

Abstract
Aggressive and infiltrative invasion is one of the hallmarks of glioblastoma. Low-density lipoprotein receptor–related protein (LRP) is expressed by glioblastoma, but the role of this receptor in astrocytic tumor invasion remains poorly understood. We show that activation of protein kinase C-α (PKC-α) phosphorylated and down-regulated LRP expression. Pretreatment of tumor cells with PKC inhibitors, phosphoinositide 3-kinase (PI3K) inhibitor, PKC-α small interfering RNA (siRNA), and short hairpin RNA abrogated phorbol 12-myristate 13-acetate–induced down-regulation of LRP and inhibited astrocytic tumor invasion in vitro. In xenograft glioblastoma mouse model and in vitro transmembrane invasion assay, LRP-deficient cells, which secreted high levels of urokinase-type plasminogen activator (uPA), invaded extensively the surrounding normal brain tissue, whereas the LRP-overexpressing and uPA-deficient cells did not invade into the surrounding normal brain. siRNA, targeted against uPA in LRP-deficient clones, attenuated their invasive potential. Taken together, our results strongly suggest the involvement of PKC-α/PI3K signaling pathways in the regulation of LRP-mediated astrocytoma invasion. Thus, a strategy of combining small molecule inhibitors of PKC-α and PI3K could provide a new treatment paradigm for glioblastomas.
Introduction
Glioblastoma multiforme are the most malignant astrocytomas, which are characterized by infiltrative growth and are resistant to conventional therapy (1). Low-density lipoprotein receptor–related protein (LRP) is a member of the LDL receptor superfamily and is expressed in brain and peripheral neurons (2–8), which is regulated both in vivo and in vitro (9, 10). This endocytic receptor is made up of a 515-kDa α-chain and an 85-kDa transmembrane β-chain (11, 12). LRP binds, internalizes a diverse array of ligands (13–16), and plays a role in signal transduction pathways that lead to cell proliferation, migration, vesicle trafficking, and cell adhesion (12, 17–22). However, the role of LRP in these pathways is not completely understood.
Glioblastomas have higher levels of protein kinase C (PKC) than nonneoplastic astrocytes (23–25). PKC is a family of serine/threonine phospholipid-dependent kinases that are mediators of cell proliferation, differentiation, apoptosis, motility, and adhesion (26, 27). The PKC family is composed of 11 isozymes that are divided into three groups based on their structures and cofactor requirements (28). The conventional PKC isoforms (PKC-α, PKC-βI/II, and PKC-γ) are activated by diacylglycerol (DAG) and phorbol esters, and they require Ca2+ for activation. The novel PKC isoforms (PKC-δ, PKC-ε, PKC-η, and PKC-θ) are activated by DAG or phorbol esters. The atypical PKC isoforms (PKC-ζ and PKC-ι/PKC-λ) are not activated by DAG or phorbol esters (29, 30).
The mechanism of PKC regulation of LRP-mediated astrocytic tumor invasion has not been previously reported. In this study, we examined the role of PKC-regulated LRP in astrocytic tumor invasion. Our data reveal that activation of PKC-α phosphorylated and down-regulated LRP, which was blocked by PKC inhibitors, small interfering RNA (siRNA), and short hairpin RNA (shRNA). Phosphoinositide 3-kinase (PI3K) inhibitor (LY 294002) also blocked phorbol 12-myristate 13-acetate (PMA)–induced down-regulation of LRP. The reduced LRP level leads to increased urokinase-type plasminogen activator (uPA) secretion. These cascades of events work in concert to drive tumor invasion in vitro and in xenograft glioblastoma mouse model, and LRP-deficient cells, which secrete high levels of uPA, extensively invaded the surrounding normal brain tissue. In contrast, uPA-deficient and LRP-overexpressing cells were less invasive. Taken together, the results strongly suggest the involvement of PKC-α/PI3K signaling pathways in the regulation of LRP-mediated astrocytoma invasion.
Materials and Methods
Antibodies and Reagents
PMA, antitubulin antibody, and type IV collagen were purchased from Sigma. Monoclonal antibody (mAb) 11H4 was purified from ascitic fluid after inoculation of hybridoma cells obtained from American Type Culture Collection (ATCC). Antibody 11H4 recognizes LRP (85 kDa) light chain (31). The full-length LRP cDNA was a generous gift from Dr. D.K. Strickland (University of Maryland). uPA-specific mAb was purchased from American Diagnostica. The specific PKC inhibitors, Gö 6976, bisindolylmaleimide, mitogen-activated protein (MAP)/ERK kinase (MEK) inhibitor (UO 126), PEA, and PI3K inhibitor LY294002 are products of Calbiochem. Anti– PKC-α, anti–PKC-β, and anti–PKC-δ antibodies were purchased from Santa Cruz Biotechnology, and phosphorylated serine-specific antibody was from Biomol. PKC-α and uPA siRNA were purchased from Dharmacon, Inc. PKC-α kinase dead constructs were a generous gift from Dr. Jae-Won Soh (Department of Chemistry, Inha University). shRNA PKC-α constructs were purchased from Sigma. The uPA inhibitor B428 was a generous gift from Dr. Galina Kuznetsor at Eisai Research Institute.
Cell Cultures and Human Samples
Human U-1242 MG cell line was kindly supplied by Dr. A.J. Yates (Ohio State University), whereas U-87 MG was obtained from ATCC. The normal human astrocytes (NHA) were obtained from Clonetics. The cell lines were originally isolated from astrocytic tumors that were designated as glioblastomas, and their characteristics were described previously by Hussaini et al. (32). All the glioblastoma samples were flash frozen in liquid nitrogen after surgical removal and then stored at −80°C until protein extraction.
Western Blot Analysis
Western blot analysis was done as previously described (33). Briefly, cell cultures were extracted with 1% Triton X-100, 50 mmol/L Tris (pH 7.5), and 150 nmol/L NaCl in the presence of 2 mmol/L EDTA, 100 μmol/L phenylmethylsulfonyl fluoride, 5 μg/mL leupeptin, and 1 Ag/mL aprotinin. The extracts were subjected to SDS-PAGE on 10% polyacrylamide slabs and then subjected to Western blotting as previously described.
Coimmunoprecipitation and Immunoblot Analysis
Immunoprecipitation studies were carried out as previously described (34). Cells were treated with either PMA in the presence or absence of pharmacologic inhibitors. Cell cultures were extracted with 1% Triton X-100. Proteins were quantitated using bicinchoninic acid assay. Cell lysate (1 mg) was incubated with LRP primary antibody overnight at 4°C. Immune complexes were collected with protein G beads and washed five times with immunoprecipitation washing buffer. The resulting immunoprecipitate was then resolved by SDS-PAGE on 10% polyacrylamide gels and then electrophoretically transferred into nitrocellulose. The immunoblot was probed with either anti-LRP or PKC-α antibody. Densitometry analysis and ImageQuant software were used to quantitate the protein bands.
Immunofluorescence and Confocal Microscopy
U-1242 MG and U-87 MG cells were plated at a density of 1 × 105 per coverslip. The cells were serum starved overnight and treated with PMA for 6 h. The cells were washed thrice with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. The cells were then permeabilized for 5 min with 0.2% Triton X-100 in PBS. Nonspecific binding was blocked by incubating cells with 3% bovine serum albumin in PBS for 1 h at room temperature. Primary antibody to LRP was diluted in blocking buffer, incubated for 1 h, and then washed off with PBS. The cells were incubated with Alexa Flour 594–conjugated goat anti-mouse IgG secondary antibody for 1 h, washed thrice, and then mounted on a slide. Cell images on the effects of PMA on LRP expression were captured using Olympus Fluoview 300 laser confocal microscopy. PKC-α and LRP colocalization was also determined after treatment with PMA.
