

Abstract
Hexavalent chromium [Cr(VI)] species such as chromates are cytotoxic. Inhalational exposure is a primary concern in many Cr-related industries and their immediate environments, and bronchial epithelial cells are directly exposed to inhaled Cr(VI). Chromates are readily taken up by cells and are reduced to reactive Cr species which may also result in the generation of reactive oxygen species (ROS). The thioredoxin (Trx) system has a key role in the maintenance of cellular thiol redox balance and is essential for cell survival. Cells normally maintain the cytosolic (Trx1) and mitochondrial (Trx2) thioredoxins largely in the reduced state. Redox western blots were used to assess the redox status of the thioredoxins in normal human bronchial epithelial cells (BEAS-2B) incubated with soluble Na2CrO4 or insoluble ZnCrO4 for different periods of time. Both chromates caused a dose- and time-dependent oxidation of Trx2 and Trx1. Trx2 was more susceptible in that it could all be converted to the oxidized form, whereas a small amount of reduced Trx1 remained even after prolonged treatment with higher Cr concentrations. Only one of the dithiols, presumably the active site, of Trx1 was oxidized by Cr(VI). Cr(VI) did not cause significant GSH depletion or oxidation indicating that Trx oxidation does not result from a general oxidation of cellular thiols. With purified Trx and thioredoxin reductase (TrxR) in vitro, Cr(VI) also resulted in Trx oxidation. It was determined that purified TrxR has pronounced Cr(VI) reducing activity, so competition for electron flow from TrxR might impair its ability to reduce Trx. The in vitro data also suggested some direct redox interaction between Cr(VI) and Trx. The ability of Cr(VI) to cause Trx oxidation in cells could contribute to its cytotoxic effects, and could have important implications for cell survival, redox-sensitive cell signaling, and the cells' tolerance of other oxidant insults.
Keywords: Chromium, Chromate, Thioredoxin, Thioredoxin reductase, Redox blot, Lung
1. Introduction
Exposure to hexavalent chromium [Cr(VI)] is associated with a wide range of toxic effects (Whiting et al., 1979; Hayes, 1982; Tsapakos et al., 1983; Cantoni and Costa, 1984; Christie et al., 1984; Levy and Venitt, 1986; Langard, 1993; Raithel et al., 1993; Ishikawa et al., 1994a; Ishikawa et al., 1994b; Deschamps et al., 1995; Taioli et al., 1995; Costa et al., 1996). The greatest human exposures are from industrial uses, including chromate pigments, zinc chromate primer paints and other corrosion inhibitors, stainless steel machining and welding, chrome plating, leather tanning, and others. These industrial uses also result in the annual release of more than 105 tons of Cr, and Cr has become a contaminant of significant concern at many sites in the United States (Gadd and White, 1993; Zhitkovich et al., 1998; EPA, 1999).
Several Cr(VI) compounds are soluble (e.g. sodium or potassium chromates) and readily enter cells via an anion carrier (Buttner and Beyersmann, 1985). Some industrially used chromates (e.g. ZnCrO4) are water-insoluble but are still implicated in toxicity. Once inside cells, Cr(VI) is reduced by a variety of chemical and enzymatic reductants (Jennette, 1982; Mikalsen et al., 1989; Shi and Dalal, 1990; Suzuki and Fukuda, 1990; Standeven and Wetterhahn, 1992; Shi et al., 1994; Myers et al., 2000; Borthiry et al., 2007), eventually to the next stable oxidation state, Cr(III). During this reduction, reactive Cr species [Cr(V) and/or Cr(IV)] are formed. These can directly cause oxidative-like damage (Sugden, 1999; Sugden et al., 2001) or they can generate ROS via redox cycling (Tsapakos et al., 1983; Standeven and Wetterhahn, 1991; Shi and Dalal, 1992; Dillon et al., 1998; Shi et al., 1999a; Shi et al., 1999b; Borthiry et al., 2007).
Inhalation of Cr-containing fumes, dusts, and particles is a prominent form of exposure, so respiratory effects of Cr (pulmonary fibrosis, chronic bronchitis, and lung cancer) are of special concern (Franchini et al., 1983; Stern, 1983; Nakagawa et al., 1984; Becker et al., 1991; Ishikawa et al., 1994a; Deschamps et al., 1995). In the lung, bronchial epithelial cells line the airways and are therefore directly exposed to inhaled chromium.
The redox balance of cellular thiols is critical for normal function and viability. While the Trx system and glutathione contribute significantly to the maintenance of cellular thiol redox balance, these systems are not in redox equilibrium with each other (Nkabyo et al., 2002; Hansen et al., 2006a). A major role of the thioredoxins is to maintain intracellular proteins in their reduced state (Arner and Holmgren, 2000), and the redox status of the thioredoxin (Trx) system in some cells may be more critical to cell survival than is glutathione.
Mammalian cells have both cytosolic (Trx1) and mitochondrial (Trx2) thioredoxins (Powis and Montfort, 2001). Each are encoded by distinct genes, but they share a conserved active site (Trp-Cys-Gly-Pro-Cys-Lys) that is cycled between the reduced (dithiol) and oxidized (disulfide) forms (Watson et al., 2003). Cells normally maintain Trx1 and Trx2 largely in the reduced state (Nordberg and Arner, 2001; Powis and Montfort, 2001; Watson et al., 2003) which is the active form. Trx2 has two Cys residues, both in the active site. Trx1 has five Cys residues, two in the active site (C32/C35), and another two in a second dithiol motif (C62/C69) (Watson et al., 2003). Of the two dithiols in Trx1, the C32/C35 active site is more readily oxidized (Watson et al., 2003).
The thioredoxins are presumed to be essential for cell survival as knockout mice lacking either Trx1 or Trx2 do not survive (Powis and Montfort, 2001; Nonn et al., 2003). Genetic suppression or inhibition of Trx results in increased ROS and apoptosis (Hansen et al., 2006a) and increased sensitivity of cells to oxidants (Chen et al., 2006), whereas overexpression of Trx2 enhances protection from oxidant-induced apoptosis (Chen et al., 2006; Hansen et al., 2006a). Factors which enhance Trx oxidation would therefore be expected to interfere with Trx activity and could decrease cell survival. Various types of oxidants can result in Trx oxidation, including t-butyl hydroperoxide, ROS and the non-specific thiol oxidant diamide (Halvey et al., 2005; Chen et al., 2006; Hansen et al., 2006a). Arsenic and mercury can cause Trx oxidation (Hansen et al., 2006b) but the mechanisms involved are not clear. The effects of Cr on the thioredoxin system have not been explored, but its redox active nature and ability to generate ROS suggest that it could have an impact.
