of genotype-by-diet interaction to both traits, hypotheses that were addressed formally in analyses described below.
TABLE 1.
Sample sizes and distributions of Lp-PLA2 activity (nmol/min/ml) and LDL cholesterol concentration (mmol/L) within and between basal and HCHF diets
| Basal Diet
|
HCHF Diet
|
|||
|---|---|---|---|---|
| Lp-PLA2 | LDL-C | Lp-PLA2 | LDL-C | |
| N | 657 | 679 | 660 | 683 |
| Mean | 6.09 | 1.07 | 7.66 | 2.15 |
| Median | 5.88 | 1.03 | 7.19 | 2.02 |
| Variance | 2.03 | 0.19 | 5.75 | 0.99 |
| Variance/Mean | 0.33 | 0.18 | 0.75 | 0.46 |
| Min value | 2.82 | 0.00 | 2.84 | 0.27 |
| Max value | 11.67 | 3.13 | 21.29 | 6.31 |
HCHF, high-cholesterol, high-fat; LDL-C, LDL-cholesterol; Lp-PLA2, lipoprotein-associated phospholipase A2.
Bivariate analyses of Lp-PLA2 activity and LDL-C concentration
Because both Lp-PLA2 activity and LDL-C concentration showed evidence of pleiotropy in baboons on a basal diet, we conducted bivariate analyses to characterize shared genetic effects in baboons on an HCHF diet. The results of the bivariate polygenic analysis for these traits are summarized in Table 2. Heritability estimates indicate that substantial additive genetic effects contribute to the residual phenotypic variance in both Lp-PLA2 activity and LDL-C levels. The estimated additive genetic correlation between Lp-PLA2 activity and LDL-C concentration indicates that both traits are influenced by substantial shared additive genetic effects, and that almost half of the additive genetic variance in each of the two traits is due to the effects of the same gene or genes. The genetic effects common to both traits account for approximately 30% and 29% of the residual phenotypic variance in Lp-PLA2 activity and LDL-C concentration, respectively. The magnitude of these shared genetic effects accounted for much of the residual phenotypic variance shared between Lp-PLA2 activity and LDL-C concentration measured in a high-fat diet (i.e., ρP2 = 0.43). However, genetic effects on phenotypic variation that are not shared by both traits also exist, because the hypothesis of complete pleiotropy between these two traits, ρG = 1, was also rejected.
TABLE 2.
Bivariate polygenic analysis of variation in Lp-PLA2 activity and LDL-C concentration in pedigreed baboons on HCHF diet
| Parameter | Lp-PLA2 | LDL-C | |
|---|---|---|---|
| h2 | 0.65 (0.07)a | 0.61 (0.07)a | |
| ρG | 0.69 (0.06) | ||
| P[ρG = 0] | 1.05 × 10−12 | ||
| P[ρG = 1] | 3.86 × 10−18 | ||
| ρG2 | 0.47 | ||
| ρE | 0.60 (0.07) | ||
| ρP | 0.65 |
h2, heritability. Maximum likelihood parameter estimates. Parentheses enclose SEM.
P << 0.0000001.
Whole-genome bivariate multipoint linkage analysis of Lp-PLA2 activity and LDL-C concentration (summarized in Table 3) found significant evidence for a pleiotropic QTL on the baboon (Papio hamadryas) equivalent of human chromosome 19 (PHA19) (plotted in Fig. 1). With very similar QTL-specific heritability estimates (i.e., 0.19 and 0.21), this pleiotropic QTL accounts for approximately 20% of the residual phenotypic variance in Lp-PLA2 activity and LDL-C levels. The estimated additive genetic correlation between Lp-PLA2 activity and LDL-C concentration at the QTL suggests that all additive genetic effects at this QTL are shared between the two traits (i.e., the QTL exerts complete pleiotropy on both traits).
TABLE 3.
