

Abstract
The Staphylococcus aureus transpeptidase sortase A (SrtA) is responsible for anchoring a range of virulence- and colonization-associated proteins to the cell wall. SrtA recognizes substrates that contain a C-terminal LPXTG motif. This sequence is cleaved following the threonine, and an amide bond is formed between the threonine and the pentaglycine cross-bridge of branched lipid II. Previous studies have implicated the β6/β7 loop region of SrtA in LPXTG recognition but have not systematically characterized this domain. To better understand the individual roles of the residues within this loop, we performed alanine-scanning mutagenesis. Val-168 and Leu-169 were found to be important for substrate recognition, and Glu-171 was also found to be important, consistent with its hypothesized role as a Ca2+-binding residue. Gly-167 and Asp-170 were dispensable for catalysis, as was Gln-172. The role of Arg-197 in SrtA has been the subject of much debate. To explore its role in catalysis, we used native chemical ligation to generate semi-synthetic SrtA in which we replaced Arg-197 with citrulline, a non-ionizable analog. This change resulted in a decrease of <3-fold in kcat/Km, indicating that Arg-197 utilizes a hydrogen bond, rather than an electrostatic interaction. Our results are consistent with a model for LPXTG recognition wherein the Leu-Pro sequence is recognized primarily by hydrophobic contacts with SrtA Val-168 and Leu-169, as well as a hydrogen bond from Arg-197. This model contradicts the previously proposed mechanism of binding predicted by the x-ray crystal structure of SrtA.
The increasing prevalence of multidrug-resistant Gram-positive bacterial infections has been a cause for great concern (1, 2). Most alarming has been the recent characterization of strains of Staphylococcus aureus which exhibit either intermediate or full resistance to vancomycin, currently the antibiotic of last resort for methicillin-resistant S. aureus infections (3-5). During pathogenesis, Gram-positive bacteria use virulence factors to aid in adherence to host endothelial tissues, evasion of the immune system, and the acquisition of iron from the local environment (6, 7). There has been a great deal of recent interest in the idea of targeting these virulence factors as a possible strategy for the development of new therapeutics for the treatment of Gram-positive infections (8, 9).
Sortases are cysteine transpeptidase enzymes found in a wide-range of Gram-positive pathogens. They catalyze the formation of a covalent linkage between surface proteins containing an LPXTG motif and the peptidoglycan layer of the cell wall (6). Many of these cell wall-anchored proteins function as virulence factors (6, 10). As such, several previous studies have implicated a role for sortases in promoting virulence in several strains of Gram-positive bacteria. Sortase knock-out strains have been shown to have reduced pathogenicity in S. aureus (11), Listeria monocytogenes (12, 13), and in several members of the Streptococcus lineage: S. gordonii (14), S. pneumonia (15), S. pyogenes (16), S. mutans (17), S. suis (18), S. sanguinis (19), and S. agalactiae (20).
In S. aureus, the majority of surface proteins are anchored by the action of sortase A (SrtA).2 SrtA recognizes substrates that contain a C-terminal cell wall-sorting motif consisting of a conserved LPXTG motif, followed by a hydrophobic domain and a positively charged tail (21, 22). The enzyme catalyzes cleavage of the LPXTG motif between the threonine and glycine residues by means of an active-site cysteine (Cys-184) (23). This leads to the formation of a covalent acyl-enzyme intermediate, which is then resolved by nucleophilic attack from the amino group of the Gly5 cross-bridge on branched lipid II, a peptidoglycan precursor (24, 25). Subsequent transglycosylation and transpeptidation reactions incorporate the protein-linked lipid II into the mature cell wall.
Although the substrate specificity requirements of SrtA have been well documented (26, 27), the exact molecular basis for this specificity remains unclear. Several recent studies have implicated the β6/β7 loop region of SrtA in LPXTG recognition (28-30). However, these studies have not systematically explored the roles of the individual residues within the β6/β7 loop and have not directly established which residues are critical for LPXTG recognition.
Much debate has occurred in the literature over the role of a conserved active-site arginine residue (Arg-197) in SrtA catalysis. It has been suggested that Arg-197 may function as a general base, helping to deprotonate either Cys-184 or the incoming Gly5 chain on branched lipid II (31, 32). Alternatively, it has been suggested that Arg-197 may play a role in stabilizing the formation of an oxyanion transition state prior to formation of the acyl-enzyme intermediate (28, 33). A recent study by Frankel et al. (33) provided evidence against the first interpretation and suggested that the role of Arg-197 was in transitionstate stabilization. However, this study was unable to discriminate whether Arg-197 acts via an electrostatic interaction or through a side-chain hydrogen bond interaction.
To better understand the determinants of substrate specificity and LPXTG recognition in SrtA, we have performed alanine-scanning mutagenesis of six key residues along the β6/β7 loop in SrtA. In addition, we have used native chemical ligation to generate a synthetic amino acid mutant of Arg-197, by replacing the arginine with a citrulline. We have characterized these mutants biophysically, and we report here the results of the mutations on the kinetic parameters kcat and Km of SrtA. Finally, we have developed a revised model for substrate recognition and catalysis in SrtA, and discuss implications for future studies.
EXPERIMENTAL PROCEDURES
General—Buffer salts were purchased from Sigma. Oligonucleotide primers were purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and were used without further purification. The pTWIN1 vector was purchased from New England Biolabs (Ipswich, MA). Escherichia coli electrocompetent DH5α cells (Invitrogen) and E. coli BL21(DE3) electrocompetent cells (Novagen) were used according to the manufacturer's recommendations. Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs. DNA sequencing was performed at the Duke University DNA Analysis Facility. Standard Fmoc-protected amino acids (Novabiochem), NH2-Gly5-OH (Bachem), t-butoxycarbonyl-Abz-OH (Bachem), and Fmoc-Dap(DNP)-OH (Bachem), were purchased and used without further purification. 5-(4-Fmoc-aminomethyl-3,5-dimethoxyphenoxy)valeric acid-methylbenzhydrylamine resin was purchased from Advanced Chemtech. Fmoc-Lys(t-butoxycarbonyl)-Wang resin was purchased from Bachem and used without further purification. Solid-phase peptide synthesis was performed on an Applied Biosystems 433A peptide synthesizer. Protein purification was performed on an AKTA fast protein liquid chromatography system equipped with a UV detector (GE Healthcare). Preparative-scale HPLC was performed on a Rainin HPXL series system equipped with a Dynamax UV-1 detector, and a preparative (50 × 250 mm, 10-15 μm) C8 column (Vydac, Inc.). Analytical HPLC was performed on an Agilent 1200 series HPLC equipped with an autosampler, fraction collector, diode array UV detector, fluorescence detector, and a fast analytical (4.6 × 50 mm, 3 μm) C18 column (Vydac, Inc.). MALDI-TOF mass spectrometry was performed using a Voyager™ mass spectrometer (Applied Biosystems). Electrospray ionization mass spectrometry was performed on an Agilent 1100 series liquid chromatography/MSD ion trap mass spectrometer, in positive ion mode.