Transfection of siRNA,shRNA PKC-α, and PKC-α KR Constructs into Glioblastoma Cells
siRNA transfections
siRNA (400 nmol/L) directed against PKC-α was transfected into both U-1242 MG and U-87 MG cells using the Amaxa Nucleofector (Amaxa) as previously reported (34). Similarly, 200 nmol/L of siRNA directed against uPA were transfected into the LRP-deficient clones (pBK-CMVLα42 and pBK-CMVLα47). The PKC-α kinase dead construct was transiently transfected into astrocytic tumor cell line with LipofectAMINE 2000 transfection reagent (Invitrogen) using 5 μg of plasmid according to the manufacturer's instructions. Lentiviral constructs of shRNA obtained from Sigma were transfected into glioblastoma cell lines according to the manufacturer's instruction. After 14 days of culture with puromycin, single-cell cloning was initiated, and clones negative for PKC-α were selected as positive. Nontargeting shRNA was used as control.
Stable Expression of LRP and uPA Antisense in Glioblastoma Cells
LRP antisense RNA expression constructs were designed for stable integration and constitutive RNA synthesis. These constructs and LRP-deficient clones were previously reported (33). For uPA antisense, uPA cDNA served as the starting template. A 637-bp restriction fragment of uPA fragment of uPA cDNA (bp 727−1,364; Genbank accession no. K02286) was excised with EcoRI. This fragment was ligated in reverse orientation into a multiple cloning site of the eukaryotic expression vector pBK-CMV. The SV40 3′ splice site and polyadenylation signal in pBK-CMV-uPA were left intact. U-1242 MG cells were transfected with 3 μg/mL of pBK-CMV-uPA or with empty vector (pBK-CMVthat did not contain a cDNA insert). U-1242 LRP-overexpressing cells were generated by transfecting full-length cDNA of LRP into the U-1242 parent cell (LRP C1 and C8). These cells were incubated for 6 h with LipofectAMINE (4 μL/mL in serum-free MEM-α). The cells were then washed twice with serum-free MEM-α and cultured in 10% fetal bovine serum–supplemented medium containing geneticin (G-418, Life Technologies-Invitrogen Corporation) at a concentration of 300 μg/mL, and single-cell cloning was initiated; positive clones were used for the experiments.
Cell Invasion Assay
Invasion was determined by the modified Boyden chamber assay (35) with an 8-μm pore size polycarbonate filter (Becton Dickinson) coated with type IV collagen (Sigma). Prepared cell suspension (300 μL; 0.5 × 106 cells per mL) in serum-free MEM-α was added to the upper compartment of each insert in the presence or absence of PKC inhibitor (Gö 6976) or siRNA directed against PKC-α. In another set of experiments, PKC-α stable knockdown clones were also used to investigate the role of PKC-α in mediating LRP-mediated cell invasion. These cells were treated with PMA for 6 h. A similar experiment was done with LRP-deficient clones using 200 nmol/L siRNA against uPA. After 6 h of incubation, the filters were fixed and stained with 0.1% crystal violet solution. The invaded cells that passed through the filter to the lower surface of the membrane were photographed with an QImaging RETIGA EXi digital camera (Canada) under a LEICA DMIRE 2 microscope. The invaded cells were counted by at least three high-power fields. Each sample was assayed in triplicate, and assays were repeated at least twice. Quantification of the invasion assay was done as described previously (36).
Xenograft Mouse Model
Adult male NOD SCID mice were purchased from The Jackson Laboratory. All animal studies were conducted at the Animal Research Core Facility at the University of Virginia School of Medicine in accordance with the institutional guidelines. The U-1242 MG luciferase-GFP cell line was prepared by transduction of parental U-1242 MG cells with lentivirus expressing both luciferase and GFP. Cells expressing GFP were sorted using the fluorescence-activated cell sorter (FACS) and grown on a three-dimensional gelfoam coated with Matrigel (BD Biosciences). Animals used for this study were anesthetized with ketamine (17.4 mg/20 g), xylazine (2.6 mg/20 g), and acepromazine and placed on a sterotactic frame. Tumor cell lines grown on gelfoam (4 × 105 cells) were then implanted into mice in their right striatum at these coordinates from the bregma: 1 mm anterior, 2 mm lateral and 4.5 mm intraparenchymal. After tumor cell implantation, mice were removed from the stereotactic apparatus, kept in separate cages, checked for signs and symptoms of neurologic deficits (seizures/ hemiparesis), and then sacrificed if positive. Animals were imaged with both bioluminescence imaging (BLI) and magnetic resonance imaging (MRI).
BLI and MRI
Two weeks after tumor implantation, mice were imaged with the IVIS 100 System (Xenogen Corporation) to record bioluminescent signal emitted from the engrafted tumor in the brain. The acquisition of emitted light from the tumor was through the IVIS 100 cooled CCD camera systems. Animals received i.p. injection of D-luciferin (Xenogen) at a dose of 330 μg/g body weight after a sufficient depth of anesthesia with xylazine/ketamine/acepromazine. Bioluminescent signals were collected for a period of 5 to 25 min after substrate injection. The average number of photons was analyzed using the Living Image Version 2.5 software.
MRI studies were done on a 4.7-T imaging system (Varian NMR System, Inc.). This system consists three-axis self-shielded magnetic field gradient, with 30 G/cm maximum gradient amplitude in all three channels. Under anesthesia by inhalation of isoflurane (1% or 2%), all animals were given i.p. injection of an MRI contrast agent, gadolinium diethylenetriamine penta-acetic acid (Magnevist; Berlex Laboratory), at a dose of 1.2 μL/g body weight and placed in a radio frequency coil with an inner diameter of 35 mm. The transverse T1-weighed images and T2-weighed images were acquired on the whole mouse brain, with a conventional spin-echo pulse sequence and a fast spin-echo sequence, respectively. The pulse repetition times and echo times were 650 and 14.54 m/s for T1-weighed images and 2,000 and 40 m/s for T2-weighed images. Other variables used were a 2.56 × 2.56 cm field of view, a 128 × 96 matrix size, in six averages, resulting in a total scan time of ∼5 min. For MRI data analysis, the coronal gadolinium-enhanced sequences were segmented, and tumor volume was calculated using the NIH image software.
Statistical Analysis
Each experiment and assay were done at least thrice. A representative assay or experiment is shown for each figure. Statistical differences for multiple comparison were determined using ANOVA followed by Dunnet's test. Data represent a mean ± SE. A P value of ≤0.05 was considered significant.
Results
Differential expression of LRP patient specimen and glioblastoma cell lines
In this study, we examined the pattern of expression of LRP protein in pilocytic astrocytoma, in glioblastoma grade IV and four well-characterized glioblastoma cell lines (U-1242 MG, U-251 MG, U-373 MG, and U-87 MG), and in NHA. Low-grade astrocytomas (pilocytic) expressed higher LRP levels, whereas the high-grade astrocytoma (glioblastoma) expressed reduced levels of LRP (Fig. 1A). This finding suggests an inverse correlation between LRP expression and astrocytoma grade. Similarly, in glioblastoma cell lines, LRP expression was reduced when compared with NHA. U-87 MG had the next highest LRP expression, followed by U-1242 MG, U-251 MG, and U-373 MG, which had the least level of LRP (Fig. 1B). In addition, we investigated the expression levels of uPA and PKC-α in patient tumor samples and in our glioma cell lines. The expressions of uPA and PKC-α were higher in patient tumor samples (Fig. 1C) and the glioblastoma cell lines (Fig. 1D) compared with NHA.