A pleiotropic QTL on baboon chromosome 19 (PHA19) influencing variation in Lp-PLA2 activity and LDL-C concentration in pedigreed baboons on HCHF diet
| Lp-PLA2, LDL-C | |
|---|---|
| LOD | 3.13 |
| Genome-wide P value | 0.019 |
| Baboon chromosome (PHA) | 19 |
| Location in baboon linkage map (cM from pter-most marker locus) | 40 |
| QTL-specific heritability (Lp-PLA2, LDL-C) | 0.19, 0.21 |
| ρQ | 0.99 |
| P[ρQ = 0] | 0.0000462 |
| P[ρQ = 1] | 0.44 |
| Orthologous human chromosome (HSA) | 19 |
| Marker loci used to interpolate support interval for QTL | D19S226, DM |
| Region in humans corresponding to QTL support interval | 19q12–q13.2 |
| Region in humans corresponding to maximum LOD in baboons | 19q12–q13.12 |
LOD, logarithm of odds; QTL, quantitative trait locus. Summary of bivariate linkage analysis results.
Fig. 1.

Bivariate multipoint linkage results [logarithm of odds (LOD) = 3.13; genome-wide P = 0.019] for the combined phenotype lipoprotein-associated phospholipase A2 activity/LDL-cholesterol concentration on PHA19, the baboon ortholog of human chromosome 19.
We defined a comprehensive support interval surrounding the QTL as the interval bounded by the locations on the baboon genetic map at which the LOD score was one LOD unit lower than the peak LOD score (“1-LOD drop” method) (34). We then used the markers flanking this support interval to ascertain the corresponding region of interest in the human genome, assuming a proportional relationship between genetic and physical distance between these markers. Thus, the region likely to harbor the QTL in baboons corresponds to an approximately 21 Mb interval in the human genome mapping to 19q12–q13.2 and containing 317 known RefSeq genes. A narrower, 10 Mb interval (19q12–q13.12) centered on the similarly interpolated location of the maximum LOD score harbors 142 RefSeq genes, including the lipolysis-stimulated protein receptor (LSR/LISCH7), three free-fatty acid receptors (GPR40/FFAR1, GPR41/FFAR3, and GPR43/FFAR2), and the LDL receptor-related protein 3 (LRP3).
Tests of genotype-by-diet interaction for Lp-PLA2 activity and LDL-C concentration
To evaluate whether genes mediate the response of Lp-PLA2 activity and LDL-C concentration to an HCHF diet, we tested for the presence of additive genetic variance in response to dietary environment for both traits. For each trait, we tested the null hypotheses σG(BASAL)2 = σG(HCHF)2 and ρG(BASAL,HCHF) = 1 (results summarized in Table 4). The estimate of ρG between the basal and HCHF diet for each trait indicates that most, but not all, total additive genetic effects were shared between both diets. Equality of additive genetic variances estimated in both the basal and the HCHF diet for each trait could not be rejected; however, tests of the null hypothesis of ρG = 1 between the basal and the HCHF diet for both Lp-PLA2 activity and LDL-C concentration did reject complete sharing of the additive genetic variance between diets for each of these two traits, thus demonstrating genotype-by-diet interaction for both traits.
TABLE 4.
Shared genetic effects between basal and HCHF diets and evidence for genotype-by-diet interaction affecting Lp-PLA2 activity and LDL-C concentration in pedigreed baboons
| Lp-PLA2 | LDL-C | |
|---|---|---|
| ρG (between basal and HCHF diets) | 0.94 (0.03) | 0.86 (0.05) |
| ρG2 (between basal and HCHF diets) | 0.89 | 0.73 |
| P[ρG = 1] | 0.0017 | 0.00001 |
| P[ρG = 0] | 2.31 × 10−25 | 4.42 × 10−19 |
| P[σG(BASAL)2 = σG(HCHF)2 | NS | NS |
Maximum likelihood parameter estimates. Parentheses enclose SEM.
