

Abstract
Cyclooxygenase (COX)-1 and -2 metabolize arachidonic acid to prostanoids and reactive oxygen species, major players in the neuroinflammatory process. While most reports focused on the inducible isoform, COX-2, the contribution of COX-1 to the inflammatory response is unclear. In the present study the contribution of COX-1 in the neuroinflammatory response to intracerebroventricular lipopolysaccharide (LPS) was investigated using COX-1 deficient (COX-1−/−) mice or wild type (COX-1+/+) mice pretreated with SC-560, a selective COX-1 inhibitor. Twenty four hours after LPS injection, COX-1−/− mice showed decreased protein oxidation and LPS-induced neuronal damage in the hippocampus compared to COX-1+/+ mice. COX-1−/− mice showed a significant reduction of microglial activation, proinflammatory mediators, and expression of COX-2, iNOS, and NADPH oxidase. The transcriptional downregulation of cytokines and other inflammatory markers in COX-1−/− mice was mediated by a reduced activation of NF-κB and STAT3. Administration of SC-560 prior to LPS injection also attenuated the neuroinflammatory response by decreasing brain levels of prostaglandin (PG)E2, PGD2, PGF2α, and thromboxane B2 (TXB2), as well as the expression of pro-inflammatory cytokines and chemokine. These findings suggest that COX-1 plays a previously unrecognized role in neuroinflammatory damage.
Keywords: Microglia, astrocytes, prostaglandin, NADPH oxidase, oxidative stress
INTRODUCTION
Two different isoforms of cyclooxygenase (COX), COX-1 and COX-2, catalyze the conversion of arachidonic acid to prostaglandins (PGs) - PGE2, PGI2, PGF2α, PGD2, and thromboxane A2 - (1) which are potent activators of a large family of G protein-coupled receptors and mediate specific biological responses in various tissues and cells (2).
Classically, COX-1 has been considered as the constitutively expressed isoform, primarily responsible for homeostatic PG synthesis, and COX-2 as the isoform induced in response to inflammatory stimuli and thus, the most appropriate target for anti-inflammatory drugs (3, 4). However, this distinction is not appropriate for the central nervous system (CNS), where COX-2 is constitutively expressed at high levels (5), and COX-1 can be up-regulated in certain pathological conditions such as global cerebral ischemia (6), N-methyl-D-aspartate induced excitotoxicity (7), traumatic brain injury (8), and in activated microglia associated with amyloid plaques (9).
Neuroinflammation, which is substantially mediated by activation of microglia (10), is a critical component of the pathogenesis of certain neurodegenerative disorders, including Alzheimer’s disease (AD) (11). Reactive microglia release inflammatory mediators such as cytokines, chemokines, prostanoids, and reactive oxygen and nitrogen species, all of which may contribute to neuronal injury (12).
Several independent epidemiological studies indicate that chronic use of non steroidal anti-inflammatory drugs (NSAIDs) can reduce the risk of developing AD (13–16), supporting the inflammatory cascade hypothesis. However, large clinical trials, mostly using selective COX-2 inhibitors, have failed to show any beneficial effect in AD patients with mild to severe symptoms (17–20). This suggests that an early treatment is crucial to stop the mechanisms underlying the disease before the onset of the symptoms and that preferential COX-1 inhibition may be more beneficial than selectively targeting COX-2. Therefore, a full understanding of the physiological, pathological, and/or neuroprotective role of COX isoforms may help to develop better therapeutic strategies for the prevention or treatment of AD.
Mice with a null mutation of the COX gene have been useful for investigating the role of each COX isoform in both physiological and pathological conditions in the CNS (21). To directly address the role of COX-1 in CNS responses to inflammatory stimuli, we assessed the effect of intracerebroventricular (icv) injection of lipopolysaccharide (LPS) on acute neuroinflammatory response in COX-1 deficient (COX-1−/−) and wild type (COX-1+/+) mice. LPS specifically activates immune response in the CNS through a Toll-like receptor 4-dependent signaling pathway that may be functionally important to determine the degree of inflammatory response through microglial activation (22). In the present study we examined the neuroinflammatory response 24 h after LPS injection based on our preliminary time course study indicating that most of the inflammatory markers, including markers for microglia, astrocytes, and cytokines peaked at 24 h. Additionally, previous reports have shown sustained inflammation at this time point after LPS injection (23, 24). We show that COX-1−/− mice are more resistant than wild-type mice to LPS-induced neuronal death and exhibit a marked reduction in inflammatory response.
MATERIALS AND METHODS
Animals and LPS administration
The study was approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with NIH guidelines on the care and use of laboratory animals. Three-month-old male COX-1 wild-type (COX-1+/+) and homozygous (COX-1−/−) mice on a C57BL/6–129/Ola genetic background were used (25). To avoid significant genetic background or stain differences between COX-1+/+ and COX-1−/− mice, all mice used in this study were obtained by heterozygous by heterozygous mating of COX-1+/− mice and therefore all contained the same strain and genetic background.