Cloning of SrtAΔN24 Constructs—The plasmid pET15bSrtAΔN24 (27) was used as a template. Primer pairs pTWIN1SrtAΔN24fwd and pTWIN1SrtAfullrev (see Table 1 for sequences) were used to PCR-amplify the sequence of SrtA(25-206) out of this vector, and primer pairs pTWIN1SrtAΔN24fwd and pTWIN1SrtAtruncrev (see Table 1) were used to PCR amplify the sequence of SrtA(25-183). In each case, the resulting DNA fragments were gel-purified, double digested with NdeI and SapI, then ligated with T4 DNA ligase into vector pTWIN1, which had been previously digested with the same enzymes, generating the plasmids pTWIN1SrtA(25-206) and pTWIN1SrtA(25-183), respectively. DNA sequencing was used to confirm that the desired sequence had been generated. The final plasmids each contained the desired SrtA sequence, fused in-frame at the C terminus to the GyrA intein from Mycobacterium xenopi and a chitin-binding domain.
TABLE 1.
Primers used in this study
| Primer | Sequence 5′ → 3′a |
|---|---|
| pTWIN1SrtAΔN24fwd | GGTGGTCATATGAAACCACATATCGATAATTATCTTCAC |
| pTWIN1SrtAfullrev | GGTGGTTGCTCTTCCGCATTTGACTTCTGTAGCTACAAAGATTTTACG |
| pTWIN1SrtAtruncrev | GGTGGTTGCTCTTCCGCAAGTAATTAATGTTAATTGTTTATCTTTACC |
| SrtA R197A | Fwd: GGTTGGGAAAAAGCTAAAATCTTTGTAGC |
| Rev: GCTACAAAGATTTAGCTTTTTTCCCAAAC | |
| SrtA R197K | Fwd: GGTTGGGAAAAAAAAAAAATCTTTGTAGC |
| Rev: GCTACAAAGATTTTTTTTTTTTCCCAAAC | |
| SrtA G167A | Fwd: CCTACAGATGTAGCAGTTCTAGATGAACAAAAAGG |
| Rev: CCTTTTTGTTCATCTAGAACTGCTACATCTGTAGG | |
| SrtA V168A | Fwd: CCTACAGATGTAGGAGCTCTAGATGAACAAAAAGG |
| Rev: CCTTTTTGTTCATCTAGAGCTCCTACATCTGTAGG | |
| SrtA L169A | Fwd: CCTACAGATGTAGGAGTTGCAGATGAACAAAAAGG |
| Rev: CCTTTTTGTTCATCTGCAACTCCTACATCTGTAGG | |
| SrtA D170A | Fwd: CCTACAGATGTAGGAGTTCTAGCTGAACAAAAAGG |
| Rev: CCTTTTTGTTCAGCTAGAACTCCTACATCTGTAGG | |
| SrtA E171A | Fwd: CCTACAGATGTAGGAGTTCTAGATGCACAAAAAGGTAAAG |
| Rev: CTTTACCTTTTTGTGCATCTAGAACTCCTACATCTGTAGG | |
| SrtA Q172A | Fwd: CCTACAGATGTAGGAGTTCTAGATGAAGCAAAAGGTAAAG |
| Rev: CTTTACCTTTTGCTTCATCTAGAACTCCTACATCTGTAGG |
Mutated codons are underlined.
Site-directed Mutagenesis—The plasmid pTWIN1SrtA(25-206) was used as a template for the introduction of single amino acid substitutions by PCR, using the QuikChange® method (Stratagene, Inc.). The forward and reverse primer pairs used to generate each of the discussed SrtA mutants are listed in Table 1. Successful mutations were confirmed by DNA sequencing.
Expression, Purification of SrtA(25-206), and Mutants—The plasmid pTWIN1SrtA(25-206) was transformed by electroporation into E. coli BL21(DE3) cells. The cells were grown at 37 °C in LB media containing 100 μg/ml ampicillin. When the A600 reached 0.5-0.6, expression was induced with 1 mm isopropyl β-d-thiogalactopyranoside, and the cells were grown for an additional 3 h at 37 °C, then were harvested by centrifugation at 3000 × g for 10 min. Cells were resuspended in buffer A (100 mm sodium phosphate, 500 mm NaCl, pH 7.0) and lysed with an EmulsiFlex-C5 high pressure homogenizer (Avestin, Inc.). The lysate was centrifuged for 45 min at 30,000 × g, and the supernatant was applied to a column of chitin beads (New England Biolabs), which had been pre-equilibrated with buffer A. The flow-through from the loading step was reapplied to the column, which was then washed with 250 ml of buffer A. Cleavage of the intein and elution of the target protein were induced by overnight recirculation (using a peristaltic pump) of buffer B (100 mm sodium phosphate, 500 mm NaCl, 40 mm dithiothreitol, pH 7.0). Fractions of flow-through from the overnight recirculation were collected, and 20-μl aliquots from each step and fraction were analyzed by SDS-PAGE. SrtA(25-206)-containing fractions were concentrated to a volume of 12 ml using an Amicon-stirred ultrafiltration cell (Millipore, Inc.) with a 5,000-NMWL filter. The concentrated protein sample was then loaded onto a HiPrep 26/60 Sephacryl S-200 gel-filtration column (GE Healthcare), which had been pre-equilibrated with buffer D (150 mm NaCl, 50 mm Tris, 5 mm CaCl2, 0.1% β-mercaptoethanol, 10% glycerol, pH 7.5), and the column was run overnight. 5-ml fractions were collected, and 20-μl aliquots of these were analyzed by 12% SDS-PAGE. Fractions containing pure SrtA(25-206) were pooled, then concentrated to at least 150 μm using a combination of Amicon-stirred ultrafiltration cells (with a 5,000-NMWL membrane) and Amicon ultracentrifugal filters (5,000-NMWL membrane). Protein concentration was determined spectrophotometrically, using the extinction coefficient ε280 = 17,420 m-1 cm-1 (same ε280 for all SrtA(25-206) mutants). Protein molecular weight was verified by electrospray ionization-mass spectrometry. Multiply charged ion states were detected, and software was used to deconvolute the spectra and obtain an accurate molecular weight for the protein (error ≤ 0.01%). All mutants (except R197Cit) were generated via the same methods.
Expression and Purification of SrtA(25-183) Protein-thioester—The plasmid pTWIN1SrtA(25-183) was used for protein expression as described above for pTWIN1SrtA(25-206), and crude protein was loaded onto a column of chitin beads. Following the wash step, the protein-thioester SrtA(25-183)-MESNA was eluted by overnight recirculation of buffer C (100 mm sodium phosphate, 500 mm NaCl, 100 mm MESNA, pH 7.0). Fractions of flow-through from the overnight recirculation were collected, and 20-μl aliquots were analyzed by 12% SDS-PAGE. Fractions containing SrtA(25-183)-MESNA were pooled, then purified by preparative-scale HPLC on a C8 column, using a linear gradient from 22-41% acetonitrile/0.1% trifluoroacetic acid. Purified SrtA(25-183)-MESNA from multiple injections was pooled, and then lyophilized to dryness. Product composition was determined by MALDI-TOF mass spectrometry (molecular mass = 18267.6 Da).
Solid-phase Synthesis of Peptides—Peptides corresponding to the C-terminal 23 residues of SrtA (residues 184-206) were synthesized by the Fmoc/piperidine strategy on preloaded Fmoc-Lys(t-butoxycarbonyl)-Wang resin on a 0.25-mmol scale. The SrtA substrate peptide Abz-LPETGG-Dap(DNP)-NH2 was synthesized using the Fmoc/piperidine strategy on 5-(4-Fmoc-aminomethyl-3,5-dimethoxyphenoxy)valeric acid-methylbenzhydrylamine resin at 0.25-mmol scale. Peptides were synthesized and purified using previously established methods (27, 30). Purified peptides were lyophilized to dryness, and product identities were confirmed by MALDI-TOF mass spectrometry.