Posterior analyses of effects of LDLR and APOE on variation in Lp-PLA2 activity and LDL-C concentration
Although they lie outside the support interval for this QTL, because substantial research has identified their effects on variation in circulating LDL-C concentration and on subsequent CVD risk, we conducted two additional series of analyses to assess the possible contributions of the genes encoding the LDL receptor (LDLR) and apo-E (APOE) to variation in Lp-PLA2 activity and LDL-C concentration. First, we performed measured genotype analyses (35) using restriction fragment-length polymorphism (RFLP) data in LDLR and APOE that were available for a subset of these same animals (n ≈ 400) (36, 37). In these analyses, we included additive and dominance effects of an AvaII polymorphism in LDLR and a BanII polymorphism in APOE in our genetic models for each of the two traits. These analyses detected significant additive genetic effects of LDLR on variation in both Lp-PLA2 activity (P = 0.0006, N = 390) and LDL-C concentration (P = 0.0020, N = 405). However, the estimated effect of LDLR on genetic variance in both traits was small, accounting for 6% or less of the additive genetic variance in each trait. Our measured genotype analyses detected no significant evidence for an effect of APOE on additive genetic variance in Lp-PLA2 activity or LDL-C concentration. Second, using marker-locus-specific estimates of IBD (based on genotype data at these same RFLP sites) for all animals with phenotype data, we conducted bivariate linkage analyses to assess whether either of these two genes contributed to the effects of a pleiotropic QTL. Our analyses yielded no significant evidence for linkage between either LDLR or APOE and a QTL influencing variation in the Lp-PLA2\LDL-C bivariate phenotype (LOD ≤ 0.19, P ≥ 0.35).
DISCUSSION
The simultaneous increases observed in Lp-PLA2 activity and LDL-C concentration in baboons fed an HCHF diet is consistent with reports by others of associations between both traits (3, 4, 7), and with what is known of the functional biological relationship between Lp-PLA2 and the LDL particle. Diet-induced increases in LDL-C concentration have been reported previously (38, 39) and the effects of increased LDL-C concentration on risk of CVD are well known (40). However, to our knowledge, this is the first report of a parallel increase in Lp-PLA2 activity, a biomarker of inflammation independently associated with increased CVD risk, in response to dietary challenge. Although details of the mechanism underlying the relationship between increased LDL-C levels and Lp-PLA2 activity are poorly understood, the parallel increase in both traits observed in baboons fed an HCHF diet suggests that the same or similar diet is likely to increase measures of both biomarkers in humans, and further exacerbate risk of CVD.
A substantial proportion of the variation in Lp-PLA2 activity and LDL-C concentration in baboons fed an HCHF diet may be attributed to the additive effects of genes, and some of these genes exert pleiotropic effects on variation in both traits. This finding is consistent with the results of our previously reported analyses from these same animals fed a basal diet containing much lower amounts of fat and cholesterol. In both studies, shared genetic effects, indicative of pleiotropy, account for substantial portions of the total additive genetic variance. However, in the present study, pleiotropic effects account for nearly twice the additive genetic variance as they did in the earlier study, i.e., approximately 47% versus 25%, respectively.
Detection of a pleiotropic QTL influencing Lp-PLA2 activity and LDL-C concentration is also consistent with the results of our earlier study in these same baboons; however, several characteristics distinguish the two QTLs. It is clear that the gene(s) underlying the QTLs detected in the two different dietary environments are different, because the QTL detected in analyses of baboons fed the HCHF diet maps to a baboon chromosomal region corresponding to HSA19q12–q13.2, whereas the QTL detected in baboons fed a basal diet maps to a baboon chromosomal region corresponding to HSA2p24.3–p23.2. Further, the QTL detected in the HCHF diet accounts for approximately 20% of the residual phenotypic variance in both traits, and all additive genetic effects at this QTL are shared between the two traits. In contrast, the QTL detected in the basal diet (9) had a smaller effect, i.e., it accounted for approximately 15% of the residual phenotypic variance in both traits, and additive genetic effects at the QTL were only partially shared between the two traits. These findings suggest the possibility that under HCHF dietary stress, Lp-PLA2 activity and LDL-C levels may be more closely regulated by a gene or genes with greater phenotypic effects than gene(s) regulating both traits in a less stressful dietary environment.