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg, i.p.) and positioned in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA). Vehicle (sterile saline, 5 μl) or LPS (E. coli serotype 0127:B8; 5 μg in 5 μl of sterile saline) was administered into the cerebral lateral ventricle using a 10 μl syringe with a fine needle (World Precision Instruments, Sarasota, FL) and a syringe pump (Stoelting, Wood Dale, IL) at a rate of 1 μl/min. This dose of LPS and time point (24 h) induced an optimal neuroinflammatory response in a preliminary pilot study and has been used to produce neuroinflammation in different laboratories (24, 26, 27). The coordinates for the sterotaxic injections were −2.3 mm dorsal/ventral, −1.0 mm lateral, and −0.5 mm anterior/posterior from the bregma (28). COX-1+/+ mice received the selective COX-1 inhibitor SC-560 (Cayman Chemical, Ann Arbor, MI; 30 mg/kg, i.p.) (29) or vehicle (40% DMSO in 0.1 M phosphate buffer, pH 7.4) once a day for 7 days. On the last day, SC-560 was given 30 min prior to LPS injection.
Tissue preparation and histology
Mice were transcardially perfused with saline followed by 4% paraformaldehyde. Brains were postfixed overnight in the same medium and placed in 30% sucrose, before sectioning (30 μm). Staining with FJB and Cresyl violet was performed using slight modifications of the technique described by (30, 31). Immunohistochemistry and double immunofluorescence were performed as previously reported (31). Mouse anti-CD11b (1:100; BD Biosciences, San Jose, CA), mouse anti-CD45 (1:100; BD Biosciences), mouse anti-FcγRII/III (1:100; BD Biosciences), and mouse anti-scavenger receptor A (SRA; 1:100; Serotec, Raleigh, NC), and biotinylated tomato lectin (TL;1:2000; Vector Laboratories, Burlingame, CA), mouse anti-glial fibrillary acidic protein (GFAP; 1:200; Sigma-Aldrich), mouse anti-nitrotyrosine (1:100; Chemicon, Temecula, CA), rabbit anti-P-p65 (1:100; Cell signaling, Beverly, MA), and rabbit anti-P-STAT3 (1:100; Cell Signaling) were used as primary antibodies.
Quantitative real-time PCR
Total RNA extraction and reverse transcription were performed as previously reported (32). Quantitative PCR was performed using the Applied Biosystems Assay-On-Demand Gene Expression protocol (32). For relative comparison of each gene, we analyzed the Ct value of real-time PCR data with the ΔΔCt method normalizing by phosphoglyceratekinase 1 (Pgk1) as an endogenous control.
Prostaglandin assay
Prostanoids were extracted by homogenization of the tissues as previously described (33) and the levels of PGE2, PGF2α and TXB2 (Oxford Biomedical, Oxford, MI), and PGD2 (Cayman Chemical) was determined using specific enzyme immunoassay (EIA) kits .
Western blot analysis
Membrane and cytosolic fractions were prepared using the Compartment Protein Extraction Kit (Chemicon) according to the manufacturer’s instructions. Western blot analyses were carried out as described previously (32). Mouse anti-p67phox, p47phox, gp91phox, Rac1 (BD Biosciences), and calnexin (Stressgen, Victoria, BC, Canada) were used as primary antibodies.
Detection of protein oxidation
The extent of protein oxidation was assessed by measuring protein carbonyl levels with an OxyBlot Protein Oxidation Detection Kit (Chemicon) according to the protocol of the manufacturer (31).
Quantitative ELISA for transcription factor activation
Nuclear proteins were prepared by using a compartmental protein extraction kit (Chemicon, Temecula) according to the manufacturer’s protocol. NF-κB and STAT3 activation was assayed using Active Motif (Carlsbad, CA, USA) ELISA-based transactivation TransAM kit containing a 96-well plate with immobilized oligonucleotides encoding an NF-κB and STAT consensus site (58).
Statistics
All data are expressed as mean ± SEM. Statistical significance was assessed with one- or two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA). Significance was taken at P < 0.05.
RESULTS
COX-1−/− mice show decreased degenerating neurons after LPS
We assessed neuronal damage in the brain at 24 h after LPS injection using the fluorescent marker Fluoro Jade B (FJB), which selectively labels injured neurons (34). LPS-injected COX-1−/− mice (Fig. 1D and H) showed fewer FJB-positive cells in the hippocampus than COX-1+/+ mice (Fig. 1B and F). However, FJB-positive neurons were not detected in the hippocampus of vehicle-injected mice of COX-1+/+ or COX-1−/− mice (Fig. 1A, C, E, and G). LPS administration caused neuronal damage in other periventricular areas of brain (data not shown).
Figure 1.

Effects of COX-1 deficiency on LPS-induced neuronal cell death. Representative photomicrographs of FJB (A-H) and Nissl staining (I-P) in the hippocampus of COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. Lower panels (E-H and M-P) show higher magnification of respective boxed area in the upper panels (A-D and I-L). Scale bars, 300 μm (A-D, I-L); 100 μm (E-H, M-P).
In adjacent sections stained with cresyl violet, a similar distribution of cell loss and gliosis was found in the hippocampus in LPS-injected COX-1+/+ mice (Fig. 1J and N) compared with vehicle-injected COX-1+/+ mice (Fig. 1I and M). Nissl staining showed that hippocampal pyramidal cells in COX-1−/− mice (Fig. 1L and P) were more preserved than in COX-1+/+ mice (Fig. 1J and N).