Generation of Semi-synthetic SrtA(25-206) Wild Type and R197Cit via Native Chemical Ligation—Native chemical ligation was carried out under standard conditions (34-36). 40.4 mg of SrtA(25-183)-MESNA was combined with 27.6 mg of SrtA(184-206) in a 20-ml scintillation vial with a rubber septum top. The vial was sealed, purged of oxygen, and kept under argon atmosphere. Degassed native chemical ligation reaction buffer (5 ml, of 6 m guanidine hydrochloride, 100 mm sodium phosphate, 20 mm 4-mercaptophenylacetic acid, 100 mm Tris(2-carboxyethyl)phosphine hydrochloride, pH 7.0) was added to the vial with a syringe, and the reaction was allowed to proceed under argon atmosphere at 40 °C. Final concentrations of the reactants were 450 μm SrtA(25-183)-MESNA and 2.0 mm SrtA(184-206). Aliquots (10 μl) of the reaction mix were removed every hour and analyzed by reversed-phase HPLC; the reaction was judged to be nearly complete after 8 h. The product SrtA(25-206) was purified by reversed-phase HPLC, using a preparative scale Jupiter™ C8 column (Vydac, Inc.), with a gradient from 22-45% acetonitrile/0.1% trifluoroacetic acid. Pooled reaction products were lyophilized to dryness. The resulting powder was resuspended in 8 ml of buffer E (25 mm sodium phosphate, pH 5.8), and incubated on ice for 4 h to re-fold. The resulting solution was applied to an SP-Sepharose fast-flow column, which had been pre-equilibrated with buffer E. SrtA(25-206) was eluted from the column using buffer E with a gradient from 0 to 500 mm NaCl. 6-ml fractions were collected, and 20-μl aliquots of these were analyzed by 12% SDS-PAGE. Fractions containing pure SrtA(25-206) were pooled and concentrated to 8-ml volume using an Amicon stirred ultrafiltration cell with a 5,000-NMWL membrane. The concentrated protein solution was dialyzed overnight at 4 °C against buffer D, using a Slide-A-Lyzer dialysis cassette with a 5,000-NWML membrane (Pierce). Recovered protein was concentrated to at least 100 μm using an Amicon ultracentrifugal filter (5,000-NMWL membrane). Protein concentration was determined spectrophotometrically using the extinction coefficient ε280 = 17,420 m-1 cm-1. The mutant protein SrtA(25-206)R197Cit was made in the same manner, except that the peptide SrtA(184-206)R197Cit was used. The final molecular weights of both semi-synthetic proteins were verified by electrospray ionization-mass spectrometry.
Thermal Stability Studies Using CD Spectroscopy—Aliquots (300 μl) of concentrated protein were dialyzed overnight at 4 °C against buffer containing 10 mm Tris (pH 7.5), 30 mm NaCl, and 5 mm CaCl2. Samples were diluted in the same buffer until final protein concentrations were 5 μm. Protein concentrations of the diluted solutions were obtained spectrophotometrically, using the extinction coefficient ε280 = 17,420 m-1 cm-1 (same for all proteins). For each protein, 2 ml was transferred to a quartz cuvette with a 1-cm path length for CD data collection. CD spectra were measured on an Aviv Instruments model 202 CD spectrometer. An initial wavelength scan was taken for recombinant SrtA(25-206), at 25 °C, from 260 to 190 nm in 1 nm steps, with a scanning speed of 4 s/nm. Two scans were taken and were averaged and corrected for background absorbance.
For thermal denaturation studies, 2-ml samples were transferred to a quartz cuvette with a 1-cm path length, and CD spectra were collected at 216 nm, in 2 °C steps from 4 to 98 °C. Samples were equilibrated for 30 s at each temperature prior to data collection, and the average CD signal at 216 nm was collected over a 20-s window. The observed signal was corrected for background absorbance and converted to mean residue ellipticity (θm, degrees cm2 dmol-1 residue-1) using
 |
(Eq.1) |
where θobs represents the observed ellipticity in degrees, c is the protein concentration in molar, d is the optical path length in centimeters, and n is the number of amino acid residues in the protein (182 in all cases).
HPLC Assays for SrtA Activity—SrtA activity was measured using the HPLC-based assay described by Kruger et al. (37). For initial activity determinations, wild-type recombinant SrtA (1 μm) and mutant enzymes (varying concentrations, ranged from 1 μm to 100 μm) were incubated with peptide substrates Abz-LPETGG-Dap(DNP)-NH2 (1.5-2.0 mm) and NH2-Gly5-OH (2 mm). The substrate Abz-LPETGG-Dap(DNP)-NH2 was used in place of the more typical Abz-LPETG-Dap(DNP)-NH2, because a recent study showed that SrtA more rapidly turns over substrates containing a glycine in the P2′ position (38). Reactions were performed in a 100-μl volume in standard assay buffer (150 mm NaCl, 5 mm CaCl2, 300 mm Tris-Cl, pH 7.5). Assays were initiated by addition of enzyme, and were run for anywhere between 15 min and 6 h at 37 °C, depending upon the activity levels. At each time point assays were quenched by removal of 80 μl of reaction mix into 40 μl of 1.2 m HCl and analyzed by HPLC.
To determine kinetic parameters for each of the mutants, assay lengths and enzyme concentrations were chosen so as to approximate the levels of activity observed for wild-type recombinant SrtA during a 7.5-min assay. Volumes of enzyme solution added to each reaction were kept below one-tenth of total assay volume. Linearity of SrtA activity was established for all of the mutants by monitoring product formation as a function of time during the assay window. Enzyme concentrations and assay lengths for kinetic analysis of each mutant SrtA were as follows: wild-type (semi-synthetic), 1 μm for 7.5 min; R197Cit, 1 μm for 20 min; R197A, 50 μm for 2.5 h; R197K, 50 μm for 2.5 h; G167A, 1 μm for 10 min; V168A, 1 μm for 75 min; L169A, 10 μm for 2 h; D170A, 1 μm for 10 min; E171A, 1 μm for 75 min; Q172A, 1 μm for 15 min.
Abz-LPETGG-Dap(DNP)-NH2 was varied from 61 μm to 13.15 mm, and the resulting data were fit to the Michaelis-Menten equation for steady-state kinetics using SigmaPlot v.8.0 (SPPS, Inc.), to obtain estimates for the kinetic parameters kcat and Km. The change in free energy of the transition state for mutants was estimated from Equation 2,
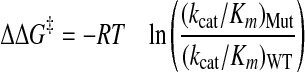 |
(Eq.2) |
where ΔΔG‡ is the change in free energy of the transition state between wild-type (WT) and mutant (Mut), R is the ideal gas constant, and T is temperature.
Modeling Studies—To perform comparative analysis, the 1.8-Å resolution x-ray structure of the LPETG-bound mutant SrtAΔN59 C184A was selected (PDB: 1T2W). This structure was solved in the absence of Ca2+. There were three molecules in the asymmetric unit of this structure. Model A was chosen for analysis, because this was the only model that crystallized with a bound LPETG peptide. Residue Ala-184 was replaced in silico with a cysteine, and the Cys-184 side chain was modeled based on its orientation in the wild-type SrtAΔN59 structure.