In conjunction with the results of our previous study of covariation in Lp-PLA2 activity and LDL-C concentration in baboons fed a basal diet, the results of this study of the same traits in baboons fed an HCHF diet provide compelling evidence for genotype-by-diet interaction in determining covariation in these two CVD risk factors. A contribution of genotype-by-diet interaction to variation in both traits may be inferred from the observation that the phenotypic variances observed for both traits differ substantially between diets, whereas the magnitude of additive genetic effects does not change between diets. However, genotype-by-diet interaction is demonstrated conclusively by the rejection of the statistical null hypothesis (and its associated biological null hypothesis of no genotype-by-diet interaction) that the residual additive genetic correlation between the same trait measured in two dietary environments equals 1. Further, the extent of the genotype-by-diet interaction effect on each trait may be inferred by examining the proportion of the additive genetic variance that is shared between measures of the trait (i.e., the squared genetic correlation) in the basal and HCHF diets. A logical conclusion drawn from finding genotype-by-diet interaction effects on both traits is that there are diet-specific additive genetic components to the variance in both Lp-PLA2 activity and LDL-C concentration. This result suggests a situation in which different genes or suites of genes contribute to variation in both traits, or, alternatively, the relative contributions of the genes involved may change as a function of exposure to increased dietary cholesterol and fat. A scenario in which the additive genetic variance in both traits is relatively stable irrespective of dietary fat and cholesterol, whereas different genes may contribute (or contribute differentially) to this variance as a function of environmental exposure, is supported further by our localization of pleiotropic QTLs for Lp-PLA2 and LDL-C to two different chromosomes. We conclude from these results that the QTL that mapped to chromosome 2p24.3–p23.2 in our previous study, and the QTL that mapped to chromosome 19q12–13.2 in this study, harbor genes that exert effects specific to the basal and HCHF diets, respectively.
In contrast to the QTL implicated on HSA2p that exerted incomplete pleiotropic effects on both traits in a basal diet (9), our results suggest that the QTL on HSA19q implicated in this study may exert complete pleiotropy on both Lp-PLA2 activity and LDL-C levels in an HCHF diet. This result is consistent with a single gene or polymorphism at this QTL with fully coordinated effects on both traits, and suggests that the gene or polymorphism detected here is part of a biological pathway that includes Lp-PLA2 and LDL in close proximity. One biological pathway that is consistent with the complete pleiotropy at the QTL on HSA19q is the LDL catabolic pathway, because LDL and Lp-PLA2 are associated at the point of LDL clearance. Genes lying at the 10 Mb region in humans that encompasses the point of maximum linkage evidence in baboons (HSA19q12–q13.12) include at least one with potential effects on LDL catabolism. LSR/LISCH7, the lipolysis-stimulated lipoprotein receptor, codes for a receptor that is differentially stimulated by free fatty acids of different chain lengths and is able to bind apoB- and apoE-containing lipoproteins (41). Furthermore, LSR is hypothesized to be an alternate pathway of LDL clearance in the liver separate from the LDLR or the LDL-receptor related protein (LRP) pathways (42). Other potentially relevant genes at HSA19q12–q13.12 include three members of the G-protein-coupled receptor family, GPR40, GPR41, and GPR43, also known as fatty-acid receptors FFAR1, FFAR3, and FFAR2, respectively. GPR40, GPR41, and GPR43 code for proteins known to be differentially activated by medium- and long-chain fatty acids (GPR40) (43) and short-chain fatty acids (GPR41 and GPR43) (44, 45). In particular, GPR40 is stimulated by ligands that are most prevalent in plasma, i.e., palmitate, oleate, stearate, linoleate, and linolenate, all components with proportions that differ significantly between the basal and the HCHF diet. Finally, LRP3, a member of the LDLR family, is also found in this region, although its function in lipoprotein metabolism, if any, is unknown (46). Interestingly, in this study, we did not detect an effect of the structural gene coding for Lp-PLA2 (PLA2G7, located at 6p21.2–p12) on either trait in either diet. Although other studies have detected effects of this gene on Lp-PLA2 activity and CVD risk (7, 8), PLA2G7 does not appear to be the primary regulator of its own enzymatic activity in this population under either dietary regimen.