The purpose of the studies reported here was to determine if Cr(VI) treatment of human bronchial epithelial cells can result in Trx oxidation. The studies support the hypothesis, demonstrating that both soluble and insoluble chromates result in Trx1 and Trx2 oxidation that is related to the Cr(VI) concentration and the time of exposure. In vitro studies with purified proteins demonstrate that there are direct redox interactions of Cr(VI) with Trx and Trx reductase (TrxR).
2. Materials and methods
2.1 Reagents
The following were purchased from Invitrogen Corp. (Carlsbad, CA): Hank’s balanced salt solution (HBSS), 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonate (AMS), and pre-cast gels (12% Bis-Tris, 16% Tris-Glycine) and matching electrophoresis and loading buffers. Phenylmethylsulfonyl fluoride (PMSF) and Tris were from Research Organics (Cleveland, OH). EDTA, guanidine-HCl, trichloroacetic acid (TCA), K2CO3, and methanol (HPLC grade) were obtained from Fisher Scientific (Hampton, NH). Sodium acetate trihydrate was from JT Baker (Phillipsburg, NJ) and glacial acetic acid was from EMD Chemicals (Gibbstown, NJ). γ-Glutamyl glutamate (γ-Glu-Glu) was obtained from MP Biochemicals (Solon, OH). Rabbit anti-human Trx1 and rabbit anti-human Trx2 were obtained from Abcam (Cambridge, MA). The Supersignal West Pico Chemiluminescent substrate was obtained from Pierce (Rockford, IL). Sodium chromate (99+%) was the highest purity available from Aldrich Chemical (Milwaukee, WI), and zinc chromate was from Pfaltz & Bauer (Waterbury, CT). Na253CrO4 was graciously provided by Dr. David H. Petering (University of Wisconsin–Milwaukee). Chromates are known carcinogens and should be handled accordingly. Purified human Trx1 (cat. no. T8690) and rat liver thioredoxin reductase (TrxR)(cat. no. T9698) were purchased from Sigma Chemical (St. Louis, MO). All other chemicals and reagents were purchased from Sigma Chemical or from the sources indicated.
2.2 Cell culture
BEAS-2B cells were obtained from the American Type Culture Collection (ATCC no. CRL-9609). They were grown at 37°C in humidified air containing 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM) with 25 mM HEPES and 4.5 g/L glucose (BioWhittaker 12-709F, Lonza, Inc.), 10% fetal bovine serum (FBS Optima, Atlanta Biologicals; or Valley Biomedical, Winchester, VA), penicillin (100 U/ml), and streptomycin (100 µg/ml). The cells were fed every 48 h, and were subcultured prior to reaching confluence using the Reagent Pak system (Clonetics, CC-5034). Normal plating density was 3000 to 5000 cells/cm2.
2.3 Trx1 redox blots
The redox status of Trx1 was determined as adapted from Halvey et al. (Halvey et al., 2005). Adherent cells (60–70% confluent) were washed once in pre-warmed HBSS, and treated with Cr(VI) in HBSS at 37°C for the indicated times. Following treatment, they were washed once in HBSS and scraped into chilled 6 M guanidine-HCl, 100 mM Tris-HCl pH 8.3, 3 mM EDTA, 0.5 % Triton X-100, 50 mM iodoacetic acid (Halvey et al., 2005). The samples were briefly sonicated on ice to shear the DNA, and guanidine and unreacted iodoacetate were removed using Microspin G25 columns (GE Healthcare, Piscataway NJ). The column eluates were mixed with an equal volume of loading buffer, separated by native PAGE (16% Tris-Glycine), transferred to nitrocellulose, blocked with 5% bovine serum albumin (BSA) and probed with anti-Trx1 and HRP-conjugated goat anti-rabbit IgG (Bio-Rad, Hercules, CA).
2.4 Trx2 redox blots
Trx2 redox status was determined according to Chen et al. (Chen et al., 2006). Cells were washed and treated with Cr(VI) as for Trx1 studies (above), but were then scraped into 10% TCA. Following incubation on ice for ≥30 min, they were briefly sonicated on ice, and centrifuged for 10 min at 4°C (12000 × g). The pellets were resuspended in ice-cold 100% acetone, iced for 30 min, and centrifuged for 15 min (12000 × g). The pellets were dried and then resuspended in 80 mM Tris-HCl pH 7, 2% SDS, 1 mM PMSF, 25 mM AMS. After incubation (15 min on ice, 10 min at 37°C) (Chen et al., 2006), the samples were centrifuged for 5 min (12000 × g), mixed with LDS loading buffer and separated by SDS-PAGE (12% Bis-Tris) under non-reducing conditions, and transferred to nitrocellulose. The blots were blocked with 5% milk (Carnation nonfat) and probed with anti-Trx2 and HRP-conjugated goat anti-rabbit IgG.
2.5 Isolation of mitochondria for Trx2 controls
Cells were harvested by scraping and centrifugation, and the pellet was washed in cold HBSS, cold STE (0.25 M sucrose, 10 mM Tris pH 7.2, 2 mM EDTA), and resuspended in a small volume of cold STE. Cells were lysed using a Teflon-on-glass Potter-Elvehjem homogenizer. The suspension was periodically examined and homogenization was considered complete when >90% of the cells were lysed. The lysate was centrifuged for 10 min at 4°C at 735 × g to pellet nuclei, large debris and unbroken cells. The supernatant fraction was transferred to a new tube and centrifuged for 10 min at 4°C (12000 × g) to pellet mitochondria. The mitochondria were washed and resuspended in STE, and stored at −20°C until use. Trx2 becomes oxidized in vitro, so the mitochondria were used directly as a reference for oxidized Trx2. To prepare a reference for reduced Trx2, the mitochondria were incubated with 20 mM dithiothreitol (DTT) for 10 min at room temperature. Both reduced and oxidized mitochondria were then mixed with TCA and processed through the AMS labeling procedure described above.