Figure 1.
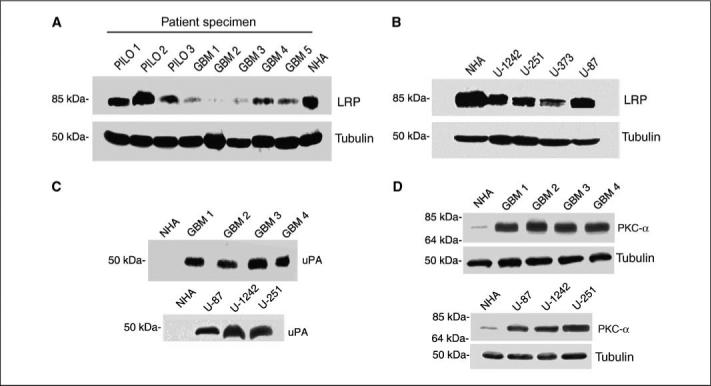
LRP, PKC-α, and uPA expression profile in astrocytoma. A, equivalent amounts (200 μgper lane) of protein were subjected to 10% SDS-PAGE and blotted with anti-LRP antibody (11H4) in patients' specimen. B, LRP expression levels detected by Western blot analysis in glioma cell lines. C, uPA expression levels in tumor samples obtained from patients and glioblastoma cell lines. D, PKC-α expression profile in patient samples and glioma cell lines.
PKC activation down-regulates LRP expression in glioblastoma cells
In addition to tyrosine phosphorylation (37), LRP cytoplasmic domain has been shown to be phosphorylated at serine residues by protein kinase A (38). We then asked the question, whether PMA, a potent activator of PKCs, can induce serine phosphorylation of LRP in glioblastomas cells. Cell lysates were immunoprecipitated with anti-LRP antibody and then probed with phosphorylated serine antibody. Treatment with PMA (100 nmol/L) for 10 and 30 min induced a robust serine phosphorylation of the LRP β-chain (Fig. 2A).
Figure 2.
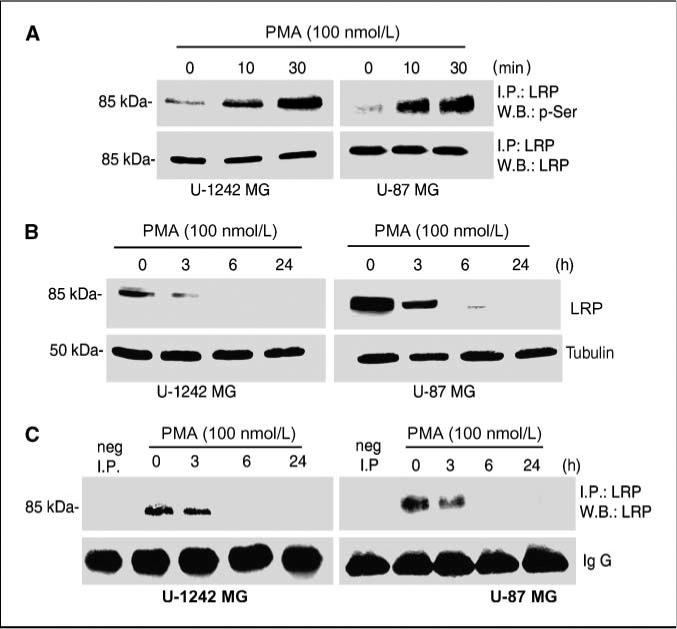
PMA regulates the expression of LRP. A, cells were serum starved after reaching 90% confluence by replacing the media with serum-free MEM-α. The cells were treated with PMA (100 nmol/L) at 10 and 30 min. Cell lysates were immunoprecipitated (I.P.) with anti-LRP and immunoblotted with phosphorylated serine antibody. Blots were stripped and reprobed for LRP. B, PMA down-regulates the expression of LRP. Cells were serum starved after reaching90% confluence by replacing the media with serum-free MEM-α. The cells were treated with PMA (100 nmol/L) at 3, 6, and 24 h. Cells were lysed using 1% Triton X-100 lysis buffer and subjected to Western blotting. The immunoblots were probed for LRP with anti-LRP antibody (11H4). Loadingcontrol was checked usingtubulin. C, cell lysates from the treatments cited above were immunoprecipitated with anti-LRP antibody usingprotein G beads. The immunoblots were probed for LRP.
LRP expression is down-regulated by epidermal growth factor in glioblastoma cells (33). In this study, we investigated the effects of PKC activation on the expression level of LRP. We used U-1242 MG and U-87 MG to determine the level of LRP total protein expression after PKC activation with PMA. The phorbol ester evoked a time-dependent down-regulation of LRP expression (Fig. 2B). The receptor was maximally down-regulated at 6 h. Immunoprecipitation study with anti-LRP antibody (11H4) agrees and confirms the Western blot analysis data (Fig. 2C). To further verify the effect of PMA on LRP expression in intact glioblastoma cell lines, laser confocal microscopy was done, and PMA down-regulated LRP in U-1242 (data not shown).
Inhibition of classic PKC isozymes and PI3K blocks LRP down-regulation
Phorbol esters interact with and activate both conventional and novel PKC isoforms; bisindolylmaleimide inhibits the activation of these PKCs, whereas Gö 6976 blocks the activities of the two classic PKCs (PKC-α and PKC-β). To determine whether classic or novel PKC isozymes are involved in LRP regulation, glioblastomas cell lines were treated with either Gö 6976 (10 μmol/L) or bisindolylmaleimide (1 μmol/L) for 60 min before PMA addition to cultures for 6 h. Both bisindolylmaleimide and Gö 6976 attenuated the down-regulation of LRP induced by PMA to basal level (Fig. 3A). Because Gö 6976 inhibits only PKC-α and PKC-βI/II, we used hispidin (10 μmol/L), a PKC-β–specific inhibitor (39). Cell cultures were treated with hispidin (10 μmol/L) for 60 min before the addition of PMA. Hispidin did not alter PMA-induced down-regulation of LRP, suggesting that PKC-β may not be the isoform mediating the effects of PMA on LRP expression (Fig. 3A).
Figure 3.