RESULTS
Mutagenesis of the β6/β7 Loop in SrtA—The recently solved x-ray crystal structure of SrtAΔN59 (28) has served as a useful guide for understanding substrate recognition in SrtA, but it has also left many questions unanswered. In particular, the role of the β6/β7 loop in LPXTG recognition has remained a mystery. Several lines of evidence suggest a role for this domain in initial substrate recognition (28-30, 39). First, it is located at the posterior of the active site cavity, in a position above the N-terminal ends of the β4 and β7 strands and above the C-terminal end of the β8 strand (Fig. 1). The β6/β7 loop is thus in an ideal position to interact with the leucine-proline at the N-terminal end of the LPXTG sorting signal, facilitating recognition. Second, the loop has been shown to be highly flexible in solution but becomes ordered upon calcium binding (29). The binding of calcium (which stimulates SrtA activity 8-fold) thus “locks” the loop into the correct conformation to allow for LPXTG binding and cleavage. Third, this loop domain differs between SrtA and SrtB, which recognizes an NPQTN sorting signal. It was shown recently that swapping the β6/β7 loop from SrtB into SrtA was sufficient to alter the specificity of SrtA by over 700,000-fold, directly implicating this loop in substrate discrimination (30).
FIGURE 1.

X-ray structure of the LPETG-bound SrtAΔN59 C184A active site. X-ray crystal structure of the active site of S. aureus SrtAΔN59 C184A (PDB: 1T2W, chain A), with a LPETG peptide bound in the active site. For clarity, we have modeled a cysteine back in place of Ala-184. Although there were three molecules in the asymmetric unit, this model (chain A) is the only one that bound a LPETG peptide. This model shows the relative positions of the β3/β4 (blue), β6/β7 (red), and β7/β8 (green) loops. Also shown are the side chains of the β6/β7-loop residues Gly-167 through Gln-172. A putative calcium-binding site (coordinated by Glu-105, Glu-108, Asp-112, and Asn-114) is indicated by the arrow. The peptide LPETG is shown in yellow. Catalytic residues His-120 and Cys-184 of SrtA are shown for reference, as is Arg-197.
Although a role for the β6/β7 loop of SrtA in LPXTG recognition appears clear, the mechanism by which it does this remains somewhat murky. Because it is highly flexible, it has been very difficult to obtain precise structural information about the residues that make up the loop. In the 2.0-Å x-ray crystal structure of SrtAΔN59 (28), there were three molecules in the asymmetric unit, and the β6/β7 loop took on a different conformation in each molecule. Although the overall r.m.s.d. for the α-carbons between the three molecules was only 0.5 Å, across the β6/β7 loop it wais 4.1 Å. Similarly, in the 1.8-Å crystal structure of SrtAΔN59 C184A (28) with an LPETG peptide bound, three molecules crystallized in the asymmetric unit, and again, in each molecule the loop took on a different conformation. r.m.s.d. for the α-carbons overall was 0.6 Å, but for the β6/β7 loop it was 2.8 Å. The average B-factors for the residues in this loop were extremely high (ranging from 91 to 136.9), and it was not possible to model the loop in two of these six models. NMR studies have shown that the loop becomes much more ordered upon the binding of a single calcium ion, implicating a role for the loop in adaptive recognition of the LPXTG substrate (29, 39). However, the exact set of residues utilized for substrate binding and cleavage remain unclear.
To better understand the importance of residues within the β6/β7 loop for substrate recognition, structural analysis was performed. Based on the x-ray structure of LPETG-bound SrtAΔN59 C184A (28), we identified residues Gly-167 through Gln-172 as potential recognition sites for LPXTG (see Fig. 1). Specifically, this model suggested that residues Val-168 and Leu-169 would be important, via hydrophobic interactions, for recognition of the hydrophobic Leu-Pro segment of the sorting signal. Gln-172 was also predicted to be important, based on a potential hydrogen bond interaction between the side chain of Gln-172 and the backbone carbonyl and amide of Leu-Pro in the crystal structure.
To test our predictions we performed alanine-scanning mutagenesis. Six site-specific amino acid mutants were generated, replacing each residue (Gly-167 through Gln-172) in turn with an alanine. Each mutant was successfully expressed in E. coli BL21(DE3) cells and was purified to ≥98% purity as judged by SDS-PAGE (data not shown).
Thermal Stability Measurements of 167-172 Mutants—To explore the effects of the alanine mutations on the stability of SrtA, we used CD spectroscopy to compare the thermal stabilities of the variant proteins with that of wild-type SrtA. Although the overall CD spectrum of SrtA does not show strong features (data not shown), the protein has been shown to be active under the conditions used for CD, and is assumed to be properly folded (33). Importantly, the observed CD signal changes both in intensity and in shape when heat-denatured, and CD has thus been validated as a tool to monitor global unfolding of this protein. For our studies, we chose to observe the CD signal at 216 nm (θ216), because the wild-type protein gives a strong negative signal at this wavelength, and this signal is altered upon heating to 98 °C. To measure the thermal stability of our mutants, measurements of θ216 were made over the range of 4 to 98 °C, and fit using a four-parameter sigmoidal fit to obtain estimates for Tm. The results are shown in Fig. 2 and Table 2. No changes were observed in either the Tm or the onset of unfolding for any of our variants, indicating that the global stability of the protein is not altered by these mutations. Although the possibility of more subtle effects on local stability cannot be ruled out, these results argue that the overall structures of the mutated proteins remain unaffected.
FIGURE 2.

Thermal denaturation studies of SrtA mutants. Plots of CD signal (converted to mean residue ellipticity, in units of deg cm2 dmol-1 residues-1) versus temperature for wild-type SrtAΔN24 and for each of the mutants generated in this study. The corresponding Tm for each mutant is shown in Table 2. Each data point was collected as the average signal over 20 s, following a 30-s equilibration at each temperature.
TABLE 2.
Tm values of SrtA mutants
| Enzyme | Tm |
|---|---|
| °C | |
| wt (rec) | 59.3 ± 0.5 |
| G167A | 58.8 ± 0.4 |
| V168A | 57.6 ± 0.3 |
| L169A | 57.5 ± 0.4 |
| D170A | 61.0 ± 0.4 |
| E171A | 57.5 ± 0.3 |
| Q172A | 58.1 ± 0.5 |
| R197A | 56.4 ± 0.5 |
| R197K | 57.3 ± 0.5 |
| wt (syn) | 58.0 ± 0.5 |
| R197Cit | 59.6 ± 0.6 |
Steady-state Kinetics of 167-172 Mutants—Once the stability of the mutants had been established, enzymatic assays were performed to explore the effects of the mutations on catalytic activity. Steady-state analysis allowed for estimation of the kinetic parameters kcat and Km for each protein. A summary of the observed kinetic parameters is shown in Table 3.
TABLE 3.