LPS-induced glial activation is decreased in COX-1−/− mice
To determine whether reduced neuronal death after LPS in COX-1−/− mice was due to a decreased glial response, we examined the expression of CD11b and CD45, markers for microglia, and GFAP, a marker for astrocytes, 24 h after LPS injection. LPS significantly increased the expression of CD11b, CD45, and GFAP mRNA compared to vehicle injected mice (P < 0.01; Fig. 2A-C). However, the induction was less in COX-1−/− mice than in COX-1+/+ mice (P < 0.05; Fig. 2A-C).
Figure 2.

Effects of COX-1 deficiency on LPS-induced expression of glial markers. Quantitative real time-PCR analysis of microglia marker CD11b (A), CD45 (B), and astrocyte marker GFAP mRNA (C) in COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. Data are mean ± SEM (n = 6). *P < 0.05, **P < 0.01, compared to vehicle-injected COX-1+/+ mice; #P < 0.05, compared to the corresponding LPS-injected COX-1+/+ mice. Effects of COX-1 deficiency on LPS-induced activation of astrocytes. (D-K) Representative photomicrographs of GFAP immunohistochemistry in the hippocampus for COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. High magnification images of GFAP immunostaining (E, G, I, and K) in the hippocampus are shown in D, F, H, and J, respectively. Scale bars, 300 μm (D, F, H, and J); 100 μm (E, G, I, and K).
Sections adjacent to those used for FJB and Nissl staining were used to determine immunoreactivity to GFAP, CD11b, CD45, Fcγ receptor II/III (FcγRII/III), and scavenger receptor A (SRA). LPS injection increased immunoreactive GFAP-positive astrocytes in the hippocampus of COX-1+/+ mice (Fig. 2F and G) compared to LPS-injected COX-1−/− mice (Fig. 2J and K). In vehicle-injected mice, only a few faintly CD11b or CD45 immunoreactive resting microglia with specifically small cell bodies, thin, and ramified processes were observed in the hippocampus of both COX-1+/+ and COX-1−/− mice (Fig. 3A and E). Intense immunoreactive CD11b- and CD45-positive microglia with enhanced staining intensity, enlarged cell bodies, and thickening of processes were observed 24 h after LPS injection in the hippocampus of COX-1+/+ mice (Fig. 3B and F). Although, increased CD11b and CD45 immunostaining intensity was observed in LPS-injected COX-1−/− mice (Fig. 3D and H) compared to vehicle-injected COX-1−/− mice (Fig. 3C and G), CD11b- and CD45-positive cells retained a resting morphology with specifically small cell bodies, thin, and ramified processes (Fig. 3D and H). Staining with other microglial markers selectively labeling phagocytic microglial phenotype (35), FcγRII/III (Fig. 3I-L) and SRA (Fig. 3M-P), gave immunostaining patterns similar to CD11b (Fig. 3A-D) and CD45 (Fig. 3E-H).
Figure 3.

Effects of COX-1 deficiency on LPS-induced activation of microglia. (A-P) Representative photomicrographs of CD11b (A-D), CD45 (E-H), FcγRII/III (I-L), and SRA (M-P) immunohistochemistry in the hippocampus for COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. The insets of A-H, J, L, N, and P show morphology of microglia at higher magnification. Scale bar, 100 μm (A-P).
Expression of inflammatory mediators is reduced in COX-1−/− mice after LPS
We determined the expression of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, and a chemokine, MCP-1, using quantitative Real-time PCR. LPS significantly increased the expression of IL-1β, IL-6, MCP-1, and TNF-α mRNA in both COX-1+/+ and COX-1−/− mice (P < 0.01; Fig. 4A). However, the mRNA levels of these cytokines were less marked in the COX-1−/− compared to COX-1+/+ mice (P < 0.05; Fig. 4A).
Figure 4.

Effects of COX-1 deficiency on LPS-induced expression of cytokines and chemokine. (A) Quantitative real time-PCR analysis of IL-1β, IL-6, MCP-1, and TNF-α mRNA for COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. Effects of COX-1 deficiency on LPS-induced brain PG levels and on the expression of enzymes in the AA metabolic pathway. COX-1+/+ and COX-1−/− mice received icv injection of LPS or vehicle 24 h before sacrifice. (B) Brain PGE2, PGD2, PGF2α, and TXB2 levels, as detected by specific immunoassay kits. (C-E) Quantitative Real-time PCR analysis of cPLA2 (C), COX-2 (D), mPGES-1 mRNA (E). Data are mean ± SEM (n = 4-6). **P < 0.01 vs. corresponding vehicle-injected COX-1+/+ mice; #P < 0.05, ##P < 0.01 vs. corresponding LPS-injected COX-1+/+ mice.