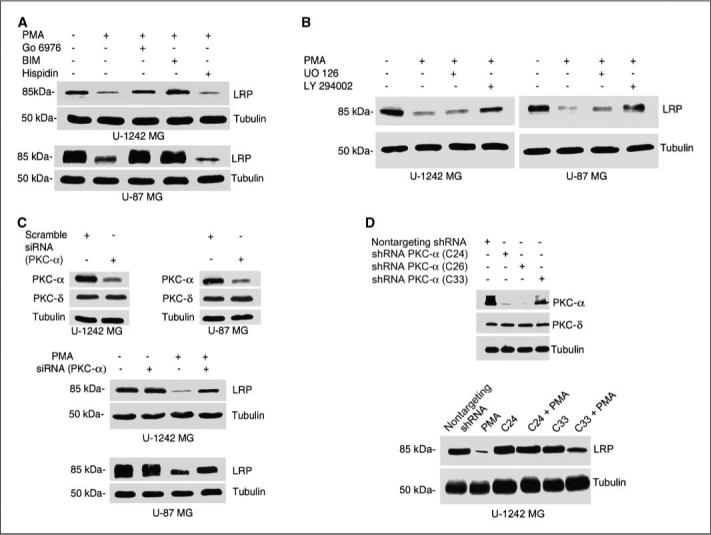
Effect of pharmacologic inhibitors on LRP expression. A, effects of Gö 6976 (a classic PKC inhibitor), bisindolylmaleimide (BIM; an inhibitor of both classic and novel PKC), and hispidin (PKC-β inhibitor) on the expression level of LRP. Serum-starved cells were treated with PKC inhibitors for 60 min, after which the cells were treated for 6 h with PMA (100 nmol/L). After washingwith ice-cold PBS, cells were solubilized in 1% Triton X-100 lysis buffer, analyzed, and immunoblotted with anti-LRP antibody. Blots were stripped and reprobed with tubulin for loading control. B, role of PI3K in PMA-induced LRP down-regulation. Serum-starved cells were treated with UO 126 (a MEK inhibitor) and LY 294002 (a PI3K inhibitor) for 60 min and then treated with PMA for 6 h. Cell lysates were electrophoresed on a 10% gel and then immunoblotted and probed for LRP. Membrane was stripped and reprobed for tubulin as loading control. C, RNA interference with siRNA against PKC-α was done as described in Materials and Methods. Cells were transiently transfected with PKC-α siRNA and treated with or without PMA for 6 h. Cells were then lysed, and cell lysates with equal concentration (200 μg per lane) were separated on 10% SDS-polyacrylamide gels and immunoblotted for LRP and tubulin. D, Western blot analysis of PKC-α shRNA knockdown glioblastoma clones. PKC-α–deficient clones abrogated PMA-induced down-regulation of LRP.
PKC activation can lead to increased phosphorylation of MAP kinase (MAPK) (32) and stimulation of the PI3K pathway (40). We investigated the involvement of these pathways in LRP regulation by PMA. Glioblastoma cells were cultured in the absence (control) or presence of the MEK inhibitor (UO 126; 10 μmol/L) or the PI3K inhibitor (LY 294002; 10 μmol/L) for 60 min, before the addition of PMA to cultures for 6 h. Cell lysates were subjected to Western blot analysis. The result shows that LY294002 inhibited PMA-induced down-regulation of LRP expression, whereas the MEK inhibitor UO 126 had no effect on the LRP regulation by PMA (Fig. 3B), suggesting that PI3K pathways may be positively involved in regulating the expression of LRP in astrocytic tumor cells. In another set of experiments, we examined whether the combination of a PKC-α inhibitor and PI3K inhibitor would be more effective than either agent alone in reversing the down-regulation of LRP. As expected, treatment with two submaximal concentrations of Gö 6976 (2.5 and 5 μmol/L) and LY294002 (2.5 and 5 μmol/L) reversed PMA-induced down-regulation of LRP expression in astrocytic tumor cell lines (data not shown).
Because pharmacologic inhibitors are not that specific (41), we designed experiments to investigate the critical role of PKC-α in mediating the down-regulation of LRP expression using siRNA silencing strategy. We first optimized the minimal siRNA PKC-α concentration that would produce the maximal silencing of the classic PKC isozyme, which was 400 nmol/L. PKC-α siRNA (400 nmol/L) was transfected into astrocytic tumor cells for 48 h, which resulted in 60% knockdown of PKC-α level in both cell lines compared with control cells transfected with nontargeting siRNA (Fig. 3C). PKC-α siRNA (400 nmol/L) was transfected into glioblastoma cell lines and treated with PMA (100 nmol/L) for 6 h. Gene silencing with the PKC-α siRNA attenuated the PMA-induced down-regulation of LRP expression (Fig. 3C).
Next, we used two different but complimentary approaches to directly investigate the role of PKC-α in mediating LRP regulation: (a) we used the kinase dead PKC-α (KR) construct, and (b) we developed stable clones of PKC-α–deficient cells by transfecting U-1242 cells with shRNA directed against PKC-α. Single cloning was initiated, and clones not expressing PKC-α were selected as positive (Fig. 3D) and used for these experiments. The kinase dead constructs of PKC-α (HA tagged) were transfected using Lipofect-AMINE 2000 according to the manufacturer's instructions. The PKC-α KR abrogated PMA-induced down-regulation of LRP (data not shown). Similarly, stable PKC-α–deficient clones (C24 and C26) generated by shRNA lentivirus infection were treated with PMA for 6 h. In clone C24, PMA failed to down-regulate LRP expression, whereas the level of the receptor in the partial knockdown clone (C33) was reduced by the phorbol esters (Fig. 3D). Taking these results together, our data suggest that PKC-α could be the putative PKC isozyme, mediating the effects of phorbol esters on LRP regulation.
PKC-α coimmunoprecipitates with LRP
The studies involving PKC inhibitors, PKC-α KR, siRNA, and shRNA, targeted against PKC-α, as reported above, suggest that the activation of classic PKC isozymes, especially PKC-α, may be mediating the down-regulation of LRP expression after PMA treatment. We next determined whether PKC-α isozymes would associate with LRP. For these studies, U-1242 MG and U-87 MG cells were cultured in the absence (control) or presence of PMA (100 nmol/L) at different time points. LRP was immunoprecipitated with anti-LRP antibody (11H4). The immunoprecipitates were subjected to Western blot analysis with anti–PKC-α and anti–PKC-βI/II antibodies. Results in Fig. 4A shows that only PKC-α, and not PKC-β, coimmunoprecipitated with LRP. To confirm these results, we carried out a reverse immunoprecipitation studies with anti–PKC-α antibody. The immunoprecipitates were immunoblotted with anti-LRP antibody. LRP coimmunoprecipitated with PKC-α (Fig. 4B). These data further suggest that PKC-α directly or indirectly associates with LRP in glioblastomas cells after treatment with PMA. Given our observation that PKC-α associates with LRP and that activation of PKC-α down-regulates the expression of LRP, we examined whether PKC-α can colocalize with LRP in intact cells using laser confocal microscopy. The red color indicates LRP expression, whereas the green color is for PKC-α staining. The merged image shows a color shift to orange yellow (Fig. 4C). This indicates that PKC-α colocalized with LRP and thus suggests a possible interaction between PKC-α and LRP in glioblastoma cell lines.
Figure 4.