2.6 HPLC Analysis of Glutathione
Adherent cells (equivalent to one T150 flask) were washed twice in pre-warmed HBSS and scraped into ice-cold 5% TCA. The samples were placed on ice and TCA-insoluble proteins were removed by centrifugation at 4°C (9900 × g for 5 min). The pellets were saved for total protein analysis (see below). The acid supernatants were stored frozen until derivatization and analysis. Samples were derivatized and analyzed by HPLC using a method developed by Fariss and Reed (Fariss and Reed, 1987), with details provided by Dr. Tak Yee Aw (Pias and Aw, 2002). γ-Glu-Glu was added to 0.5 ml sample as an internal standard. Then, 0.05 ml 80 mM iodoacetic acid was added followed by 0.34 ml of 1 M K2CO3 to raise the pH to ≥8.0. After 60 min incubation at room temperature, 0.125 ml of 6% 1-fluoro-2,4-dinitrobenzene (in ethanol) was added, followed by 0.35 ml 1 M K2CO3 (to raise the pH to 10). Samples were incubated overnight in the dark at 4°C. Following centrifugation (9900 × g) at 4°C for 20 min at 11,000 rpm, an aliquot of each supernatant was filtered through a 0.45 µm Acrodisc syringe filter, and analyzed by HPLC. For HPLC, a Sphereclone NH2 5 µ column (250 × 4.6 mm) with a 4.6 mm guard column was used and absorbance was monitored at 365 nm. The mobile phase (flow rate of 1.5 ml per min) was 90% solvent A (80% methanol) and 10% solvent B (2% sodium acetate in 64% methanol) for the first five min. During the next 10 min, a linear gradient was applied ending at 1% solvent A and 99% solvent B. 1% A/99% B was maintained for 10 min, and then a gradient was applied to return and equilibrate the column to the starting conditions. The retention times of the peaks for GSH and GSSG in the samples were compared to those of GSH and GSSG standards that were derivatized in the same manner. GSH eluted at 11.8 to 12 min, whereas GSSG eluted 2 min later. Concentrations were determined from peak area relative to the standard curves, and were normalized per µg of protein.
For protein analysis, the TCA-insoluble pellets (from above) were dissolved in 0.1 M NaOH for ≥5 days at room temperature and then protein was determined by a modified Lowry method, with bovine serum albumin as the standard (Myers and Myers, 1997).
2.7 Electron Spin Resonance for Cr(V)
Cr(V) generation by purified enzymes was examined using direct detection by ESR in a buffer system with a final concentration of 0.15 M KCl/2.5 mM potassium phosphate pH 7.35. The NADPH-generating mix (Myers and Myers, 1998) (which maintains NADPH at a constant level of 0.2 mM) and purified Trx and TrxR were added to pre-warmed buffer and incubated for 5 min at 37°C. The enzymes and Na2CrO4 were added followed by incubation at 37°C for the indicated times. The reaction was stopped by immersion in liquid nitrogen (77 K), and samples were stored at 77 K, typically for less than one week, until analysis by ESR. It was previously shown that Cr(V) is stable for at least several months at 77 K (Jannetto et al., 2001).
Samples were quickly thawed, placed in a quartz flat cell and ESR spectra were obtained without delay at room temperature using a Bruker EMX spectrometer. Instrument settings are indicated in the results. ESR spectra were confirmed in replicate experiments. The g values were determined by comparison to the 2,2,-diphenyl-1-picrylhydrazyl radical (DPPH) which has a g value of 2.0036.
2.8 Electron Spin Resonance Spin Trapping
The spin trap 5-Diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO) was from Oxis International (Portland, OR) or Alexis Biochemicals (Lausen, Switzerland). The generation of reactive oxygen species (ROS) in Cr(VI)-exposed cells was assessed at fixed time points using ESR spin trapping. The procedures are similar to that described previously for experiments with microsomal enzymes (Borthiry et al., 2007). The cells were grown in DMEM medium as described above, harvested by scraping into medium, and centrifuged for 5 min at 100 × g. The cell pellet was washed in LHC-9 medium (Invitrogen) and resuspended in a small volume of LHC-9. It has been previously shown that LHC-9 medium does not reduce Cr(VI) (Borthiry et al., 2008). Cell density was determined by counting an aliquot using a hemacytometer. Aliquots of the cell suspension were incubated at 37°C in LHC-9 medium containing DEPMPO (14 mM) with and without Cr(VI) (400 µM). After the indicated times, the samples were immersed and stored in liquid nitrogen. Freezing and thawing does not change the ESR spectra of DEPMPO adducts (Frejaville et al., 1995; Tordo, 1998). For ESR analysis, each sample was quickly thawed, placed in a quartz flat cell and ESR spectra were obtained without delay at room temperature using a Bruker EMX spectrometer. Instrument settings are indicated in the results. The spectrum for each sample was stable during the ESR analysis time. The hyperfine couplings were measured from the ESR scan data, and were confirmed using the simulation software WinSim (version 0.95) (National Institutes of Environmental Health Sciences) (Duling, 1994).
3. Results
3.1 Sodium chromate results in Trx2 oxidation
The redox status of the mitochondrial form (Trx2) was examined by redox western blot. Trx2 has a single dithiol (the active site). Reduced Trx2 migrates slower in these gels because AMS forms covalent bonds with the two –SH groups of the active site thereby increasing the mass of Trx2 (Hansen et al., 2006a). If the active site is oxidized, AMS cannot bind and Trx2 runs according to its native mass. Untreated cells were compared to those exposed to various concentrations of Na2Cr(VI)O4 for between 10 and 180 min. Representative redox blots and the percent of Trx2 that is oxidized are shown in Fig. 1A,B. In untreated cells, all of Trx2 was in the reduced form (–SH,–SH) at all timepoints, i.e. it migrated coincident with Trx2 in DTT-treated mitochondria. Cr(VI) exposure resulted in Trx2 oxidation in BEAS-2B cells, and this oxidation was dependent on both the Cr(VI) concentration and the duration of exposure (Fig. 1B). While 10 µM Cr(VI) for 30 min did not oxidize Trx2, exposure for 90 or 180 min resulted in 12 to 28% oxidation (Fig. 1B). Higher Cr(VI) (50 µM) caused Trx2 oxidation more quickly, with 86% oxidation apparent at 10 min, and 30 min or longer converting 100% of Trx2 to the oxidized form (Fig. 1B), i.e. it migrated coincident with Trx2 in air-oxidized mitochondria. In a single trial, 100 µM Cr(VI) (100 µM) resulted in complete Trx2 oxidation at all timepoints, including the 10 min exposure (not shown).
Fig. 1.