We also measured whether brain levels of PGE2, a major proinflammatory PG (36), and other PGs, PGD2, PGF2α, and TXB2, were altered by COX-1 deficiency 24 h after LPS injection. As shown in Fig. 4B, in vehicle-injected mice, there were no significant differences in PGE2, PGD2, PGF2α, and TXB2 levels between COX-1+/+ and COX-1−/− mice. Following LPS injection, levels of brain PGE2, PGD2, PGF2α, and TXB2 were significantly increased in COX-1+/+ mice as compared to vehicle-injected COX-1+/+ mice (P < 0.01; Fig. 4B). However, brain PGE2, PGD2, PGF2α, and TXB2 levels after LPS were significantly attenuated in COX-1−/− mice compared to COX-1+/+ mice (P < 0.05, P < 0.01, respectively; Fig. 4B). Then, we determined mRNA levels of enzymes involved in PG synthesis: PLA2, COX-2, and terminal PGE2 synthases using quantitative real time-PCR. Brain mRNA levels of cPLA2, COX-2, and mPGES-1 were increased in LPS-injected COX-1+/+ mice (P < 0.01; Fig. 4C-E). However, cPLA2, COX-2, and mPGES-1 mRNA expression were significantly decreased in LPS-injected COX-1−/− mice (Fig. 4C-E). The expression of sPLA2 and mPGES-2 mRNA was similar in COX-1+/+ and COX-1−/− mice after LPS injection (data not shown).
To determine whether selective pharmacological inhibition of COX-1 in the brain could recapitulate some of the changes observed in the COX-1−/− mice, we measured expression of proinflammatory mediators after LPS in COX-1+/+ mice pre-treated with a selective COX-1 inhibitor, SC-560, or vehicle. Pretreatment of COX-1+/+ mice with SC-560 reduced the mRNA expression of IL-1β, IL-6, MCP-1, and TNF-α in COX-1+/+ mice (P < 0.05, P < 0.01, respectively; Fig. 5A), similarly to the decrease level observed in the COX-1−/− mice. Furthermore, pretreatment with SC-560 significantly inhibited LPS-induced PGE2, PGD2, PGF2α, and TXB2 production in the COX-1+/+ mice (P < 0.01, Fig. 5B).
Figure 5.

Effects of SC-560 on LPS-induced gene expression of proinflammatory cytokines and on brain prostaglandin levels. COX-1+/+ mice were treated for 7 days with SC-560 (30 mg/kg, i.p.) or vehicle before LPS injection. (A) Quantitative Real-time PCR analysis of IL-1β, IL-6, MCP-1, and TNF-α mRNA. (B) Immunoassay of brain PGE2, PGD2, PGF2α, and TXB2 levels. Data are mean ± SEM (n = 6–9). **P < 0.01 vs. corresponding vehicle-injected COX-1+/+ mice; #P < 0.05, ##P < 0.01 vs. corresponding LPS-injected COX-1+/+ mice.
Inducible nitric oxide synthase (iNOS), NADPH oxidase, and myeloperoxidase (MPO), major sources of reactive oxygen species in the inflammatory process, are expressed in glial cells, and may be involved in the neuroinflammatory injury. Therefore, we examined gene transcription and protein expression of ROS-generating enzymes in COX-1+/+ and COX-1−/− mice 24 h after LPS injection using quantitative real time-PCR and Western blotting. In response to LPS, COX-1−/− mice showed a less marked increase in iNOS and MPO mRNA levels compared to LPS-injected COX-1+/+ mice (P < 0.01; Fig. 6A and B). Similarly, mRNA levels of NADPH oxidase cytosolic component p47phox and membrane component gp91phox were significantly elevated at 24 h after LPS injection in both genotypes compared with vehicle-injected controls (P < 0.01; Fig. 6C and D). However, COX-1−/− mice showed less of an increase in the expression of p47phox and gp91phox mRNA than COX-1+/+ mice (P < 0.05, P < 0.01, respectively; Fig. 6C and D). Immunolocalization of p67phox revealed that microglia expressed this protein (Fig. 6E). By Western blotting we confirmed that components of NADPH oxidase were reduced after LPS, in COX-1−/− mice compared to COX-1+/+ mice (Fig. 6F). NADPH oxidase activation is accompanied by the translocation of its cytosolic component p67phox from the cytosol to the membrane, which leads to ROS production (37). LPS injection increased translocation of p67phox to the membrane in both COX-1+/+ and COX-1−/− mice (P < 0.01; Fig. 6G and H). However, the increase was lower in COX-1−/− mice compared to COX-1+/+ mice (P < 0.05; Fig. 6G and H).
Figure 6.

Effects of COX-1 deficiency on LPS-induced expression of ROS-generating enzymes. (A-D) Quantitative Real-time PCR analysis of iNOS (A), MPO (B), p47phox (C), and gp91phox mRNA (D) for COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. (E) Immunofluorescence photomicrographs of p67phox immunoreactivity within the activated microglia. Scale bars, 25 μm. (F) Expression of NADPH oxidase components in COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. (G) Activation of NADPH oxidase by LPS. Representative immunoblot of translocation of p67phox to the membrane in COX-1+/+ and COX-1−/− mice that received icv injection of LPS or vehicle 24 h before sacrifice. (H) Quantification of p67phox levels expressed as the ratio of membrane to total. Data are mean ± SEM (n = 6). **P < 0.01 vs. corresponding vehicle-injected COX-1+/+ mice; #P < 0.05, ##P < 0.01 vs. corresponding LPS-injected COX-1+/+ mice.