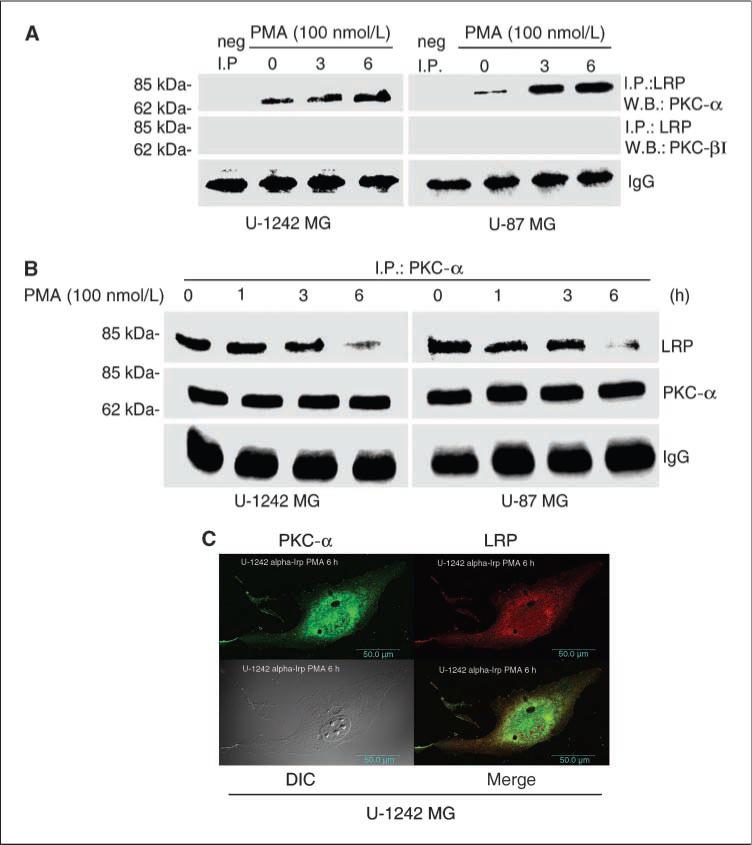
Localization of LRP and PKC-α in glioblastoma cells. A, LRP coimmunoprecipitate with PKC-α. Cells grown to 80% to 100% confluence were serum starved and treated with PMA at the indicated time periods. Cells were washed with ice-cold PBS and lysed with Triton X-100 lysis buffer. Cell lysates were immunoprecipitated with anti-LRP antibody (11H4) and then Western blotted with anti–PKC-α and anti–PKC-β antibodies. B, a reversed immunoprecipitation was carried out using anti–PKC-α antibody and then immunoblotted and probed for LRP. C, confocal microscopy showing the colocalization of both PKC-α and LRP in U-1242 MG cells. After treatment with PMA for 6 h, cells were prepared for immunofluorescent confocal microscopy by incubating with primary antibodies to PKC-α (Santa Cruz) and LRP (11H4). After washing off the primary antibodies, the cells were incubated with Alexa Flour 594–conjugated goat antimouse IgG secondary antibody/Alexa Flour 598–conjugated goat antirabbit IgG secondary antibody. Green, PKC-α; red, LRP; yellow, overlap/merge.
PKC activation increases cell migration in astrocytic tumor cells
We designed experiments to investigate the biological significance of down-regulating LRP in glioblastoma cell line. PMA treatment for 6 h increased cell invasion by 2-fold. The increase in cell invasion was blocked by Gö 6976, PKC-α, and PKC-β inhibitor (Fig. 5A, top). To further determine the involvement of PKC-α, we used siRNA characterized in Fig. 3C in our invasion assay. The result revealed that cells transfected with siRNA directed against PKC-α decreased PMA-induced increase in invasion (Fig. 5A). Furthermore, we tested the effect of knocking down PKC-α using shRNA in astrocytic tumor invasion. Stable cells lines deficient in PKC-α inhibited PMA-induced cell invasion (Fig. 5A). Collectively, these data suggest that PKC-α is a key mediator of LRP-mediated astrocytic tumor invasion.
Figure 5.
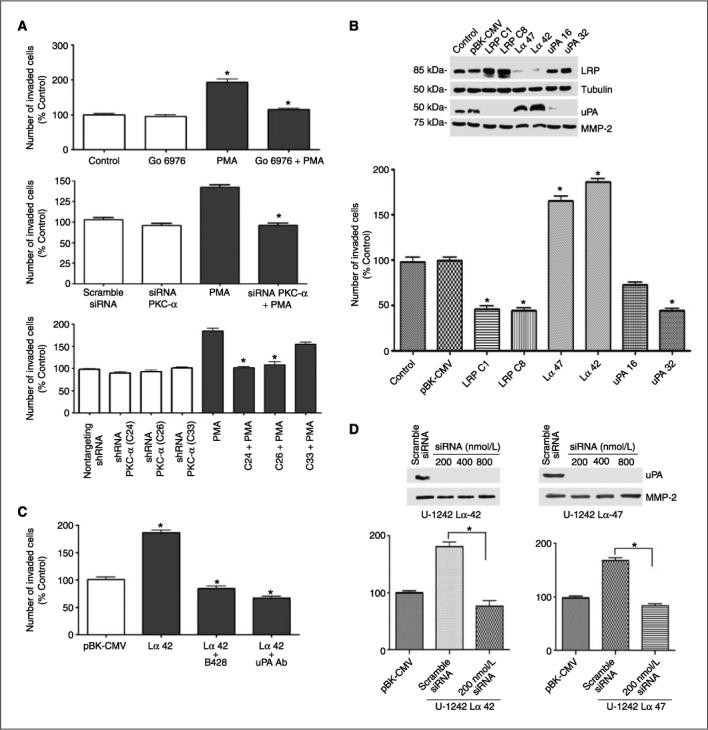
PKC activation increases cell invasion. A, in vitro invasion assay using the modified Boyden chamber assay. PMA increases invasion of glioblastoma cells by ∼2-fold, and the increase in invasion was attenuated by Gö 6976 (a classic PKC inhibitor). siRNA PKC-α abrogated PMA-induced increase in cell invasion by ∼60%. Stable PKC-α–deficient clones derived from shRNA transfection abrogated PMA-induced increases in cell invasion. B, down-regulation of LRP correlates with increase in uPA secretion. Western blot analysis of LRP-deficient, LRP-overexpressing, and uPA-deficient cells in U-1242 clones. In vitro invasion assay of U-1242 MG LRP-deficient clones (U-1242 Lα42 and U-1242 Lα47), 1242 wild type, empty vector (U-1242-PBK-CMV), LRP-overexpressing clones, and uPA knockdown clones (U-1242-uPA-32) usingthe modified Boyden Chamber assay coated with type IV collagen. C, in vitro invasion assay of LRP-deficient clone in the presence of uPA-specific inhibitor (20 μmol/L) and neutralizing antibody (25 μg/mL). D, effect of siRNA silencing of uPA in LRP-deficient clones (U-1242 Lα42 and U-1242 Lα47). siRNA targeting uPA attenuated the in vitro invasive phenotype of LRP-deficient clones. Results are averages of three independent experiments. Differences in means for transfected versus controls were statistically significant (P < 0.05).
Because PMA interacts with eight different PKC isozymes, we directly tested the biological roles of LRP in glioblastoma invasion. U-1242 MG cells were transfected with antisense construct to LRP. An empty-vector construct was used as a control. Western blot analyses showed that LRP knockdown clones (U-1242 Lα 42 and U-1242 Lα 47) secreted high uPA levels and LRP-overexpressing clones (U-1242 LRP-C1 and U-1242 LRP-C8) secreted low levels of uPA (Fig. 5B). Next, we investigated the potential invasive phenotypes of U-1242 MG LRP knockdown clones (pBK-CMVLα42 and pBK-CMV-Lα47), LRP-overexpressing clones (C1 and C8), empty vector, and uPA-deficient cells in vitro. The LRP-deficient clones invaded the extracellular matrix more than the empty vector. In contrast to LRP-overexpressing clones, uPA-deficient clones showed the least in vitro invasive phenotype (Fig. 5B). There was an inverse correlation between the expression of LRP and uPA in the clones. Experiments were designed to determine the role of uPA in promoting the aggressive invasion observed with the LRP-deficient clone. In this study, we used a specific uPA inhibitor, B428, at a concentration of 20 μmol/L and a neutralizing antibody to uPA at 25 μg/mL. Notably, both uPA inhibitor and the neutralizing antibody attenuated the invasive phenotype in LRP-deficient clone (Fig. 5C). Furthermore, 200 nmol/L of siRNA targeting urokinase abrogate the increased in vitro invasion of LRP-deficient clones (Fig. 5D).