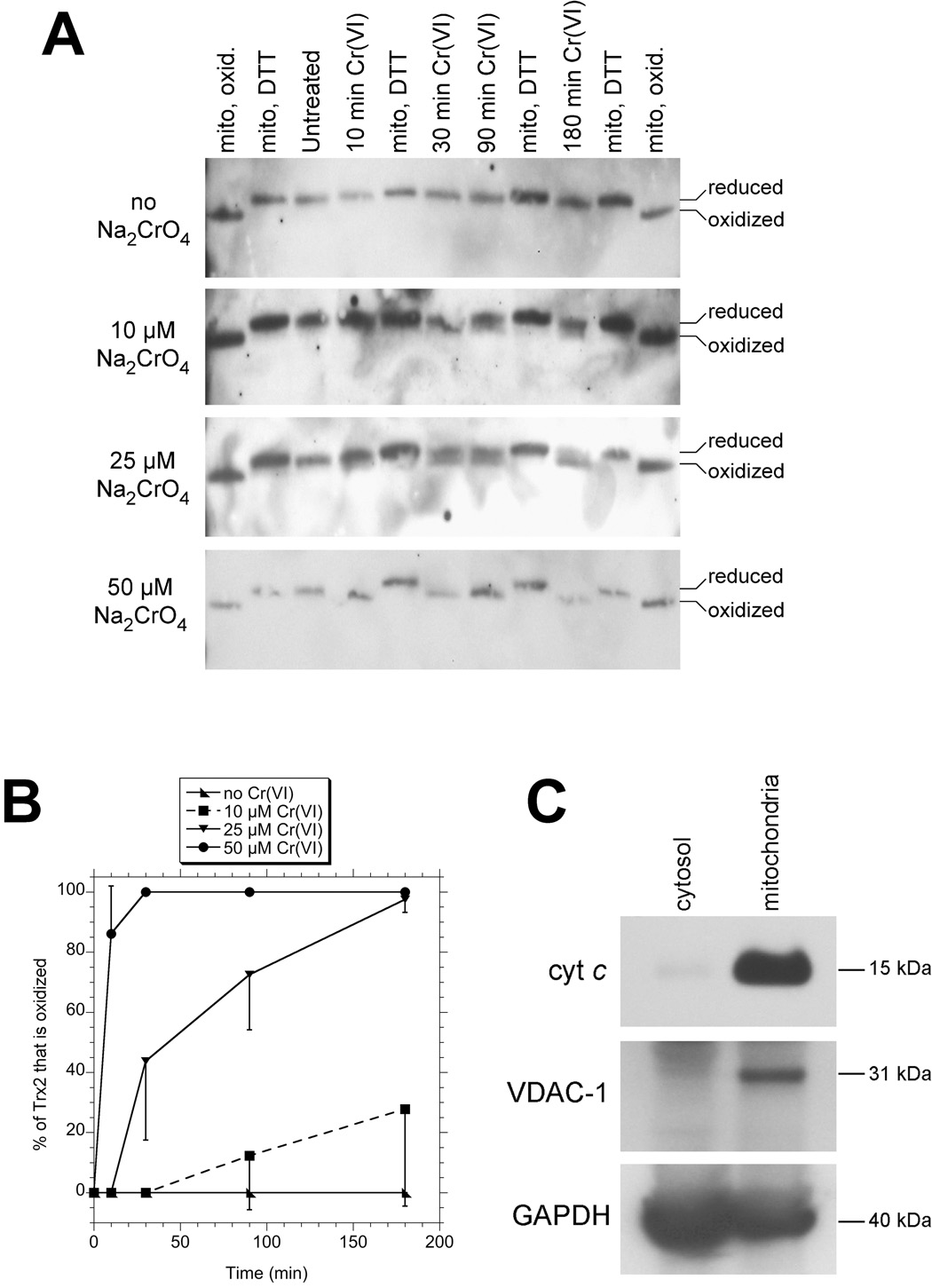
A: Redox western blots of Trx2. BEAS-2B cells were either untreated or treated with the indicated concentrations (left) of NaCr(VI)O4 for the indicated times (top). The cells were then washed once in HBSS and immediately processed for Trx2 redox status. Standards for the migration of oxidized and reduced Trx2 are included, and consist of isolated mitochondria that had either been stored frozen (“mito, oxid.”) (Trx2 oxidizes in storage), or had been pre-treated with DTT which reduces Trx2 (“mito, DTT”). Oxidized Trx2 migrated at 12 kDa consistent with its known mass. The blots shown are representative of triplicate independent experiments.
B: Percent (mean ± S.D.) of total Trx2 that is oxidized as determined by densitometry of redox blots from triplicate experiments. The error bars are shown in one direction to avoid visual confusion.
C: Western blots of purified mitochondrial and cytosolic fractions showing the distribution of the mitochondrial markers cytochrome c and VDAC-1, and for the loading control GAPDH which is found in various cellular locations including the cytosol and mitochondria (Tarze et al., 2007).
The authenticity of the purified mitochondria used as standards in the Trx2 redox blots was verified by western blots. The purified mitochondria contained the mitochondrial markers VDAC-1 and cytochrome c, whereas these markers were absent in the post-12000 × g supernatant fraction (cytosol) (Fig. 1C).
3.2 Sodium chromate results in Trx1 oxidation
The effect of Na2Cr(VI)O4 treatment on the redox status of Trx1 (cytosolic form) in cells was also analyzed by redox western blot. Iodoacetate covalently labels the –SH groups of reduced Trx1; the resulting negative charge increases migration in native PAGE relative to oxidized Trx1. Reduced Trx1 refers to the form in which both dithiols are reduced and represents the active form. Reduced Trx1 migrates the fastest because it binds iodoacetate at both dithiols. The active site dithiol is the more easily oxidized, so partially oxidized Trx1 largely represents oxidation of the active site C32/C35 (Watson et al., 2003), whereas the fully oxidized form migrates the slowest because both dithiols (C32/C35, C62/C69) are oxidized.
Untreated cells were compared to those exposed to various concentrations of Na2Cr(VI)O4 for 30, 90, or 180 min. Representative redox blots are shown in Fig. 2A,B and the percent of Trx1 that is partially oxidized (one dithiol oxidized) is shown in Fig. 2D. In untreated cells, >95% of Trx1 was in the reduced state (Fig. 2A,D). Cr(VI) treatment caused increases in partially oxidized Trx1 which were dependent on both the Cr(VI) concentration and time of exposure (Fig. 2D). Even with high Cr(VI) for extended time (100 µM for 180 min), only the partially oxidized form was seen (Fig. 2B). This is in contrast to acrolein which can generate both partially and fully oxidized Trx1 (Fig. 2C). These acrolein results are consistent with those previously reported for human endothelial cells (Szadkowski and Myers, 2007). For each Cr(VI) concentration, the majority of Trx1 oxidation had occurred by 30 min with further smaller increases observed at later times (Fig. 2D).