Two well-studied genes on chromosome 19, APOE and LDLR, exert significant effects on LDL-C concentration in humans, and have been shown to influence risk of CVD in multiple studies (as reviewed in Ref. 12). Additionally, recent genome-wide association analyses have detected significant association of single-nucleotide polymorphisms in these two genes with LDL-C and coronary artery disease (47, 48). However, the fact that polymorphisms in neither gene were linked to a QTL influencing the bivariate phenotype, coupled with the fact that both genes map outside the estimated support interval for our QTL, leads us to conclude that neither LDLR nor APOE are responsible for the pleiotropic QTL that we have detected on the baboon ortholog of human chromosome 19. This conclusion does not exclude these genes as contributors to covariation in these traits in baboons or, by extension, humans; indeed, our observation that polymorphism in baboon LDLR is significantly associated with variation in both LDL-C concentration and Lp-PLA2 activity supports such a contribution. However, in the HCHF dietary environment to which these baboons have been exposed, the effect of polymorphism in LDLR on covariation in these two traits is relatively small, compared with that of the pleiotropic QTL, and contributes primarily to the additive genetic (polygenic) background. That we detected no similar contribution of polymorphism in APOE to the additive genetic background in these two traits may be attributable to the fact that baboons (and other cercopithecoid primates) appear to be monomorphic for the specific APOE allele associated with variation in lipoprotein metabolism and CVD risk in humans (49–51). This result may also be due to the possibility that the APOE polymorphisms analyzed in this study are either not functional or not in linkage disequilibrium with functional polymorphisms that may contribute to variation in LDL-C concentration and/or Lp-PLA2 activity in these baboons. Nevertheless, neither our linkage nor association analyses provide any evidence that either APOE or LDLR is responsible for the effect of the pleiotropic QTL localized in this study.
Localizing QTLs with effects on Lp-PLA2 activity and LDL-C concentration is an efficient first step toward identifying a set of positional candidate genes for further analysis. In this study, we have described a QTL, detected in the HCHF diet and corresponding to 19q12–q13.2, that appears to exert complete pleiotropy on covariation in Lp-PLA2 activity and LDL-C concentration in a nonhuman primate model for the genetics of atherosclerosis risk factors. To our knowledge, this is the first report of a QTL affecting both Lp-PLA2 activity and LDL-C concentration detected in an HCHF dietary environment. We have also demonstrated the contribution of genotype-by-diet interaction to covariation in Lp-PLA2 activity and LDL-C concentration measured in both basal and HCHF dietary environments. Knowledge of the genes regulating both traits under the controlled dietary conditions possible with a nonhuman primate model with close genetic and physiological similarity to humans will ultimately improve our understanding of how Lp-PLA2 activity and LDL-C concentration contribute to increased risk of human cardiovascular disease.
Acknowledgments
For technical contributions and support, the authors thank Ms. T. Baker, Ms. S. Birnbaum, Mr. J. Bridges, Ms. C. Jett, Mr. P.H. Moore, Jr., Ms. D.E. Newman, Dr. K.S. Rice and the SNPRC veterinary and animal care staff, Ms. M.L. Sparks, and Ms. J.F. VandeBerg.
Published, JLR Papers in Press, March 11, 2008.
Footnotes
This study was made possible by research grants from the National Institutes of Health (P01 HL-028972, R01 HL-068922, R01 RR-008781); a base grant from the National Center of Research Resources (NCRR) to the Southwest National Primate Research Center (SNPRC; P51 RR-013986); and was conducted in facilities constructed with support from NCRR Research Facilities Improvement Program Grants (C06 RR-014578, C06 RR-13556, C06 RR-15456, C06 RR-017515).