In vivo tumor implantation
To extrapolate our in vitro data in vivo, we implanted the LRP-deficient and uPA-deficient clones shown in Fig. 5A and B into NOD SCID mice. U-1242 MG, U-1242 pBK-CMV, U-1242 pBK-CMV-Lα42, and U-1242 uPA 32 were infected with lentivirus expressing luciferase and GFP genes. The luciferase-tagged cells were sorted using the FACS. The results showed an average of 80% to 95% infection in these cells (results not shown). The glioblastoma cells were cultured in a three-dimensional gelfoam and then implanted into mice, and tumor growth was monitored using BLI and MRI.
Bioluminescence scan of mice 7 weeks postimplantation revealed that U-1242 MG LRP-deficient cells (pBK-CMV-Lα42) emitted higher number of photons per second (1.3 × 108, n = 5) than the uPA-deficient clone (1.73 × 106, n = 5; Fig. 6A, top). Using another noninvasive technique, mice were injected with gadolinium (0.25 mmol/kg body weight), the brains were imaged using the MRI, and tumor volume was calculated. Our data clearly show that LRP-deficient cells (pBK-CMV-Lα42) had a significantly larger tumor volume (20 mm3) and had more gadolinium-enhanced tumor area compared with both control and vector control groups, whereas the uPA knockdown clone had the least intracranial tumor invasion, the least tumor volume (4 mm3), and the least gadolinium enhancement (Fig. 6A, bottom). Brain slices from the mice used above were harvested and stained with H&E. Our data show that LRP-deficient cells (pBK-CMV-Lα42 and pBK-CMV-Lα47) extensively invaded the surrounding brain tissue (Fig. 6C) compared with empty vector control and the wild type (Fig. 6B). Figure 6D showed that LRP-overexpressing and uPA-deficient clones had the least tumor invasion and size. These data further support our in vitro findings and strengthens the hypothesis that LRP plays a key role in astrocytic tumor invasion.
Figure 6.
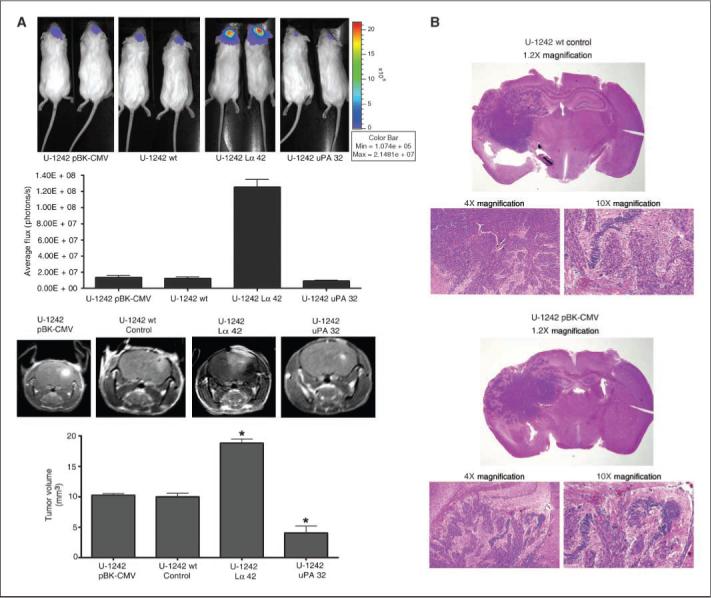
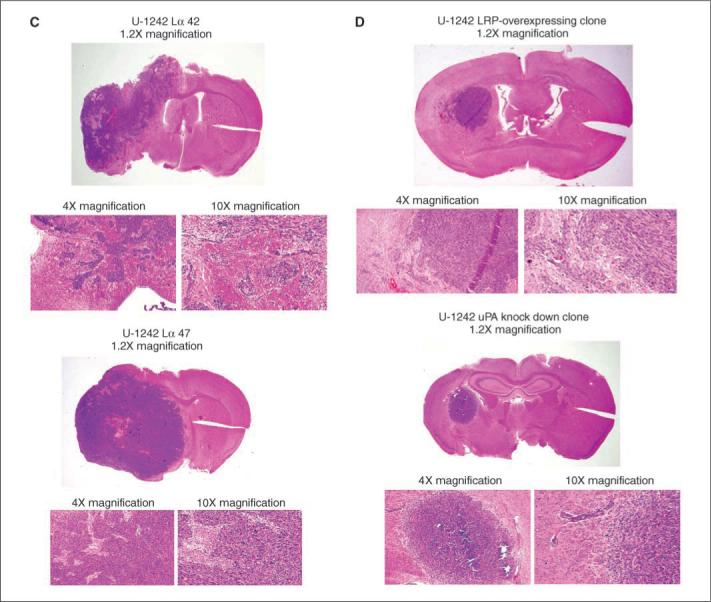
In vivo intracranial tumor invasion. A, top, cells were transduced to express both GFP and luciferase usinga lentiviral transfection. GFP-positive cells were grown on three-dimensional gelfoam and implanted into the brain of NOD SCID mice using stereotactic coordinates. BLI was done on animals using the IVIS 100 System. The intensity of emitted photons was calculated using the Living Image 2.50 software. The scale bars indicate the photon efflux. Bottom, coronal intracranial images of tumor growth in the brain were also captured with MRI (Varian). Tumor volume was calculated and represented as histogram representing each group. The MRI images are T1-weighed gadolinium-enhanced sequences. Differences in mean tumor volume for LRP-deficient and uPA-deficient clones versus empty vector were statistically significant (P < 0.05). B–D, H&E stain of 1242 wild type, empty vector, and LRP-deficient, LRP-overexpressing, and uPA-deficient clones implanted in mice.
Discussion
The aggressive propensity of glioblastoma to infiltrate surrounding brain tissue results in distant foci within the central nervous system that renders this tumor surgically incurable (42). The significance of LRP regulation and increased PKC activity has not been previously explored. In this paper, we hypothesize that changes in LRP expression can alter the levels of its ligands and thereby exert significant biological effects on various cellular responses, particularly those related to migration and invasion.
Our study using two well-characterized glioblastomas cell lines (U-1242 MG and U-87 MG) showed that treatment with PMA induced a time-dependent phosphorylation of LRP at serine residue and then down-regulated the level of LRP after long-term treatment. We have identified PKC-α as the putative PKC isozyme responsible for mediating the down-regulation of LRP expression using pharmacologic inhibitors. Stable and transient gene silencing of PKC-α abrogated PMA-induced down-regulation of LRP. Additional experiments suggest that PKC-α coimmunoprecipitated with LRP in glioblastoma cell lines after treatment with PMA. PKC-α is a widely expressed serine/threonine kinase that is activated by a variety of stimuli, and it plays a very important role in cellular proliferation, apoptosis, differentiation, migration, and motility (43, 44). The expression levels of PKC-α correlate with increased malignancy and invasive phenotype in lung carcinoma, as well as human glioblastoma multiforme (45–47). Thus, the association between PKC-α and LRP β-chain expression provides a new paradigm for dissecting the role of PKC in modulating the functions of LRP in glioblastomas.