LPS-induced oxidative stress is reduced in COX-1−/− mice
A significant part of LPS-induced neurotoxic process is mediated by oxidative damage (38), which can be evaluated by assessing protein carbonyls and nitrotyrosine levels (39). Therefore, we investigated whether protein carbonyls and nitrotyrosine levels were altered in COX-1−/− mice 24 h after LPS injection. In vehicle-injected mice, brain levels of protein carbonyls (Fig. 7A and B) were similar between COX-1+/+ and COX-1−/− mice. Levels of protein carbonyls (Fig. 7A and B) were significantly increased in LPS-injected compared to vehicle-injected COX-1+/+ mice. However, in LPS-injected COX-1−/− mice there were no significant change in protein carbonyls (Fig. 7A and B) compared to vehicle-injected COX-1−/− mice. Sections adjacent to those used for FJB and Nissl staining in Fig. 1 were used to determine nitrotyrosine immunoreactivity in the hippocampus. Nitrotyrosine immunoreactivity was increased in the hippocampus of LPS-injected COX-1+/+ mice (Fig. 7D) compared to vehicle-injected COX-1+/+ mice (Fig. 7C). In contrast, very few nitrotyrosine immunoreactive cells were detected in the hippocampus of LPS-injected COX-1−/− mice (Fig. 7F).
Figure 7.

Effects of COX-1 deficiency on LPS-induced oxidative stress. COX-1+/+ and COX-1−/− mice received icv injection of LPS or vehicle 24 h before sacrifice. (A) Representative immunoblot of protein carbonyls. (B) Quantification of protein carbonyls. Data are mean ± SEM (n = 4). **P < 0.01 vs. corresponding vehicle-injected COX-1+/+ mice; #P < 0.05, ##P < 0.01 vs. corresponding LPS-injected COX-1+/+ mice. (C-F) Representative photomicrographs of nitrotyrosine immunohistochemistry in the hippocampus. Scale bar, 100 μm.
LPS-induced microglial transcription factor activation is decreased in COX-1−/− mice
Activation of NF-κB and signal STAT3 transcription factors plays a critical role in the production of inflammatory mediators by activated microglia (40). To determine the transcriptional mechanism underlying the decreased inflammatory response to LPS observed in the COX-1−/− mice, we determined NF-κB p65 and STAT3 binding activity. NF-κ binding activity was significantly increased in LPS-injected COX-1+/+ compared to vehicle-injected COX-1+/+ mice (P < 0.01; Fig. 8A), but was not increased in LPS-injected COX-1−/− mice (Fig. 8A). STAT3 binding activity was also significantly increased 24 h after LPS injection in both COX-1+/+ and COX-1−/− mice compared to the vehicle-injected COX-1+/+ mice (P < 0.01; Fig. 8B). However, the increase in STAT3 activation in COX-1−/− mice was less than in COX-1+/+ mice (P < 0.01; Fig. 8B). The action of STAT signaling may be very transient, as its activation can induce the negative regulator, such as suppressors of cytokine signaling (SOCS), and exert a control on STAT phosphorylation (41). Increased expression of SOCS3 mRNA after LPS administration was not significantly different between COX-1+/+ and COX-1−/− mice (data not shown).
Figure 8.
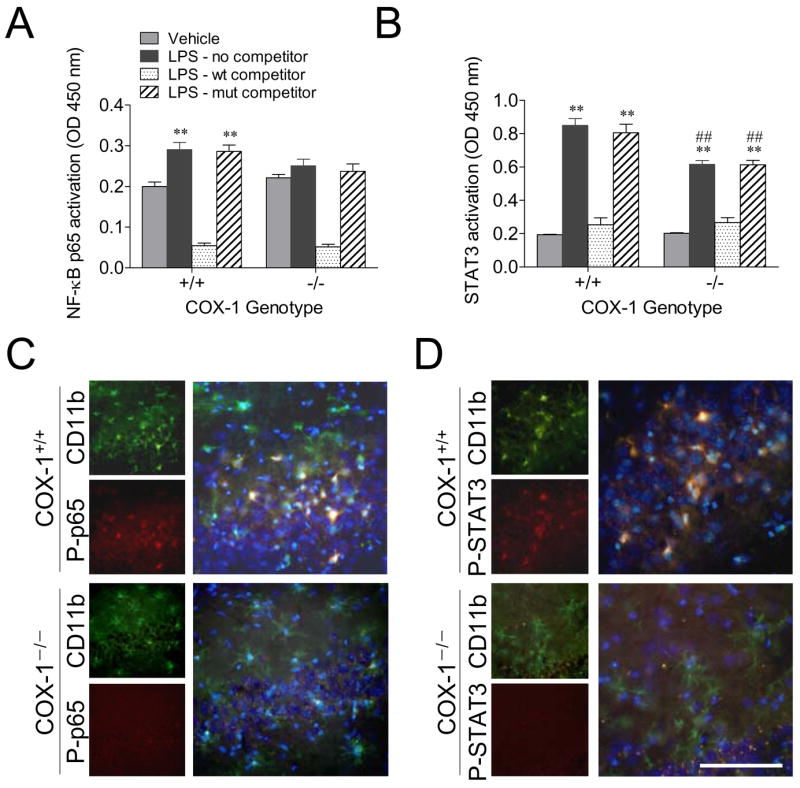
Effects of COX-1 deficiency on LPS-induced activation of transcription factors NF-κB and STAT3. COX-1+/+ and COX-1−/− mice received icv injection of LPS or vehicle 24 h before sacrifice. ELISA-based immunoassay of NF-κ p65 (A) and STAT3 DNA binding activity (B). Data are mean ± SEM (n = 6). **P < 0.01 vs. corresponding vehicle-injected COX-1+/+ mice; #P < 0.05, ##P < 0.01 vs. corresponding LPS-injected COX-1+/+ mice. Immunofluorescence photomicrographs of P-p65 (C) and P-STAT3 immunoreactivity (D) within the activated microglia in the hippocampus, stained with anti-CD11b (green) and anti-P-STAT3 (red). Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm (C and D).