Fig. 2.
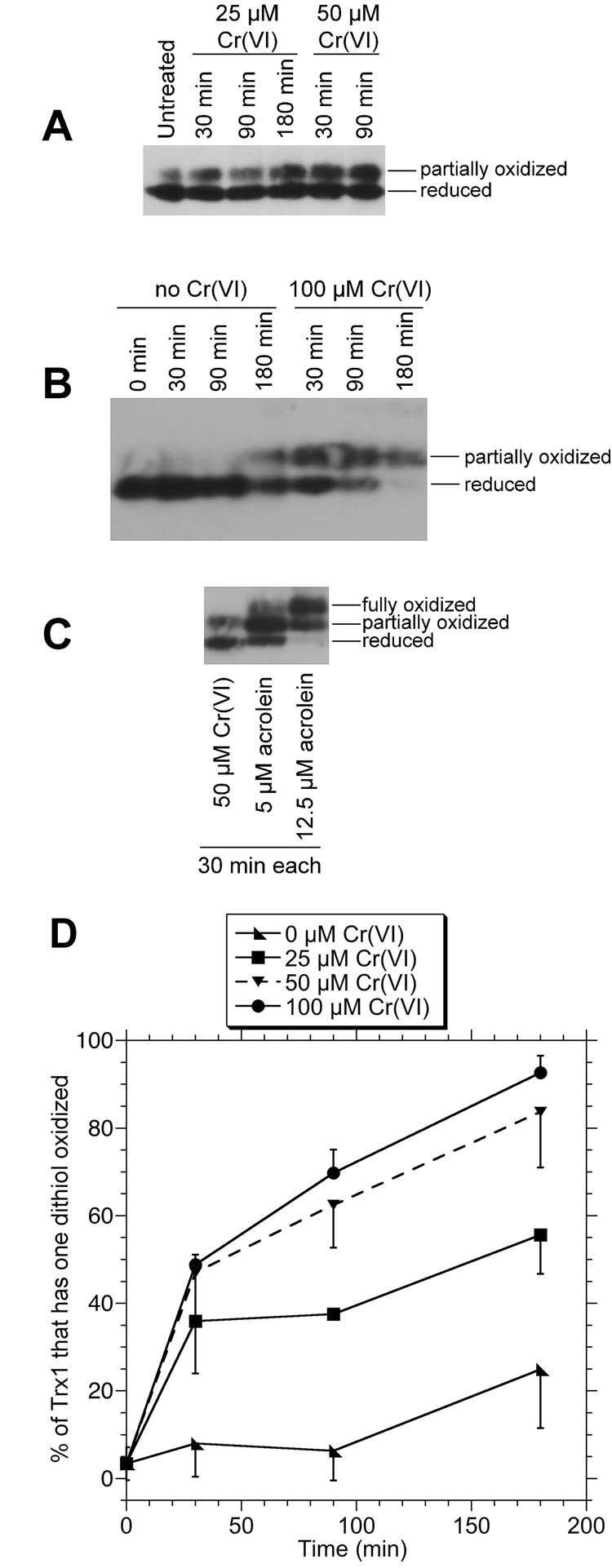
A,B: Redox western blots of Trx1 in BEAS-2B cells treated with the indicated concentrations of Na2Cr(VI)O4 for the indicated times, washed once in HBSS and immediately processed for Trx1 redox status. C: Redox western blot of cells treated for 30 min with either 50 µM Na2Cr(VI)O4 or acrolein (5 or 12.5 µM). D: Percent (mean ± S.D.) of total Trx1 that has one dithiol oxidized as determined by densitometry of the redox blots from triplicate independent experiments. The error bars are shown in one direction to avoid visual confusion. For some, the error bars are smaller than the points as shown.
3.3 Trx oxidation by an insoluble chromate
The data to this point were generated with Na2CrO4, which is highly water soluble. Insoluble forms of chromate (e.g. ZnCrO4) are also used industrially and therefore occupational exposure is of concern. The potential for ZnCrO4 to mediate Trx oxidation was therefore examined. With 50 µM ZnCrO4, some of the Trx2 was oxidized after just 10 min, most was oxidized after 30 min, and Trx2 was completely oxidized after 90 min and longer (Fig. 3A). For Trx1, progressive increases in Trx1 oxidation were observed over time, although a small percentage of Trx1 remained reduced even after 180 min with 50 µM ZnCrO4 (Fig. 3B). As with Na2CrO4, Trx1 was only converted to the partially oxidized form in ZnCrO4-treated cells, in contrast to acrolein which also generates fully oxidized Trx1 (Fig. 3B). The effects of ZnCrO4 are due to the chromate and not the Zn because 100 µM ZnCl2 causes no detectable oxidation of Trx1 or Trx2 (Hansen et al., 2006b).
Fig. 3.