Kinetic parameters of SrtA alanine scan mutants
| Enzyme | kcat | Km | kcat/Km | Activity decrease from wt |
|---|---|---|---|---|
| s−1 | mM | M−1s−1 | -fold | |
| wt (rec) | 1.10 ± 0.06 | 8.76 ± 0.78 | 125 ± 18 | |
| G167A | 1.13 ± 0.16 | 8.70 ± 2.0 | 129 ± 48 | n.c.a |
| V168A | 0.15 ± 0.01 | 6.56 ± 0.64 | 22.7 ± 3.6 | 5.5 |
| L169A | 1.23 × 10−2 ± 0.01 × 10−2 | 9.14 ± 0.15 | 1.35 ± 0.14 | 93 |
| D170A | 1.09 ± 0.13 | 8.13 ± 1.7 | 133 ± 43 | n.c. |
| E171A | 0.16 ± 0.01 | 6.74 ± 0.69 | 23.1 ± 3.6 | 5.4 |
| Q172A | 1.13 ± 0.11 | 12.7 ± 1.9 | 89.3 ± 22 | 1.4 |
n.c., no change.
For the mutants G167A and D170A, no change in the kinetic parameters was observed. Indeed, for these variants both kcat and Km were within experimental error of the values for wild-type enzyme, suggesting that these residues do not play a role in substrate recognition or catalysis.
By contrast, mutation of either Val-168 or Leu-169 to alanine was detrimental to catalysis (Fig. 3). The mutations caused a 7.3-fold and an 89-fold decrease in kcat, respectively. Interestingly, in both cases the value of Km was unchanged from the wild type. Loss of a substrate-binding residue would normally be expected to cause an increase in Km, yet that is not observed here. The 93-fold decrease in kcat/Km observed for the L169A mutant is larger than those seen for individual mutations within the active-site β7 strand itself and is the largest observed for a non-catalytic residue (33). Although the effects of the Val-168 and Leu-169 mutations are confined to kcat, this does not rule out a role for these residues in substrate recognition. A recent study showed that kcat effects on specificity can be observed in enzymes that require a conformational rearrangement to actively align catalytic residues prior to catalysis (40), and such a rearrangement has been proposed for SrtA (28, 39). It is possible that the roles of Val-168 and Leu-169 are more than a simple initial binding of LPXTG; instead, we theorize that they are responsible for positioning the substrate such that it is properly oriented for nucleophilic attack by the active site Cys-184.
FIGURE 3.

Kinetics of SrtA alanine scan mutants. The bar graph shows the measured catalytic efficiency kcat/Km for each of the alanine scan mutants. The corresponding kinetic parameters kcat and Km are shown in Table 3.
Mutation of Glu-171 to alanine resulted in a 6.9-fold decrease in kcat. A previous study has implicated this residue in calcium binding and, thus, the stabilization of the β6/β7 loop (29). A potential calcium-binding site was identified, consisting of Glu-171 and several residues on the β3/β4 loop (Glu-105, Glu-108, Asp-112, and Asn-114). In that study, the authors observed a 4.8-fold decrease in kcat upon mutation of Glu-171 to alanine, and our results are consistent with this. Calcium binding has been shown to have a stimulatory effect on SrtA catalysis, increasing the rate of the enzyme by ∼8-fold. The mutant E171A is impaired in catalysis by about the same amount. Given that mutation of Glu-171 to alanine has previously been shown to reduce sensitivity to Ca2+ (29), the data collectively suggest that the effect of this mutation is to impair or abolish calcium binding by the enzyme. If so, then the remainder of the β6/β7 loop would remain highly flexible, preventing efficient substrate binding and cleavage. Interestingly, in the structural model of SrtAΔN59 C184A bound to LPETG, Glu-171 is pointing out toward bulk solvent, away from both the peptide and the putative calcium-binding site (see Fig. 1). Our results, as well as those of the previous study (29), argue that this cannot be the correct orientation for this residue.
Regarding Gln-172, mutation to alanine has no effect on kcat, and only a negligible effect on Km, arguing against an important role for this residue in substrate recognition. In the LPETG-bound structure of SrtAΔN59 C184A, this residue is pointing into the active site, and appears to be well positioned to make one or two hydrogen bonds to the backbone of the Leu-Pro amide bond in the peptide. However, the kinetics of the mutant argues against the existence of such a hydrogen bond.
Mutagenesis of Arg-197—In addition to residues along the β6/β7 loop, a conserved arginine within the active site (Arg-197) has been demonstrated to be important for catalysis (31). Although this residue has been shown to be important for LPXTG cleavage, its mechanism of action has been the subject of some controversy. Three distinct roles for this residue have been proposed: 1) that it acts as a general base, helping to deprotonate either Cys-184 or the incoming secondary nucleophile (i.e. Gly5) for catalysis (28, 31, 32), 2) that it helps to stabilize the formation of an oxyanion-like transition state, via electrostatic interactions (28, 33), or 3) that it utilizes a hydrogen bond interaction to bind to and orient the LPXTG substrate for catalysis (shown here). A previous study provided evidence arguing against the first possibility (33). The pH-rate profile of SrtA was found to be unaffected by mutation of Arg-197 to alanine or lysine, suggesting that Arg-197 does not participate in one of the important ionizations for catalysis (33). However, an unanswered question is whether Arg-197 acts primarily via electrostatic interactions or via specific hydrogen bond interactions with its side chain guanidino group.
Native Chemical Ligation to Generate Arg-197Cit Mutant—To discriminate between these two possibilities, we generated a mutant protein where Arg-197 was replaced by a citrulline residue (Fig. 4). Citrulline is an isosteric analog of arginine, which differs only in the replacement of the guanidino group of arginine by a urea group in citrulline. It thus mimics closely the shape of arginine and retains much of its hydrogen-bonding capability, as both residues contain a terminal -NH2 group, which can act as a hydrogen-bond donor. Unlike arginine, citrulline does not carry a formal positive charge at neutral pH, and it cannot participate in electrostatic interactions. Several previous studies have shown that citrulline can be an effective mimic for arginine and can be used to discriminate specific interactions (41-45).
FIGURE 4.

Structures of arginine and citrulline.
To generate the point-mutant R197Cit, we used the technique of native chemical ligation as described by Dawson et al. (34). To maintain consistency in our results, we also made wild-type SrtA by this method. Hereafter, we will refer to fully recombinant SrtA as “SrtA rec,” and semi-synthetic SrtA as “SrtA syn,” to avoid confusion. In addition to the R197Cit mutant, we also generated recombinant point mutants R197A and R197K, for comparison.
Thermal Stability of Arg-197 Mutants—All of the Arg-197 mutants were initially evaluated for thermal stability by CD, as described above for the β6/β7-loop mutants. Results of the CD denaturation studies are shown in Fig. 2. Neither the onset of unfolding nor the Tm was affected by any of the mutations, indicating that the mutants remain folded.
Kinetics of Arg-197 Mutants—The kinetics for the Arg-197 mutants are shown in Table 4. Mutation of Arg-197 to either alanine or lysine led to a sharp decrease in kcat, resulting in a near-total loss of activity. Overall, the variant R197A had a 960-fold decrease in efficiency (kcat/Km) from the wild type, and R197K had a 690-fold decrease in kcat/Km. These decreases are in the same range as those observed previously for these mutations (33).
TABLE 4.