To characterize the cellular localization of activated NF-κ p65 and STAT3, double-immunofluorescence staining was performed with a combination of antibodies against P-p65 and CD11b or P-STAT3 and CD11b. Increased immunofluorescence of P-p65 (Fig. 8C) and P-STAT3 (Fig. 8D) was seen in the hippocampus, consistent with Western blot analyses (data not shown). Merged images of immunofluorescence on the same sections revealed that LPS-activated NF-κ p65 and STAT3 were colocalized within activated microglia in both wild-type and COX-1−/−, indicating that activation of these transcription factors that mediated the transcription of inflammatory mediators occurred specifically in microglial cells (Fig. 8B and D).
DISCUSSION
We report that 24 h after LPS injection COX-1 genetic deletion attenuates microglial and astrocyte activation, reduces proinflammatory cytokines expression, decreases protein carbonyls and nitrotyrosine levels, as well as the activation of NADPH oxidase, expression of iNOS and MPO, and prevents the loss of neuronal cells in the hippocampus. These data suggest that COX-1 facilitates activation of glial cells and supports inflammatory processes that evolve in neuronal damage. Selective pharmacological inhibition of COX-1 with SC-560 prior to LPS injection also significantly reduced LPS-induced prostaglandin production as well as the mRNA expression of pro-inflammatory cytokines.
LPS-induced neurodegeneration was likely mediated by activated microglia and increased proinflammatory cytokines and chemokines. The direct involvement of microglial activation and the subsequent release of inflammatory mediators in the process of neurodegeneration are supported by in vitro evidence that LPS does not induce cell death in neuron-enriched culture condition and that cell-free supernatant from LPS-stimulated microglia cultures induces significant cell death in pure neuronal culture (42, 43). In this regard, cytokines such as IL-1β and TNF- α are thought to contribute to neuronal death in models of acute CNS injury as well as in chronic neurodegenerative disease (44).
The signal transduction cascade of TNF-α, IL-1β, and IL-6 has a point of convergence in the upstream NF-κ-inducing kinase and STAT3. Upon induction of these signal transduction pathways, NF-κ and STAT3 translocate to the nucleus, where they induce transcription of many genes critical in the inflammatory response, including cytokines and chemokines (40, 45). When microglia are stimulated by LPS, the cascade of intracellular signaling events lead to NADPH oxidase and iNOS activation and the subsequent release of oxygen radicals and NO in activated microglia. The induction of O2−, as well as NO, from activated microglia can enhance the production of more potent free radicals such as peroxynitrite (ONOO−). In addition, O2− and NO act as potent cell signaling molecules and amplify production of LPS-induced TNF-α and PGE2 by up-regulation of COX-2 (38). These initial effects combined with the activation of secondary signaling cascades activate a robust immune response that consequently causes neuronal injury and death (46). Therefore, the inhibition of microglial production of reactive oxygen and nitrogen species may be neuroprotective. We show that genetic deletion of COX-1 significantly reduces LPS-induced expression of both O2− and NO-forming enzymes. Indeed, the levels of nitrotyrosine and protein carbonyls, which are biomarkers of oxidative stress, were reduced in LPS-injected COX-1−/− compared to COX-1+/+ mice.
In the past decade, COX-2 has been viewed as the major player in inflammation and in inflammatory processes associated with certain neurodegenerative diseases, including AD. This has led to the preferential use of selective COX-2 inhibitors to reduce chronic inflammation and pain and to avoid undesired side effects of NSAIDs (3). Although, the precise role and contribution of COX-1 to the inflammatory cascade in neurodegenerative diseases has not been clearly established, we have recently shown that COX-1 mRNA expression was selectively upregulated in rat hippocampus during normal aging, possibly increasing the susceptibility of the aging brain to neuroinflammation (47). Interestingly, significant COX-1-dependent PGE2 production is also reported in several experimental animal models of brain injury such as global cerebral ischemia and NMDA-induced excitotoxicity (6, 7, 29). In addition, COX-1-expressing microglia accumulated in ischemic and traumatic lesions (8, 48), as well as in amyloid plaques of AD patients (9). In the present study, we found that LPS-induced PGE2 production and the mRNA expression of enzymes involved in the release and metabolism of AA, including cPLA2, COX-2, and mPGES-1, are significantly reduced in COX-1−/− mice. While one could speculate that the reduced neuroinflammatory response to LPS may be caused by a decreased COX-2 expression and PGE2 production, ongoing studies from our laboratory indicate that COX-2−/− mice, in contrast, show increased glial activation and inflammatory markers after LPS injection compared to their wild types (Aid et al., unpublished observations). Importantly, icv LPS did not induce increases in the mPGES-1 mRNA or COX-2 immunoreactivity in neurons (49), supporting the idea that COX-2 activation and PGE2 production occurs mainly in non-neuronal cells such as microglia in COX-1+/+ mice. These results suggest that COX-1 activity can exacerbate LPS-induced production of PGE2 by microglia. In turn, augmented PGE2 secretion may act in an autocrine and paracrine manner to further potentiate microglial function. Overall, these data suggest that COX-1 plays a key role in the neuroinflammatory response.