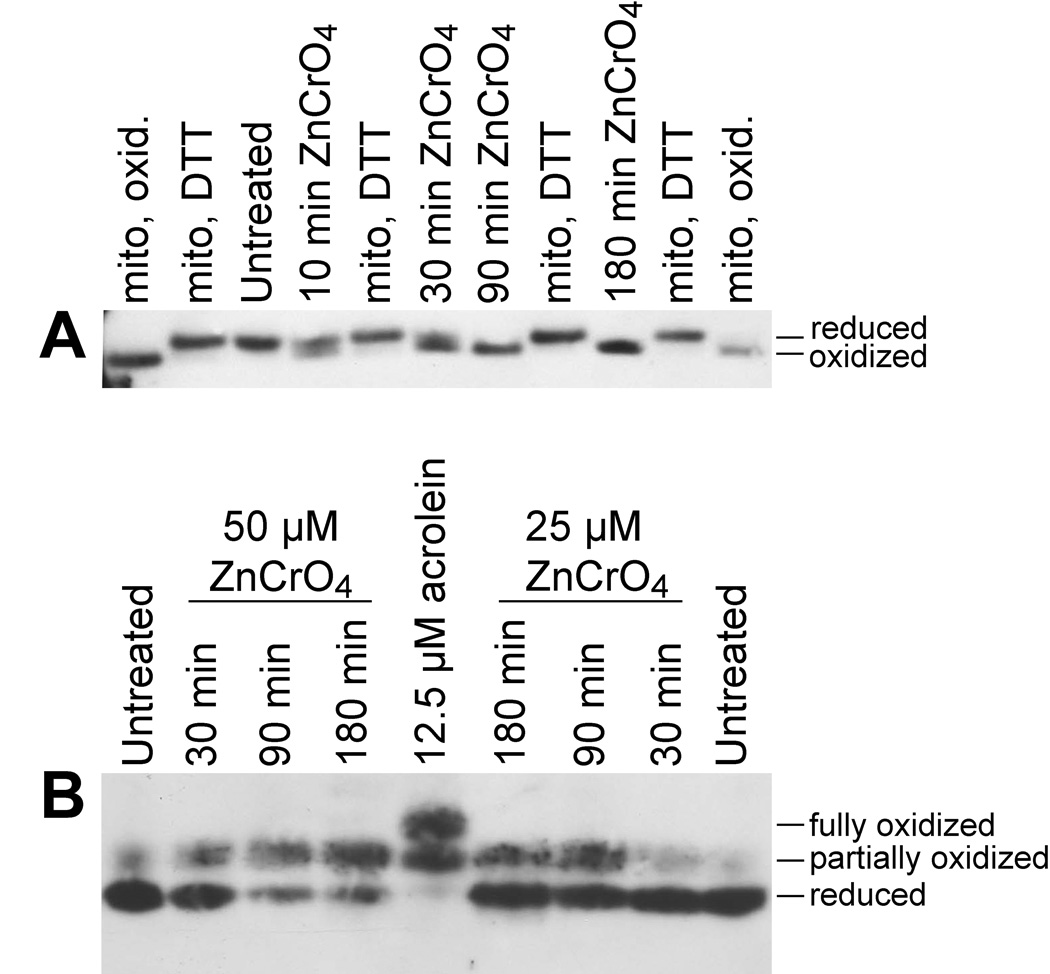
Redox western blots of Trx2 (A) or Trx1 (B) in BEAS-2B cells that were either untreated or treated with ZnCr(VI)O4. In A, cells were treated with 50 µM ZnCr(VI)O4 for the indicated times, and standards for the migration of oxidized Trx2 (“mito, oxid.”) and reduced Trx2 (“mito, DTT”) are included as described in Fig. 1. In B, cells were treated with 25 or 50 µM ZnCr(VI)O4 for the indicated times. Cells treated with 12.5 µM acrolein are included in the center lane to demonstrate migration of the partially and fully oxidized forms of Trx1. The blots shown are representative of replicate experiments.
3.4 Cr(VI) and GSH levels
To determine whether Cr(VI) has the capacity to deplete cellular thiols in general in these cells, the cellular content of reduced glutathione (GSH) was examined in cells treated with Na2CrO4 for various times. None of the Cr(VI) treatments significantly changed the GSH levels (Fig. 4). The levels of oxidized glutathione (GSSG) remained below detection limits in these cells. As a positive control, the nonspecific thiol oxidant diamide did deplete cellular GSH by almost 80% (Fig. 4). Diamide did not generate significant GSSG, consistent with the known ability of diamide to yield GS-protein adducts in cells (Soderdahl et al., 2003). Since Cr(VI) did not decrease GSH levels in these cells, the oxidation of Trx1 and Trx2 cannot be explained as the result of a general oxidation of cellular thiols.
Fig. 4.

Relative levels of GSH in cells exposed to various concentrations of Na2Cr(VI)O4 or to diamide as indicated. The average GSH concentration in untreated cells was 46 nmol per mg protein. **, P <0.01 vs. the other treatments.
3.5 Direct interaction with Cr(VI)
The oxidation of thioredoxins in cells could either result from a direct interaction of Cr(VI) with the thioredoxin system or an indirect consequence of enhanced oxidative stress. In order to determine if a direct interaction is possible, in vitro studies were conducted with commercially available purified TrxR and human Trx1. In the absence of added reductant, purified Trx1 was a mix of reduced and partially oxidized forms (Fig. 5, lane a). Incubation with TrxR plus NADPH resulted in reduction of Trx1 as expected (Fig. 5, lane b), and confirmed the activity and expected interactions of these proteins. The addition of increasing amounts of Cr(VI) resulted in increased oxidation of one dithiol of Trx1 (Fig. 5, lanes c–e). Trx1 oxidation was especially prominent in lanes d and e in which the Cr(VI) concentration exceeded the concentration of NADPH and therefore exceeded the capacity of NADPH to potentially support regeneration of the reduced form of Trx1.
Fig. 5.
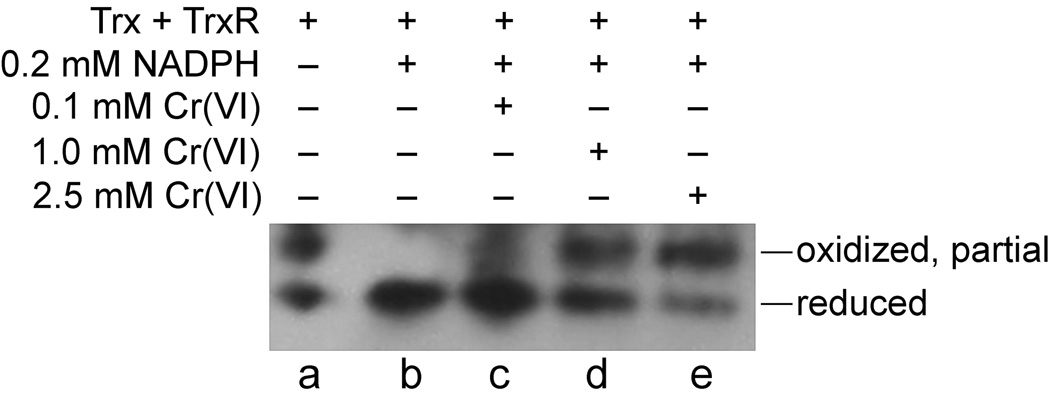
Cr(VI) causes oxidation of purified Trx in vitro as demonstrated by redox western blot for Trx1. Samples were incubated for 1 hr at 37°C and then analyzed for Trx1 redox status. The total reaction volume was 0.050 ml and 0.015 U of purified Trx and TrxR were included. NADPH (0.2 mM) and various concentrations of Na2Cr(VI)O4 were included as indicated.