Kinetic parameters of SrtA Arg 197 mutants
| Enzyme | kcat | Km | kcat/Km | Activity decrease from wt rec (syn) |
|---|---|---|---|---|
| s−1 | mm | m−1s−1 | -fold | |
| wt (rec) | 1.10 ± 0.06 | 8.76 ± 0.78 | 125 ± 18 | |
| R197A | 6.28 × 10−4 ± 0.60 × 10−5 | 4.69 ± 0.12 | 0.13 ± 0.01 | 960 |
| R197K | 1.90 × 10−3 ± 0.20 × 10−4 | 10.4 ± 0.19 | 0.18 ± 0.01 | 690 |
| wt (syn) | 0.67 ± 0.05 | 11.2 ± 1.4 | 59.7 ± 11 | 2.1 |
| R197Cit | 0.29 ± 0.01 | 13.7 ± 1.2 | 21.1 ± 2.7 | 5.9 (2.8) |
In the case of the wild-type enzyme made by native chemical ligation (SrtA syn), there was a clear decrease in activity compared with the fully recombinant wild-type enzyme (SrtA rec). kcat was decreased by 1.6-fold in SrtA syn, and Km was increased slightly, leading to an overall 2.1-fold decrease in kcat/Km for this enzyme. Native chemical ligation was performed under denaturing conditions (6 m guanidine hydrochloride), following which the protein was refolded, and therefore it is not surprising that these harsh conditions caused a loss of some activity in SrtA syn. For the mutant R197Cit, which was also generated by native chemical ligation, we therefore compared the kinetics of the mutant with SrtA syn, rather than SrtA rec, to control for the effects of the native chemical ligation reaction.
R197Cit was found to be active, and the kinetics were similar to those of SrtA syn. The mutant R197Cit had a small decrease in kcat (2.3-fold lower than that of SrtA syn), and a negligible increase in Km, for an overall 2.8-fold decrease in kcat/Km. These data argue against an electrostatic interaction between Arg-197 and the substrate. If an electrostatic interaction were important (either for substrate binding or for stabilizing the formation of an oxyanion-like transition state), then citrulline, which carries no formal positive charge, would be a poor substitute for arginine. Indeed, mutation of Arg-197 to lysine, which carries a positive charge, resulted in a 690-fold decrease in kcat/Km.
Citrulline, unlike lysine, accurately mimics the steric shape of arginine. It is also capable, like arginine, of forming hydrogen bonds via its terminal -NH2 group. Therefore the fact that the R197Cit mutant leads to such small decreases in SrtA activity is strong evidence for a hydrogen-bonding role for Arg-197. Given that the effect of the R197Cit mutant (as well as the other Arg-197 mutants) is predominately on kcat and not Km, the hydrogen bond is likely not important for the initial SrtA-LPXTG binding event. Rather, the Arg-197 hydrogen bond is more likely playing a role by orienting the substrate in such a manner as to bring the catalytic residues (Cys-184 and His-120) into proper alignment for substrate cleavage.
Comparative Structural Analysis of SrtAΔN59—Given the inconsistencies encountered between the existing x-ray crystal structures of SrtAΔN59 and the mutagenesis data obtained in this study and others, we turned to the original NMR structural model of SrtAΔN59 (PDB: 1IJA) (39). Although NMR structures are generally of lower resolution than x-ray crystal structures, they often take into better account the flexibility of surface loops. Also, the NMR model was obtained in the presence of 20 mm CaCl2, whereas none of the x-ray structures were solved in the presence of calcium (28, 39). Based on the above observations, we concluded that the NMR structure was a more appropriate model for the β6/β7 loop of SrtA.
To compare the differences between the NMR model and the available x-ray structures, we selected two structures for comparison, which are shown in Fig. 5. On the left is shown the A chain from the x-ray structure of the LPETG-bound SrtAΔN59 C184A mutant (PDB: 1T2W). For ease of comparison, we have modeled a cysteine back in place of Ala-184 in the active site. On the right is the NMR model of native SrtAΔN59, which was determined in the presence of Ca2+, but in the absence of a LPETG peptide. Overall, the NMR model of SrtA is similar to the x-ray structure. Both structures take on the canonical “sortase-like” β-barrel fold. The r.m.s.d. for α-carbons between the two structures is a somewhat high 2.1 Å. However, within the “catalytic core” of β-strands β1, β2, β4, β5, β6, β7, and β8, it falls to just 0.8 Å. The majority of the differences between the two structures lie in the conformation of surface-exposed loops. In particular, the β6/β7 loops differ greatly between the two models, with an α-carbon r.m.s.d. of 3.7 Å. Interestingly, in the NMR model the β6-β7 loop is shifted closer to the active site, where the α-carbon of residue Val-168 is 3.1 Å closer to the α-carbon of Cys-184 in the NMR model relative to the x-ray structure, and that of Leu-169 is 4.2 Å closer. The closer position of this loop leads to a more compact active site. For the NMR model, the active site cavity contains a volume of 456 Å3, compared with a cavity of 525 Å3 in the x-ray structure.
FIGURE 5.

Comparative structural analysis of SrtAΔN59. Top: the x-ray crystal structure of LPETG-bound SrtAΔN59 C184A (PDB: 1T2W, chain A) is shown alongside an NMR model of native, unliganded SrtAΔN59 (PDB: 1IJA). For ease of comparison, a cysteine has been modeled back in place of Ala-184 in the x-ray structure. Note the different backbone conformation of the β6/β7-loops in the two models. In the NMR model, the Cα atoms of Val-168 and Leu-169 are 3.1 Å and 4.2 Å closer to Cys-184 than in the x-ray structure, respectively. The LPETG peptide from the x-ray structure is shown in stick format, for reference. Bottom: space-filling models of the x-ray and NMR structures of SrtAΔN59, highlighting the smaller active site of the NMR model (456 Å3, compared with 525 Å3 in the x-ray structure).
In addition to the different position of the β6/β7 loop, the orientation of several key side chains differs in the NMR model. Most strikingly, the side chain of Gln-172, which pointed into the active site in the x-ray structure, is rotated up and away from the active site. Instead of being oriented to make a hydrogen bond to the backbone of the substrate LPETG peptide, it is now pointing out to solvent. Likewise, the orientation of Glu-171 is altered from the x-ray structure; in the x-ray structure it pointed away from the active site, but in the NMR model it points toward the active site and toward the putative calcium-binding site made up of residues Glu-105, Glu-108, Asp-112, and Asn-114 (29). In both of these cases, the position of the side chain in the NMR model is more consistent with the biochemical data than the position observed in the x-ray structure.
Another important difference between the two structures is the relative position of Arg-197. In the x-ray structure, it occupies a position far from the β6-β7 loop, and is located relatively near to the catalytic Cys-184. The distance from the η-nitrogen of Arg-197 to the γ-carbon of Val-168 is 10.3 Å, whereas the distance to the γ-sulfur of Cys-184 is only 8.1 Å. Arg-197 occupies an equivalent structural position in the NMR model. Because the β6/β7 loop is shifted closer to the active site in this structure, it puts the η-nitrogen of Arg-197 only 4.9 Å from the γ-carbon of Val-168, and it is now 10.0 Å from the γ-sulfur of Cys-184 (see Fig. 5). This relative repositioning places Arg-197 in closer proximity to Val-168 and Leu-169, two residues that have been shown to be important for substrate recognition.
Taken together, the NMR model is more consistent with the available biochemical data than the existing x-ray structure, at least concerning the positioning of the β6/β7 loop. This may reflect the fact that this loop is inherently flexible in SrtA (29, 39), and together it highlights some of the limitations of x-ray crystal structures for functional information.