The activation of PI3K/Akt and Ras/RAF/MEK/MAPK cascades leads to the regulation of numerous transcription factors and expression of genes involved in cell proliferation, drug resistance, inflammation, migration, and decreased rates of apoptosis, which are hallmarks of malignant gliomas. Our data showed that the use of LY294002, a PI3K inhibitor, reversed PMA-induced down-regulation of LRP. This suggests a role for PI3K pathway in regulating LRP expression and function in astrocytic tumor cells. Overexpression of epidermal growth factor receptor and mutation of PTEN have been associated with astrocytoma progression, and both of these activate the PI3K/Akt pathway. Thus, targeting this pathway could be of potential therapeutic importance.
Invasion through the extracellular matrix is an important step in tumor invasion. We, therefore, investigated the role of LRP in mediating astrocytic tumor invasive growth in vitro and in vivo. Our data show that down-regulating LRP expression correlates with increased amount of uPA secreted into the culture medium with a resultant increase in glioblastoma cell invasion in vitro. These data suggest that decrease in LRP expression and increase in uPA protein and activity could be associated with glioblastoma invasion. To directly dissect the role of LRP and uPA in driving astrocytic tumor invasion, we generated LRP and uPA knockdown clones and investigated their invasive phenotypes both in vitro and in vivo. LRP knockdown clones, which secretes high amount of uPA into conditioned medium, invaded the extracellular matrix more that the empty vector, U-1242 MG wild-type cells, LRP-overexpressing clones, and uPA-deficient clone in vitro. Figure 6A using BLI in NOD SCID mice showed that the glioma cell line deficient in LRP had increased number of photons, whereas uPA-deficient clone expressed lower number of photons. We also imaged animals using MRI scan and showed that LRP-deficient cells had more gadolinium enhancement in the area of tumor growth. The H&E stain sections of mice brains clearly revealed that LRP-deficient cells (pBK-CMV-Lα42 and pBK-CMV-Lα47; Fig. 6C) invaded more into the surrounding brain regions, whereas the LRP-overexpressing clones and uPA-deficient clone did not infiltrate into distant brain tissue (Fig. 6D). We, thus, suggest that reduced LRP expression and increased levels of secreted uPA drive astrocytoma invasion, a major hallmark of malignant gliomas. In addition, siRNA against uPA, uPA-specific inhibitor, and the neutralizing antibody decreased the propensity of LRP-deficient clone to invade the extracellular matrix in vitro. Other workers had reported that uPA is a critical element in tumor biology, especially that it controls cell motility, tissue remodeling, and bioavailability of angiogenic factors (48). Thus, inhibition of uPA could regulate cell invasion and affects the bioavailability of angiogenic factors, which are essential in tumor invasion.
Despite recent advances in the understanding of molecular mechanism of astrocytoma progression, glioblastomas are surgically incurable and refractory to classic chemotherapy and radiotherapy. Developing novel therapeutic approaches for clinical treatment remains a major challenge. Our results provide insight into the mechanism by which PKC and the PI3K pathways can regulate LRP expression and astrocytic tumor invasion. The inhibition of both the PKC-α and PI3K pathways and uPA may be useful in the design of new therapeutic interventions aimed at altering the invasive phenotype of astrocytic tumors.
Acknowledgments
We thank Ruoya Ho (Somlyo Laboratory), supported by NIH PO1HL 48807, for confocal microscopy assistance and Rene Jack Roy and Dr. Stuart S. Berr (Department of Radiology) for excellent technical assistance on MRI and BLI.
Grant support: NIH grants NS035122 and CA090851 (I.M. Hussaini) and Farrow Fellowship (S. Amos).
References
- 1.Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70:217–28. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- 2.Herz J, Hamann U, Rogne S, Mylebost O, Gausepohl H, Stanley KK. Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 1988;7:4119–27. doi: 10.1002/j.1460-2075.1988.tb03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moestrup SK, Gliemann J, Pallesen G. Distribution of the α2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 1992;269:375–82. doi: 10.1007/BF00353892. [DOI] [PubMed] [Google Scholar]
- 4.Bu G, Maksymovitch EA, Geuze H, Schwarz AL. Subcellular localization and endocytic function of low density lipoprotein receptor-related protein in human glioblastomas cells. J Biol Chem. 1994;269:29874–82. [PubMed] [Google Scholar]
- 5.Yamamoto M, Sawaya R, Mohanam S, et al. Expression and localization of urokinase-type plasminogen activator receptor in human gliomas. Cancer Res. 1994;54:5016–20. [PubMed] [Google Scholar]
- 6.Yamamoto M, Ikeda K, Ohshima K, Tsugu H, Kimura H, Tomonaga M. Expression and cellular localization of low-density lipoprotein receptor-related protein/α2-macroglobulin receptor in human glioblastoma in vivo. Brain Tumor Pathol. 1998;15:23–30. doi: 10.1007/BF02482097. [DOI] [PubMed] [Google Scholar]
- 7.Maletinska L, Blakely EA, Bjornstad KA, Deen DF, Knoff LJ, Forte TM. Human glioblastoma cell lines: levels of low-density lipoprotein receptor and low-density lipoprotein receptor-related protein. Cancer Res. 2000;60:2300–3. [PubMed] [Google Scholar]
- 8.Lopes MB, Bogaev CA, Gonias SL, VandenBerg SR. Expression of α2-macroglobulin receptor/low density lipoprotein receptor-related protein is increased in reactive and neoplastic glial cells. FEBS Lett. 1994;338:301–5. doi: 10.1016/0014-5793(94)80288-2. [DOI] [PubMed] [Google Scholar]
- 9.Brown MD, Banker GA, Hussaini IM, Gonias SL, VandenBerg SR. Low density lipoprotein receptor-related protein is expressed early and becomes restricted to a somatodendritic domain during neuronal differentiation in culture. Brain Res. 1997;747:313–7. doi: 10.1016/s0006-8993(96)01321-2. [DOI] [PubMed] [Google Scholar]
- 10.Gonias SL, LaMarre J, Crookston DJ, et al. α2-Macroglobulin and the α2-macroglobulin receptor/LRP. A growth regulatory axis. Ann N Y Acad Sci. 1994;737:273–90. doi: 10.1111/j.1749-6632.1994.tb44318.x. [DOI] [PubMed] [Google Scholar]
- 11.Herz J, Kowal RC, Goldstein JL, Brown MS. Proteolytic processing of the 600 kd low density lipoprotein receptor-related protein (LRP) occurs in a trans-Golgi compartment. EMBO J. 1990;9:1769–76. doi: 10.1002/j.1460-2075.1990.tb08301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strickland DK, Gonias SL, Argraves WS. Diverse roles of the LDL receptor family. Trends Endocrinol Metab. 2002;13:66–74. doi: 10.1016/s1043-2760(01)00526-4. [DOI] [PubMed] [Google Scholar]
- 13.Kounnas MZ, Henkin J, Argraves WS, Strickland DK. Low density lipoprotein receptor-related protein/α2-macroglobulin receptor mediates cellular uptake of prourokinase. J Biol Chem. 1993;268:21862–7. [PubMed] [Google Scholar]
- 14.Hahn-Dantona E, Ruiz JF, Bornstein P, Strickland DK. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J Biol Chem. 2001;276:15498–503. doi: 10.1074/jbc.M100121200. [DOI] [PubMed] [Google Scholar]