To further explore the possible redox interactions between the thioredoxin system and Cr(VI), experiments were conducted that examined the ability of TrxR and/or Trx to reduce Cr(VI). The one-electron reduction product, Cr(V), is commonly used as an indicator for Cr(VI) reduction. Cr(V) is a d1 paramagnetic species that can be directly detected by ESR spectroscopy by its sharp line spectrum (g = 1.979) at conventional X-band frequency. NADPH is a slow chemical reductant of Cr(VI) and generated a small Cr(V) signal (Fig. 6A), consistent with previous results (Borthiry et al., 2007). When purified TrxR and Trx were included, the intensity of the Cr(V) signal was markedly increased (Fig. 6B), indicating prominent Cr(VI)-reducing activity. To determine if both proteins are required, experiments were conducted in which each protein was omitted. TrxR, in the absence of Trx, generated a large Cr(V) signal (Fig. 6C) that was even somewhat in excess of that obtained with both proteins. This indicates that TrxR alone has prominent Cr(VI)-reducing activity. Conversely, in the absence of TrxR, Trx generated a considerably smaller Cr(V) signal (Fig. 6D); however, this signal was larger than that with NADPH alone, suggesting some ability of Trx to reduce Cr(VI). Since Trx cannot accept electrons directly from NADPH, the absence of TrxR would prevent regeneration of reduced Trx to support continued activity. This small amount of Trx-mediated Cr(VI) reduction is therefore likely due to the fraction of purified Trx that is already reduced (see Fig. 5). Omission of NADPH greatly diminished the Cr(V) signal (Fig. 6E) confirming that the vast majority of the enzyme (TrxR plus Trx) activity is NADPH-dependent. The ESR signal was not observed in the absence of Cr(VI) (Fig. 6F). When isotopically pure Na253CrO4 (53Cr is a stable isotope) was substituted for Na2CrO4, the signal was split into four lines (separation of about 17.4 G) (Fig. 6G), which is typical for the hyperfine coupling for 53Cr(V) and is due to the nuclear spin of I = 3/2 for 53Cr. Traces F and G in Fig. 6 therefore confirm that the ESR signals in traces A through E are due to Cr(V). Overall, these ESR findings are consistent with those in Fig. 5 in demonstrating that the thioredoxin system is capable of direct redox interactions with Cr(VI).
Fig. 6.
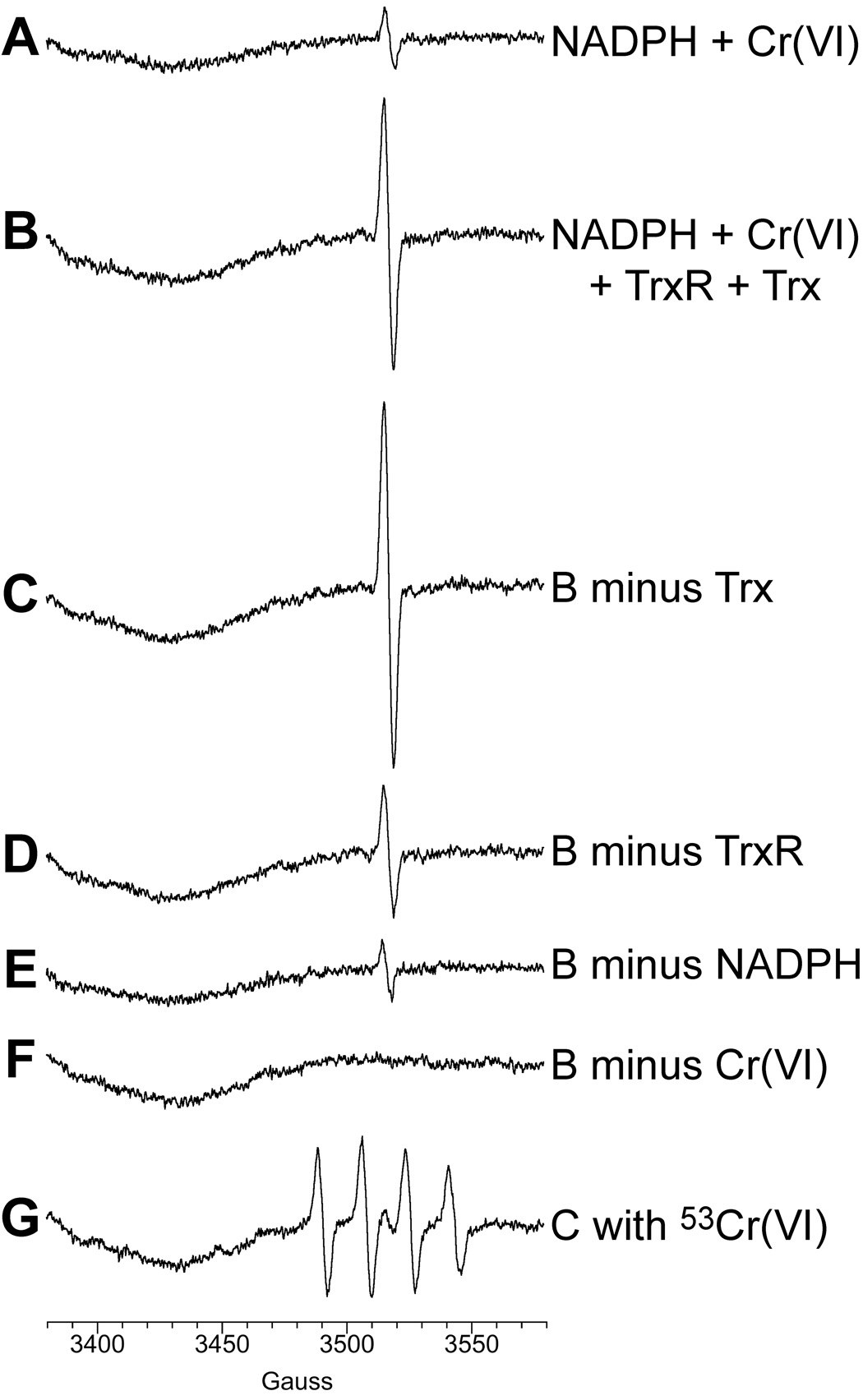
Purified thioredoxin reductase (TrxR) and thioredoxin (Trx) generate Cr(V) from Na2Cr(VI)O4 (100 µM) as demonstrated by ESR spectra of Cr(V) following incubation for 5 min at 37°C. The total reaction volume was 0.3 ml and contained Trx and/or TrxR (0.09 U each) as indicated. Representative ESR spectra obtained at room temperature are shown. Instrument settings were: 5 G modulation amplitude, 50 mW microwave power, 6.32 × 104 receiver gain, 40.96 msec time constant, 9.76 GHz microwave frequency, sweep width = 200 G, field set = 3480 G, modulation frequency = 100 kHz, scan time = 42 sec; number of scans, 9.