DISCUSSION
Alanine-scanning mutagenesis is a powerful tool for exploring the roles of specific residues in binding and catalysis. In our study, mutation of the hydrophobic residues Val-168 and Leu-169 resulted in 5.5- and 93-fold decreases in kcat/Km, respectively. These correspond to increases in the free energy of the transition state (ΔΔG‡) of 1.1 and 2.8 kcal/mol. Although some debate exists over the exact strength of a hydrophobic interaction in a protein, a generally agreed upon value is ∼0.7 kcal per additional methylene group involved (46). In the case of a leucine to alanine mutation, the loss of three methylene groups would be expected to add 2.1 kcal/mol to the free energy, close to the value observed for the L169A mutant. Similarly, the V168A mutation would be predicted to increase the free energy by 1.4 kcal/mol, close to our calculated value of 1.1 kcal/mol. Thus, the effects of our mutations are consistent with a model were Val-168 and Leu-169 both make hydrophobic interactions with the leucine of the substrate LPXTG sequence.
An Expanded Model for Substrate Recognition by SrtA—Recent studies have shed light on the molecular basis for substrate recognition in sortases, but many questions have remained unanswered. Although the importance of the β6/β7 loop in LPXTG recognition has been established (29, 30), the exact mechanism by which it does so has not been shown, nor have the contributions of individual residues within the β6/β7 loop been investigated in a systematic way. Additionally, the role of Arg-197 in catalysis by SrtA has remained somewhat a mystery. A recent study was able to rule out a role for this residue in proton-transfer events and suggested a role for Arg-197 in stabilizing the formation of an oxyanion transition state (33); nonetheless, this study was unable to distinguish whether Arg-197 acts via an electrostatic interaction or a hydrogen bond interaction.
In the current study, we have used a combination of alanine-scanning mutagenesis and steady-state kinetic measurements to explore the roles of six individual residues within the β6/β7 loop. We have provided evidence that the LPXTG sorting signal of potential SrtA substrates is recognized primarily by hydrophobic interactions between residues Val-168 and Leu-169, which interact with the bulky, apolar Leu-Pro residues of the sorting signal. Although Val-168 and Leu-169 have previously been suggested as potential recognition sites, to our knowledge this study is the first to directly show evidence for the importance of these hydrophobic residues. Additionally, we have provided evidence that argues that residue Gln-172 does not make a critical hydrogen-bonding interaction with the substrate. Finally, we have used a native chemical ligation strategy to generate the semi-synthetic mutant R197Cit, and used this to provide evidence against the existence of an electrostatic interaction involving Arg-197 during substrate recognition.
Taken together, these results are consistent with an expanded model for LPXTG recognition and cleavage based on a reverse protonation catalytic mechanism (47). LPXTG-containing substrates are recognized primarily by van der Waals interactions between the bulky, hydrophobic Leu-Pro residues and the side chains of Val-168 and Leu-169. Meanwhile, the bound conformation is stabilized via a hydrogen bond from Arg-197. Although the exact position of this hydrogen bond cannot be determined from our results, one possible site would be the backbone carbonyl between the proline and the X position of the LPXTG motif. Alternatively, Arg-197 could hydrogen bond with the carbonyl of the scissile Thr-Gly bond and thus stabilize the oxyanion transition state of the enzyme. It remains unclear whether this hydrogen bond is utilized by the enzyme solely to position the substrate within the active site, or if it also acts to facilitate a distortion of the geometry of the peptide substrate in preparation for catalysis. Regardless, these interactions collectively combine to stabilize a specific LPXTG-bound conformation of the substrate, very likely in an activated form for catalysis.
Once bound, cleavage of the substrate is achieved through a reverse protonation mechanism as described previously (47). Nucleophilic attack by the Cys-184 thiolate generates a tetrahedral intermediate. The His-120 imidazolium then protonates the leaving group (glycine) nitrogen of the substrate, leading to formation of a covalent acyl-enzyme thioester and an unprotonated His-120 imidazole. In a minimalist view, this His-120 imidazole (or an as yet unidentified basic residue) acts to deprotonate the incoming N-terminal amine of the Gly5 chain of branched lipid II. This primary amine then serves as a nucleophile and attacks the acyl-enzyme thioester, forming a second tetrahedral intermediate. Protonation of this intermediate would then produce the transpeptidation product and regenerate the active site of SrtA for another round of catalysis.
With the data from this study, we are able to add to our understanding of how SrtA discriminates LPXTG substrates from other sequences. However, we still do not fully understand all of the elements of substrate recognition. In particular, it remains to be discovered how the pentaglycine substrate lipid II binds to SrtA, and what role (if any) the carbohydrate or anionic pyrophosphate residues of lipid II play in facilitating binding and transpeptidation. We do not yet clearly understand the exact pattern of hydrogen bonds that stabilize the bound conformation of LPXTG prior to acylation by Cys-184, nor do we yet have a full view of the dynamics of SrtA as the enzyme transitions between substrate recognition, chemical catalysis, and product release. Additional structural and mechanistic studies are ongoing, and will increase our understanding of molecular recognition within the sortase family of transpeptidases.
Acknowledgments
We thank Prof. Terrence Oas and Yu-Chu Chang for assistance with CD experiments and Dr. Dawn Schmidt for critical evaluation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant AI46611. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: SrtA, sortase A; Fmoc, N-(9-fluorenyl)methyloxycarbonyl; Abz, aminobenzoic acid; Dap, diaminopropionic acid; DNP, dinitrophenol; HPLC, high pressure liquid chromatography; MALDI-TOF, matrix-assisted laser desorption ionization time of flight; MESNA, sodium 2-mercaptoethanesulfonate; MPAA, 4-mercaptophenylacetic acid; NMWL, nominal molecular weight limit; r.m.s.d., root mean square deviation.