References
- 1.Lavi S., J. P. McConnell, C. S. Rihal, A. Prasad, V. Mathew, L. O. Lerman, and A. Lerman. 2007. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidylcholine in the coronary circulation: association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation. 115 2715–2721. [DOI] [PubMed] [Google Scholar]
- 2.Koenig W., D. Twardella, H. Brenner, and D. Rothenbacher. 2006. Lipoprotein-associated phospholipase A2 predicts future cardiovascular events in patients with coronary heart disease independently of traditional risk factors, markers of inflammation, renal function, and hemodynamic stress. Arterioscler. Thromb. Vasc. Biol. 26 1586–1593. [DOI] [PubMed] [Google Scholar]
- 3.Persson M., J. A. Nilsson, J. J. Nelson, B. Hedblad, and G. Berglund. 2007. The epidemiology of Lp-PLA(2): distribution and correlation with cardiovascular risk factors in a population-based cohort. Atherosclerosis. 190 388–396. [DOI] [PubMed] [Google Scholar]
- 4.Guerra R., B. Zhao, V. Mooser, D. Stafforini, J. M. Johnston, and J. C. Cohen. 1997. Determinants of plasma platelet-activating factor acetylhydrolase: heritability and relationship to plasma lipoproteins. J. Lipid Res. 38 2281–2288. [PubMed] [Google Scholar]
- 5.Tew D. G., C. Southan, S. Q. J. Rice, G. M. Lawrence, H. Li, H. F. Boyd, K. Moores, I. S. Gloger, and C. H. Macphee. 1996. Purification, properties, sequencing, and cloning of a lipoprotein-associated, serine-dependent phospholipase involved in the oxidative modification of low-density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 16 591–599. [DOI] [PubMed] [Google Scholar]
- 6.Macphee C. H., K. E. Moores, H. F. Boyd, D. Dhanak, R. J. Ife, C. A. Leach, D. S. Leake, K. J. Milliner, R. A. Patterson, K. E. Suckling, et al. 1999. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem. J. 338 479–487. [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang S. Y., H. Shibata, K. Karino, B. Y. Wang, S. Kobayashi, J. Masuda, and T. Nabika. 2007. Comprehensive evaluation of genetic and environmental factors influencing the plasma lipoprotein-associated phospholipase A2 activity in a Japanese population. Hypertens. Res. 30 403–409. [DOI] [PubMed] [Google Scholar]
- 8.Jang Y., O. Y. Kim, S. J. Koh, J. S. Chae, Y. G. Ko, J. Y. Kim, H. Cho, T. S. Jeong, W. S. Lee, J. M. Ordovas, et al. 2006. The Val279Phe variant of the lipoprotein-associated phospholipase A2 gene is associated with catalytic activities and cardiovascular disease in Korean men. J. Clin. Endocrinol. Metab. 91 3521–3527. [DOI] [PubMed] [Google Scholar]
- 9.Vinson A., M. C. Mahaney, L. A. Cox, J. Rogers, J. L. VandeBerg, and D. L. Rainwater. 2008. A pleiotropic QTL on 2p influences serum Lp-PLA2 activity and LDL cholesterol concentration in a baboon model for the genetics of atherosclerosis risk factors. Atherosclerosis. 196 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kammerer C. M., D. L. Rainwater, L. A. Cox, J. L. Schneider, M. C. Mahaney, J. Rogers, and J. L. VandeBerg. 2002. Locus controlling LDL cholesterol response to dietary cholesterol is on baboon homologue of human chromosome 6. Arterioscler. Thromb. Vasc. Biol. 22 1720–1725. [DOI] [PubMed] [Google Scholar]
- 11.Masson L. F., and G. McNeill. 2005. The effect of genetic variation on the lipid response to dietary change: recent findings. Curr. Opin. Lipidol. 16 61–67. [DOI] [PubMed] [Google Scholar]
- 12.Loktionov A. 2003. Common gene polymorphisms and nutrition: emerging links with pathogenesis of multifactorial chronic diseases. J. Nutr. Biochem. 14 426–451. [DOI] [PubMed] [Google Scholar]
- 13.Ye S. Q., and P. O. Kwiterovich, Jr. 2000. Influence of genetic polymorphisms on responsiveness to dietary fat and cholesterol. Am. J. Clin. Nutr. 72 (Suppl.): 1275–1284. [DOI] [PubMed] [Google Scholar]
- 14.Shi Q., G. B. Hubbard, R. S. Kushwaha, D. Rainwater, C. A. Thomas III, M. M. Leland, J. L. VandeBerg, and X. L. Wang. 2007. Endothelial senescence after high-cholesterol, high-fat diet challenge in baboons. Am. J. Physiol. Heart Circ. Physiol. 292 H2913–H2920. [DOI] [PubMed] [Google Scholar]
- 15.Cheng M. L., S. C. Woodford, J. L. Hilburn, and J. L. VandeBerg. 1986. A novel system for storage of sera frozen in small aliquots. J. Biochem. Biophys. Methods. 13 47–51. [DOI] [PubMed] [Google Scholar]
- 16.Allain C. C., L. S. Poon, C. S. Chan, W. Richmond, and P. C. Fu. 1974. Enzymatic determination of total serum cholesterol. Clin. Chem. 20 470–475. [PubMed] [Google Scholar]
- 17.Lipid Research Clinics Program. 1974. Manual of Laboratory Operations. Vol. 1: Lipid and Lipoprotein Analysis (DHEW publication NIH 75–628). US Government Printing Office, Washington, DC.