- 15.Nykajaer A, Petersen CM, Moller B, et al. Purified αa2-macroglobulin receptor/LDL receptor-related protein binds urokinase plasminogen activator inhibitor type-1 complex. Evidence that the α2-macroglobulin receptor mediates cellular degradation of urokinase receptor-bound complexes. J Biol Chem. 1992;267:14543–6. [PubMed] [Google Scholar]
- 16.Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689–95. doi: 10.1172/JCI1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goretzki L, Mueller B. Low density lipoprotein related protein (LRP) interacts with a GTP bound protein. Biochem J. 1998;336:381–6. doi: 10.1042/bj3360381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J. Clin Invest. 2001;108:779–84. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver AM, Hussaini IM, Mazar A, Henkin J, Gonias SL. Embryonic fibroblasts that are genetically deficient in low density lipoprotein receptor-related protein demonstrate increased activity of the urokinase receptor system and accelerated migration on vitronectin. J Biol Chem. 1997;272:14372–9. doi: 10.1074/jbc.272.22.14372. [DOI] [PubMed] [Google Scholar]
- 20.Webb DJ, Nguyen DH, Gonias SL. Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and matrigel invasion. J Cell Sci. 2000;113:123–34. doi: 10.1242/jcs.113.1.123. [DOI] [PubMed] [Google Scholar]
- 21.Lutz C, Nimpf J, Jenny M, et al. Evidence of functional modulation of the MEKK/JNK/cJun signaling cascade by the low density lipoprotein receptor-related protein (LRP). J Biol Chem. 2000;277:43143–51. doi: 10.1074/jbc.M204426200. [DOI] [PubMed] [Google Scholar]
- 22.Boucher P, Gotthardt M, Li W-P, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–32. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 23.Todo T, Shitara N, Nakamura H, Takakura K, Ikeda K. Immunohistochemical demonstration of protein kinase C isozymes in human brain tumors. Neurosurgery. 1991;3:399–403. doi: 10.1097/00006123-199109000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Couldwell WT, Uhm JH, Antel JP, Yong VW. Enhanced protein kinase C activity correlates with the growth rate of malignant gliomas in vitro. Neurosurgery. 1991;29:880–6. doi: 10.1097/00006123-199112000-00013. [DOI] [PubMed] [Google Scholar]
- 25.Da Rocha AB, Mans DR, Regner A, Schwartsmann G. Targeting protein kinase C: new therapeutic opportunities against high-grade malignant gliomas? Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- 26.Dempsey EC, Newton AC, Mochly-Rosen D, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 27.Parker PJ, Murray-Rust J. PKC at a glance. J Cell Sci. 2004;117:131–2. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- 28.Jaken S. Protein kinase C isozymes and substrates. Curr Opin Cell Biol. 1996;8:168–73. doi: 10.1016/s0955-0674(96)80062-7. [DOI] [PubMed] [Google Scholar]
- 29.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 30.Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308:693–8. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- 31.Kowal RC, Herz J, Goldstein JL, Esser V, Brown MS. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc Natl Acad Sci U S A. 1989;86:5810–4. doi: 10.1073/pnas.86.15.5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hussaini IM, Karns LR, Vinton G, et al. Phorbol 12-myristate 13-acetate induces protein kinase ceta-specific proliferative response in astrocytic tumor cells. J Biol Chem. 2000;27:22348–54. doi: 10.1074/jbc.M003203200. [DOI] [PubMed] [Google Scholar]
- 33.Hussaini IM, Brown MD, Karns LR, et al. Epidermal growth factor differentially regulates low density lipoprotein receptor-related protein gene expression in neoplastic and fetal human astrocytes. Glia. 1999;25:71–84. [PubMed] [Google Scholar]
- 34.Amos S, Martin PM, Polar GA, Parsons SJ, Hussaini IM. Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase C δ/c-Src pathways in glioblastoma cells. J Biol Chem. 2005;280:7729–38. doi: 10.1074/jbc.M409056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao YG, Xiao AZ, Newcomer RG, et al. Activation of pro-gelatinase B by endometase/matrilysin-2 promotes invasion of human prostate cancer cells. J Biol Chem. 2003;278:15056–64. doi: 10.1074/jbc.M210975200. [DOI] [PubMed] [Google Scholar]
- 36.Mohan PM, Chintala SK, Mohanam S, et al. Adenovirus-mediated delivery of antisense gene to urokinase-type plasminogen activator receptor suppresses glioma invasion and tumor growth. Cancer Res. 1999;59:3369–73. [PubMed] [Google Scholar]
- 37.Loukinova E, Ranganathan S, Kuznetsov S, et al. Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP). Evidence for integrated co-receptor function between LRP and the PDGF. J Biol Chem. 2002;277:15499–506. doi: 10.1074/jbc.M200427200. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, van Kerkhof P, Marzolo MP, Strous GJ, Bu G. Identification of a major cyclic AMP-dependent protein kinase A phosphorylation site within the cytoplasmic tail of the low-density lipoprotein receptor-related protein: implication for receptor-mediated endocytosis. Mol Cell Biol. 2001;21:1185–95. doi: 10.1128/MCB.21.4.1185-1195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonindard C, Bergonzi C, Denier C, et al. Synthetic hispidin, a PKC inhibitor, is more toxic toward cancer cells than normal cells in vitro. Cell Biology and Toxicology. 1997;13:141–53. doi: 10.1023/a:1007321227010. [DOI] [PubMed] [Google Scholar]
- 40.Aeder SE, Martin PM, Soh JW, Hussaini IM. PKCD mediates glioblastomas cell proliferation through the Akt and mTOR signaling pathways. Oncogene. 2004;23:9062–9. doi: 10.1038/sj.onc.1208093. [DOI] [PubMed] [Google Scholar]
- 41.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kreth FW, Warnke PC, Scheremet R, Ostertag CB. Surgical resection and radiation therapy versus biopsy and radiation therapy in the treatment of glioblastoma multiforme. J Neurosurg. 1993;78:762–6. doi: 10.3171/jns.1993.78.5.0762. [DOI] [PubMed] [Google Scholar]
- 43.Engers R, Mrzyk S, Springer E, et al. Protein kinase C in human renal cell carcinomas: role in invasion and differential isoenzyme expression. Br J Cancer. 2000;82:1063–9. doi: 10.1054/bjoc.1999.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan M, Li P, Sun M, Yin G, Yu D. Upregulation and activation of PKC a by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC a and Src inhibitors. Oncogene. 2006;25:3286–95. doi: 10.1038/sj.onc.1209361. [DOI] [PubMed] [Google Scholar]
- 45.Basu A, Weixel K, Saijo N. Characterization of the protein kinase C signal transduction pathway in cisplatin-sensitive and -resistant human small cell lung carcinoma cells. Cell Growth Differ. 1996;7:1507–12. [PubMed] [Google Scholar]
- 46.Mawrin C, Diete S, Treuheit T, et al. Prognostic relevance of MAPK expression in glioblastoma multiforme. Int J Oncol. 2003;23:641–8. [PubMed] [Google Scholar]
- 47.Cho KK, Mikkelsen T, Lee YJ, Jiang F, Chopp M, Rosenblum ML. The role of protein kinase Cα in U-87 glioma invasion. Int J Dev Neurosci. 1999;17:447–61. doi: 10.1016/s0736-5748(99)00054-4. [DOI] [PubMed] [Google Scholar]
- 48.Gondi CS, Lakka SS, Yanamandra N, et al. Expression of antisense uPAR and antisense uPA from a bicistronic adenoviral construct inhibits glioma cell invasion, tumor growth, and angiogenesis. Oncogene. 2003;22:5967–75. doi: 10.1038/sj.onc.1206535. [DOI] [PubMed] [Google Scholar]