References
- 1.Klevens, R. M., Morrison, M. A., Fridkin, S. K., Reingold, A., Petit, S., Gershman, K., Ray, S., Harrison, L. H., Lynfield, R., Dumyati, G., Townes, J. M., Craig, A. S., Fosheim, G., McDougal, L. K., and Tenover, F. C. (2006) Emerg. Infect. Dis. 12 1991-1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klevens, R. M., Morrison, M. A., Nadle, J., Petit, S., Gershman, K., Ray, S., Harrison, L. H., Lynfield, R., Dumyati, G., Townes, J. M., Craig, A. S., Zell, E. R., Fosheim, G. E., McDougal, L. K., Carey, R. B., and Fridkin, S. K. (2007) J. Am. Med. Assoc. 298 1763-1771 [DOI] [PubMed] [Google Scholar]
- 3.Appelbaum, P. C. (2007) Int. J. Antimicrob. Agents 30 398-408 [DOI] [PubMed] [Google Scholar]
- 4.Appelbaum, P. C. (2006) Clin. Microbiol. Infect. 12 Suppl. 2, 3-10 [DOI] [PubMed] [Google Scholar]
- 5.Appelbaum, P. C. (2006) Clin. Microbiol. Infect. 12 Suppl. 1, 16-23 [DOI] [PubMed] [Google Scholar]
- 6.Marraffini, L. A., Dedent, A. C., and Schneewind, O. (2006) Microbiol. Mol. Biol. Rev. 70 192-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maresso, A. W., and Schneewind, O. (2006) Biometals 19 193-203 [DOI] [PubMed] [Google Scholar]
- 8.Alksne, L. E., and Projan, S. J. (2000) Curr. Opin. Biotechnol. 11 625-636 [DOI] [PubMed] [Google Scholar]
- 9.Clatworthy, A. E., Pierson, E., and Hung, D. T. (2007) Nat. Chem. Biol. 3 541-548 [DOI] [PubMed] [Google Scholar]
- 10.Mazmanian, S. K., Ton-That, H., and Schneewind, O. (2001) Mol. Microbiol. 40 1049-1057 [DOI] [PubMed] [Google Scholar]
- 11.Mazmanian, S. K., Liu, G., Jensen, E. R., Lenoy, E., and Schneewind, O. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 5510-5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garandeau, C., Reglier-Poupet, H., Dubail, I., Beretti, J. L., Berche, P., and Charbit, A. (2002) Infect. Immun. 70 1382-1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bierne, H., Mazmanian, S. K., Trost, M., Pucciarelli, M. G., Liu, G., Dehoux, P., Jansch, L., Garcia-del Portillo, F., Schneewind, O., and Cossart, P. (2002) Mol. Microbiol. 43 869-881 [DOI] [PubMed] [Google Scholar]
- 14.Bolken, T. C., Franke, C. A., Jones, K. F., Zeller, G. O., Jones, C. H., Dutton, E. K., and Hruby, D. E. (2001) Infect. Immun. 69 75-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paterson, G. K., and Mitchell, T. J. (2006) Microbes Infect. 8 145-153 [DOI] [PubMed] [Google Scholar]
- 16.Barnett, T. C., and Scott, J. R. (2002) J. Bacteriol. 184 2181-2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee, S. F., and Boran, T. L. (2003) Infect. Immun. 71 676-681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osaki, M., Takamatsu, D., Shimoji, Y., and Sekizaki, T. (2002) J. Bacteriol. 184 971-982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaguchi, M., Terao, Y., Ogawa, T., Takahashi, T., Hamada, S., and Kawabata, S. (2006) Microbes Infect. 8 2791-2796 [DOI] [PubMed] [Google Scholar]
- 20.Lalioui, L., Pellegrini, E., Dramsi, S., Baptista, M., Bourgeois, N., Doucet-Populaire, F., Rusniok, C., Zouine, M., Glaser, P., Kunst, F., Poyart, C., and Trieu-Cuot, P. (2005) Infect. Immun. 73 3342-3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischetti, V. A., Pancholi, V., and Schneewind, O. (1990) Mol. Microbiol. 4 1603-1605 [DOI] [PubMed] [Google Scholar]
- 22.Schneewind, O., Model, P., and Fischetti, V. A. (1992) Cell 70 267-281 [DOI] [PubMed] [Google Scholar]
- 23.Ton-That, H., Mazmanian, S. K., Alksne, L., and Schneewind, O. (2002) J. Biol. Chem. 277 7447-7452 [DOI] [PubMed] [Google Scholar]
- 24.Ruzin, A., Severin, A., Ritacco, F., Tabei, K., Singh, G., Bradford, P. A., Siegel, M. M., Projan, S. J., and Shlaes, D. M. (2002) J. Bacteriol. 184 2141-2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry, A. M., Ton-That, H., Mazmanian, S. K., and Schneewind, O. (2002) J. Biol. Chem. 277 16241-16248 [DOI] [PubMed] [Google Scholar]
- 26.Mazmanian, S. K., Ton-That, H., Su, K., and Schneewind, O. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 2293-2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruger, R. G., Otvos, B., Frankel, B. A., Bentley, M., Dostal, P., and McCafferty, D. G. (2004) Biochemistry 43 1541-1551 [DOI] [PubMed] [Google Scholar]
- 28.Zong, Y., Bice, T. W., Ton-That, H., Schneewind, O., and Narayana, S. V. (2004) J. Biol. Chem. 279 31383-31389 [DOI] [PubMed] [Google Scholar]
- 29.Naik, M. T., Suree, N., Ilangovan, U., Liew, C. K., Thieu, W., Campbell, D. O., Clemens, J. J., Jung, M. E., and Clubb, R. T. (2006) J. Biol. Chem. 281 1817-1826 [DOI] [PubMed] [Google Scholar]
- 30.Bentley, M. L., Gaweska, H., Kielec, J. M., and McCafferty, D. G. (2007) J. Biol. Chem [DOI] [PubMed]
- 31.Marraffini, L. A., Ton-That, H., Zong, Y., Narayana, S. V., and Schneewind, O. (2004) J. Biol. Chem. 279 37763-37770 [DOI] [PubMed] [Google Scholar]
- 32.Liew, C. K., Smith, B. T., Pilpa, R., Suree, N., Ilangovan, U., Connolly, K. M., Jung, M. E., and Clubb, R. T. (2004) FEBS Lett. 571 221-226 [DOI] [PubMed] [Google Scholar]
- 33.Frankel, B. A., Tong, Y., Bentley, M. L., Fitzgerald, M. C., and McCafferty, D. G. (2007) Biochemistry 46 7269-7278 [DOI] [PubMed] [Google Scholar]
- 34.Dawson, P. E., Muir, T. W., Clark-Lewis, I., and Kent, S. B. (1994) Science 266 776-779 [DOI] [PubMed] [Google Scholar]
- 35.Hackeng, T. M., Mounier, C. M., Bon, C., Dawson, P. E., Griffin, J. H., and Kent, S. B. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 7845-7850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson, P. E. (1997) Methods Enzymol. 287 34-45 [DOI] [PubMed] [Google Scholar]
- 37.Kruger, R. G., Dostal, P., and McCafferty, D. G. (2004) Anal. Biochem. 326 42-48 [DOI] [PubMed] [Google Scholar]
- 38.Pritz, S., Wolf, Y., Kraetke, O., Klose, J., Bienert, M., and Beyermann, M. (2007) J. Org. Chem. 72 3909-3912 [DOI] [PubMed] [Google Scholar]
- 39.Ilangovan, U., Ton-That, H., Iwahara, J., Schneewind, O., and Clubb, R. T. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 6056-6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai, Y. C., and Johnson, K. A. (2006) Biochemistry 45 9675-9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Metanis, N., Brik, A., Dawson, P. E., and Keinan, E. (2004) J. Am. Chem. Soc. 126 12726-12727 [DOI] [PubMed] [Google Scholar]
- 42.Metanis, N., Keinan, E., and Dawson, P. E. (2005) J. Am. Chem. Soc. 127 5862-5868 [DOI] [PubMed] [Google Scholar]
- 43.Jantz, D., and Berg, J. M. (2003) J. Am. Chem. Soc. 125 4960-4961 [DOI] [PubMed] [Google Scholar]
- 44.Kienhofer, A., Kast, P., and Hilvert, D. (2003) J. Am. Chem. Soc. 125 3206-3207 [DOI] [PubMed] [Google Scholar]
- 45.Imparl, J. M., Senshu, T., and Graves, D. J. (1995) Arch. Biochem. Biophys. 318 370-377 [DOI] [PubMed] [Google Scholar]
- 46.Tanford, C. (1962) J. Am. Chem. Soc. 84 4240-4247 [Google Scholar]
- 47.Frankel, B. A., Kruger, R. G., Robinson, D. E., Kelleher, N. L., and McCafferty, D. G. (2005) Biochemistry 44 11188-11200 [DOI] [PubMed] [Google Scholar]