The results from epidemiological data indicating that NSAIDs are effective in preventing or delaying the onset of AD combined with the failure of COX-2 selective inhibitors in clinical trials in AD patients with moderate to severe AD suggest that an early treatment is crucial to stop the mechanisms underlying the disease before the onset of the symptoms or that COX-2 selective inhibitors are not effective in delaying the progression of AD. In this regard, an intriguing hypothesis is that the protective effects of NSAIDs may be related to COX-1 rather than COX-2 inhibition. Supporting this hypothesis, COX-1 selective inhibitors (SC-560 and valeryl salicylate), but not COX-2 selective inhibitors (SC-236 and DuP-697), reduce Aβ1–42-induced PGs production and neurotoxicity in postmortem human microglia and in murine cortical neurons (50, 51). Furthermore, a small double blind, placebo-controlled study with indomethacin, a preferential COX-1 inhibitor (52), appeared to protect mild to moderately impaired AD patients from cognitive decline (53). Interestingly, COX-1 is prominently expressed by microglia in rodent and human brain (9, 54) and appears to be increased in AD brain (9) and in activated microglia in association with amyloid deposits (50). In contrast, COX-2 has not been detected in microglia and astrocytes in AD (55). These combined data suggest that COX-2 may not be the exclusive COX isoform responsible for pathophysiological consequences in neurodegenerative diseases, especially in AD, but that COX-1 also plays a critical role in the process of neuroinflammation and neurodegeneration.
In summary, we show that, likely because of its predominant expression in microglia, COX-1 facilitates activation of glial cells and supports inflammatory processes and that genetic deletion of COX-1 significantly attenuates the neuroinflammatory response, oxidative stress and neuronal damage in response to LPS. Therefore, COX-1 may represent a viable therapeutic target to treat neuroinflammation.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. We thank Drs. Saba Aid, Christopher D. Toscano, and Miles Herkenham for experimental suggestions and critical comments, and Dr. Alan B. Zonderman for statistical help.
References
- 1.Langenbach R, Loftin C, Lee C, Tiano H. Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem Pharmacol. 1999;58:1237–1246. doi: 10.1016/s0006-2952(99)00158-6. [DOI] [PubMed] [Google Scholar]
- 2.Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002:68–69. 535–556. doi: 10.1016/s0090-6980(02)00054-0. [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- 4.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U S A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al-Dalain S, Martinez G, Leon OS, Springer JE. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem. 2003;86:545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pepicelli O, Fedele E, Berardi M, Raiteri M, Levi G, Greco A, Ajmone-Cat MA, Minghetti L. Cyclo-oxygenase-1 and -2 differently contribute to prostaglandin E2 synthesis and lipid peroxidation after in vivo activation of N-methyl-D-aspartate receptors in rat hippocampus. J Neurochem. 2005;93:1561–1567. doi: 10.1111/j.1471-4159.2005.03150.x. [DOI] [PubMed] [Google Scholar]
- 8.Schwab JM, Beschorner R, Meyermann R, Gozalan F, Schluesener HJ. Persistent accumulation of cyclooxygenase-1-expressing microglial cells and macrophages and transient upregulation by endothelium in human brain injury. J Neurosurg. 2002;96:892–899. doi: 10.3171/jns.2002.96.5.0892. [DOI] [PubMed] [Google Scholar]
- 9.Yermakova AV, Rollins J, Callahan LM, Rogers J, O’Banion MK. Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol. 1999;58:1135–1146. doi: 10.1097/00005072-199911000-00003. [DOI] [PubMed] [Google Scholar]
- 10.McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 11.Nelson PT, Soma LA, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- 12.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 13.in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 14.Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, Pericak-Vance MA, Saunders AM. Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging. 1995;16:523–530. doi: 10.1016/0197-4580(95)00049-k. [DOI] [PubMed] [Google Scholar]
- 15.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 16.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 17.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. Jama. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 18.Scharf S, Mander A, Ugoni A, Vajda F, Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology. 1999;53:197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- 19.Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 20.Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, Reines SA. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30:1204–1215. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 21.Bosetti F. Arachidonic acid metabolism in brain physiology and pathology: lessons from genetically altered mouse models. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem. 2003;87:1518–1526. doi: 10.1046/j.1471-4159.2003.02120.x. [DOI] [PubMed] [Google Scholar]
- 24.Montine TJ, Milatovic D, Gupta RC, Valyi-Nagy T, Morrow JD, Breyer RM. Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J Neurochem. 2002;83:463–470. doi: 10.1046/j.1471-4159.2002.01157.x. [DOI] [PubMed] [Google Scholar]