- 18.Rainwater D. L., C. M. Kammerer, M. C. Mahaney, J. Rogers, L. A. Cox, J. L. Schneider, and J. L. VandeBerg. 2003. Localization of genes that control LDL size fractions in baboons. Atherosclerosis. 168 15–22. [DOI] [PubMed] [Google Scholar]
- 19.Cox L. A., M. C. Mahaney, J. L. VandeBerg, and J. Rogers. 2006. A second-generation genetic linkage map of the baboon (Papio hamadryas) genome. Genomics. 88 274–281. [DOI] [PubMed] [Google Scholar]
- 20.Almasy L., and J. Blangero. 1998. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blangero J., J. T. Williams, and L. Almasy. 2001. Variance component methods for detecting complex trait loci. Adv. Genet. 42 151–181. [DOI] [PubMed] [Google Scholar]
- 22.Mahaney M. C., J. Blangero, A. G. Comuzzie, J. L. VandeBerg, M. P. Stern, and J. W. MacCluer. 1995. Plasma HDL cholesterol, triglycerides, and adiposity. A quantitative genetic test of the conjoint trait hypothesis in the San Antonio Family Heart Study. Circulation. 92 3240–3248. [DOI] [PubMed] [Google Scholar]
- 23.Curtis D., and P. C. Sham. 1994. Using risk calculation to implement an extended relative pair analysis. Ann. Hum. Genet. 58 151–162. [DOI] [PubMed] [Google Scholar]
- 24.Heath S. C. 1997. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ott, J. 1999. Analysis of Human Genetic Linkage. 3rd edition. The Johns Hopkins University Press, Baltimore, MD.
- 26.Amos C., M. de Andrade, and D. Zhu. 2001. Comparison of multivariate tests for genetic linkage. Hum. Hered. 51 133–144. [DOI] [PubMed] [Google Scholar]
- 27.Feingold E., P. O. Brown, and D. Siegmund. 1993. Gaussian models for genetic linkage analysis using complete high-resolution maps of identity by descent. Am. J. Hum. Genet. 53 234–251. [PMC free article] [PubMed] [Google Scholar]
- 28.Almasy L., T. D. Dyer, and J. Blangero. 1997. Bivariate quantitative trait linkage analysis: pleiotropy versus co-incident linkages. Genet. Epidemiol. 14 953–958. [DOI] [PubMed] [Google Scholar]
- 29.Blangero J. 1993. Statistical genetic approaches to human adaptability. Hum. Biol. 65 941–966. [PubMed] [Google Scholar]
- 30.Eisen E. J., and J. E. Legates. 1966. Genotype-sex interaction and the genetic correlation between the sexes for body weight in Mus musculus. Genetics. 54 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodman L. A. 1960. On the exact variance of products. J. Am. Stat. Assoc. 55 708–713. [Google Scholar]
- 32.Bohrnstedt G. W., and A. S. Goldberger. 1969. On the exact covariance of products of random variables. J. Am. Stat. Assoc. 64 1439–1442. [Google Scholar]
- 33.Lynch, M., and B. Walsh. 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Inc., Sunderland, MA.