- 25.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 26.Delgado M, Ganea D. Vasoactive intestinal peptide prevents activated microglia-induced neurodegeneration under inflammatory conditions: potential therapeutic role in brain trauma. Faseb J. 2003;17:1922–1924. doi: 10.1096/fj.02-1029fje. [DOI] [PubMed] [Google Scholar]
- 27.Milatovic D, Zaja-Milatovic S, Montine KS, Shie FS, Montine TJ. Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immune response in vivo. J Neuroinflammation. 2004;1:20. doi: 10.1186/1742-2094-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Academic, San Diego, Calif; London: 2001. [Google Scholar]
- 29.Moore AH, Olschowka JA, Williams JP, Okunieff P, O’Banion MK. Regulation of prostaglandin E2 synthesis after brain irradiation. Int J Radiat Oncol Biol Phys. 2005;62:267–272. doi: 10.1016/j.ijrobp.2005.01.035. [DOI] [PubMed] [Google Scholar]
- 30.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 31.Choi SH, Lee da Y, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci. 2005;25:4082–4090. doi: 10.1523/JNEUROSCI.4306-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi SH, Langenbach R, Bosetti F. Cyclooxygenase-1 and -2 enzymes differentially regulate the brain upstream NF-kappa B pathway and downstream enzymes involved in prostaglandin biosynthesis. J Neurochem. 2006;98:801–811. doi: 10.1111/j.1471-4159.2006.03926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell WS. Reversed-phase high-pressure liquid chromatography of arachidonic acid metabolites formed by cyclooxygenase and lipoxygenases. Anal Biochem. 1985;148:59–69. doi: 10.1016/0003-2697(85)90628-1. [DOI] [PubMed] [Google Scholar]
- 34.Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 35.Herber DL, Maloney JL, Roth LM, Freeman MJ, Morgan D, Gordon MN. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia. 2006;53:382–391. doi: 10.1002/glia.20272. [DOI] [PubMed] [Google Scholar]
- 36.Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- 37.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 38.Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 39.Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 40.Kim HY, Park EJ, Joe EH, Jou I. Curcumin suppresses Janus kinase-STAT inflammatory signaling through activation of Src homology 2 domain-containing tyrosine phosphatase 2 in brain microglia. J Immunol. 2003;171:6072–6079. doi: 10.4049/jimmunol.171.11.6072. [DOI] [PubMed] [Google Scholar]
- 41.Planas AM, Gorina R, Chamorro A. Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem Soc Trans. 2006;34:1267–1270. doi: 10.1042/BST0341267. [DOI] [PubMed] [Google Scholar]
- 42.Jeohn GH, Kong LY, Wilson B, Hudson P, Hong JS. Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol. 1998;85:1–10. doi: 10.1016/s0165-5728(97)00204-x. [DOI] [PubMed] [Google Scholar]
- 43.Suzumura A, Takeuchi H, Zhang G, Kuno R, Mizuno T. Roles of glia-derived cytokines on neuronal degeneration and regeneration. Ann N Y Acad Sci. 2006;1088:219–229. doi: 10.1196/annals.1366.012. [DOI] [PubMed] [Google Scholar]
- 44.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 45.Rummel C, Sachot C, Poole S, Luheshi GN. Circulating interleukin-6 induces fever through a STAT3-linked activation of COX-2 in the brain. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1316–1326. doi: 10.1152/ajpregu.00301.2006. [DOI] [PubMed] [Google Scholar]
- 46.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 47.Aid S, Bosetti F. Gene expression of cyclooxygenase-1 and Ca(2+)-independent phospholipase A(2) is altered in rat hippocampus during normal aging. Brain Res Bull. 2007;73:108–113. doi: 10.1016/j.brainresbull.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwab JM, Brechtel K, Nguyen TD, Schluesener HJ. Persistent accumulation of cyclooxygenase-1 (COX-1) expressing microglia/macrophages and upregulation by endothelium following spinal cord injury. J Neuroimmunol. 2000;111:122–130. doi: 10.1016/s0165-5728(00)00372-6. [DOI] [PubMed] [Google Scholar]
- 49.Xia Y, Yamagata K, Krukoff TL. Differential expression of the CD14/TLR4 complex and inflammatory signaling molecules following i.c.v. administration of LPS. Brain Res. 2006;1095:85–95. doi: 10.1016/j.brainres.2006.03.112. [DOI] [PubMed] [Google Scholar]
- 50.Hoozemans JJ, Veerhuis R, Janssen I, van Elk EJ, Rozemuller AJ, Eikelenboom P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer’s disease. Brain Res. 2002;951:218–226. doi: 10.1016/s0006-8993(02)03164-5. [DOI] [PubMed] [Google Scholar]
- 51.Bate C, Veerhuis R, Eikelenboom P, Williams A. Neurones treated with cyclo-oxygenase-1 inhibitors are resistant to amyloid-beta1-42. Neuroreport. 2003;14:2099–2103. doi: 10.1097/00001756-200311140-00018. [DOI] [PubMed] [Google Scholar]
- 52.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, et al. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 53.Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 54.Hoozemans JJ, Rozemuller AJ, Janssen I, De Groot CJ, Veerhuis R, Eikelenboom P. Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol (Berl) 2001;101:2–8. doi: 10.1007/s004010000251. [DOI] [PubMed] [Google Scholar]
- 55.Hoozemans JJ, Veerhuis R, Rozemuller AJ, Eikelenboom P. Non-steroidal anti-inflammatory drugs and cyclooxygenase in Alzheimer’ disease. Curr Drug Targets. 2003;4:461–468. doi: 10.2174/1389450033490902. [DOI] [PubMed] [Google Scholar]