- 34.Dupuis J., and D. Siegmund. 1999. Statistical methods for mapping quantitative trait loci from a dense set of markers. Genetics. 151 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boerwinkle E., R. Chakraborty, and C. F. Sing. 1986. The use of measured genotype information in the analysis of quantitative phenotypes in man. I. Models and analytical methods. Ann. Hum. Genet. 50 181–194. [DOI] [PubMed] [Google Scholar]
- 36.Hixson J. E., C. M. Kammerer, L. A. Cox, and G. E. Mott. 1989. Identification of LDL receptor gene marker associated with altered levels of LDL cholesterol and apolipoprotein B in baboons. Arterioscler. Thromb. Vasc. Biol. 9 829–835. [DOI] [PubMed] [Google Scholar]
- 37.Cole S. A., S. Chen, C. J. Isaacson, and J. E. Hixson. 1997. BanII RFLP at the apolipoprotein E (APOE) locus in baboons. Anim. Genet. 28 319. [PubMed] [Google Scholar]
- 38.Hegsted D. M., L. M. Ausman, J. A. Johnson, and G. E. Dallal. 1993. Dietary fat and serum lipids: an evaluation of the experimental data. Am. J. Clin. Nutr. 57 875–883. [DOI] [PubMed] [Google Scholar]
- 39.Mensink R. P., and M. B. Katan. 1992. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler. Thromb. 12 911–919. [DOI] [PubMed] [Google Scholar]
- 40.Schaefer E. J. 2002. Lipoproteins, nutrition, and heart disease. Am. J. Clin. Nutr. 75 191–212. [DOI] [PubMed] [Google Scholar]
- 41.Mann C. J., J. Khallou, O. Chevreuil, A. A. Troussard, L. M. Guermani, K. Launay, B. Delplanque, F. T. Yen, and B. E. Bihain. 1995. Mechanism of activation and functional significance of the lipolysis-stimulated receptor. Evidence for a role as chylomicron remnant receptor. Biochemistry. 34 10421–10431. [DOI] [PubMed] [Google Scholar]
- 42.Bihain B. E., and F. T. Yen. 1998. The lipolysis stimulated receptor: a gene at last. Curr. Opin. Lipidol. 9 221–224. [DOI] [PubMed] [Google Scholar]
- 43.Briscoe C. P., M. Tadayyon, J. L. Andrews, W. G. Benson, J. K. Chambers, M. M. Eilert, C. Ellis, N. A. Elshourbagy, A. S. Goetz, D. T. Minnick, et al. 2003. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278 11303–11311. [DOI] [PubMed] [Google Scholar]
- 44.Le Poul E., C. Loison, S. Struyf, J. Y. Springael, V. Lannoy, M. E. Decobecq, S. Brezillon, V. Dupriez, G. Vassart, J. Van Damme, et al. 2003. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 278 25481–25489. [DOI] [PubMed] [Google Scholar]
- 45.Brown A. J., S. M. Goldsworthy, A. A. Barnes, M. M. Eilert, L. Tcheang, D. Daniels, A. I. Muir, M. J. Wigglesworth, I. Kinghorn, N. J. Fraser, et al. 2003. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 278 11312–11319. [DOI] [PubMed] [Google Scholar]
- 46.Ishii H., D. H. Kim, T. Fujita, Y. Endo, S. Saeki, and T. T. Yamamoto. 1998. cDNA cloning of a new low-density lipoprotein receptor-related protein and mapping of its gene (LRP3) to chromosome bands 19q12-q13.2. Genomics. 51 132–135. [DOI] [PubMed] [Google Scholar]
- 47.Willer C. J., S. Sanna, A. U. Jackson, A. Scuteri, L. L. Bonnycastle, R. Clarke, S. C. Heath, N. J. Timpson, S. S. Najjar, H. M. Stringham, et al. 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 40 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kathiresan S., O. Melander, C. Guiducci, A. Surti, N. P. Burtt, M. J. Rieder, G. M. Cooper, C. Roos, B. F. Voight, A. S. Havulinna, et al. 2008. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 40 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hixson J. E., L. A. Cox, and S. Borenstein. 1988. The baboon apolipoprotein E gene: structure, expression, and linkage with the gene for apolipoprotein C-1. Genomics. 2 315–323. [DOI] [PubMed] [Google Scholar]
- 50.Hanlon C. S., and D. C. Rubinsztein. 1995. Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis. 112 85–90. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg R. B. 1994. Identification of functional domains in the plasma apolipoproteins by analysis of inter-species sequence variability. J. Lipid Res. 35 2212–2222. [PubMed] [Google Scholar]