3.6 Spin Trapping for Radical Species in Cells
In addition to direct interactions with Cr(VI), it is possible that some of the Trx oxidation in cells results from other reactive species such as ROS. Spin trapping was therefore used to detect radical species by ESR. DEPMPO can trap and stabilize hydroxyl radical as DEPMPO/•OH and superoxide as DEPMPO/•OOH, giving ESR spectra characteristic of each (Frejaville et al., 1995). In addition, it can trap glutathionyl radical as DEPMPO/•SG (Karoui et al., 1996).
The spin trapping data for BEAS-2B cells are shown in Fig. 7. Multiline spectra were observed in the presence of Cr(VI) plus DEPMPO but not when Cr(VI) was omitted (Fig. 7A). When cells are incubated with just Cr(VI), and not DEPMPO, the single line spectrum for Cr(V) was observed (g = 1.979) (Fig. 7B), matching the signal previously reported for these cells (Borthiry et al., 2008). This Cr(V) signal overlaps the high-field end of the multiline spectrum of the DEPMPO adducts observed in Fig. 7A, and therefore increases the intensity of this component of the signal. Using the hyperfine coupling constants for DEPMPO/•OH (aP = 47.1 G, aH = 13.2 G, aN = 14.1 G) and DEPMPO/•SG (aP = 45.8 G, aH = 14.9 G, aN = 14.1 G) (Karoui et al., 1996), simulation of the multiline spectra indicate that essentially the entire signal can be accounted for by three species: DEPMPO/•OH, DEPMPO/•SG, and Cr(V). The majority of the spectral lines for DEPMPO/•OH and DEPMPO/•SG overlap, but the two adducts can be discerned by the central portion of the spectra. The relative levels of these two DEPMPO adducts remained fairly constant over the times examined (5, 10, 20 and 40 min), with 44 to 55% of the signal attributed to DEPMPO/•OH and 31 to 42% attributed to DEPMPO/•SG. Cr(V) declined somewhat over time, accounting for 23% of the signal at 5 min vs. 11% at 40 min.
Fig. 7.
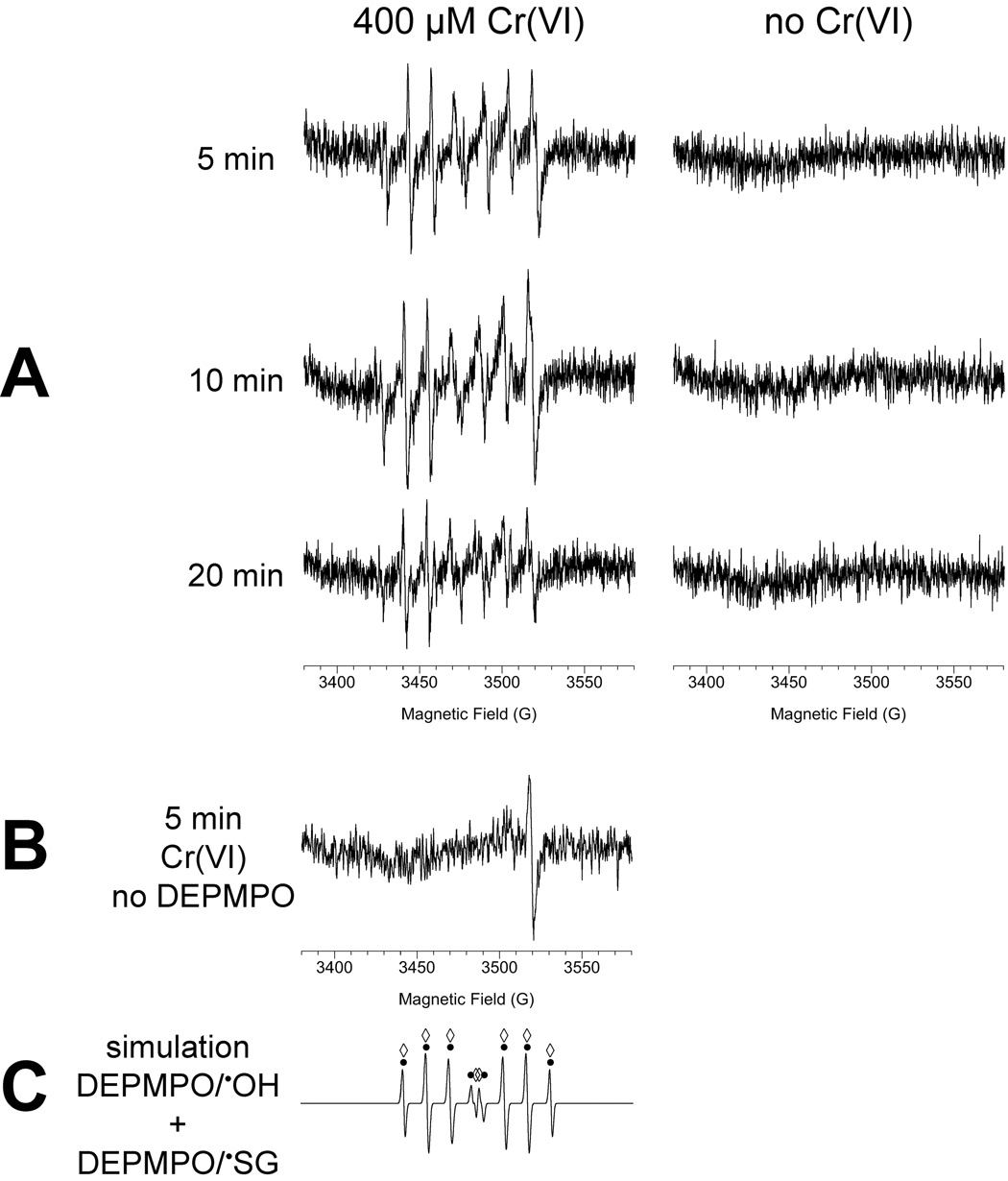
A: ESR spectra of BEAS-2B cells incubated with the spin trap DEPMPO (14 mM) in the presence (left) or absence (right) of 400 µM Cr(VI) for the indicated times. For each, the total reaction volume was 0.25 ml and contained 5 × 106 cells. Similar spectra were also obtained after 40 min incubation (not shown). B: The spectrum obtained when cells are incubated for 5 min with Cr(VI) in the absence of DEPMPO. C: Computer simulation of a spectrum for equal parts of DEPMPO/•OH and DEPMPO/•SG. The signal corresponding to DEPMPO/•OH is indicated by black dots above the spectrum, whereas that for DEPMPO/•SG is indicated by open diamonds. Instrument settings were: 1 G modulation amplitude, 20 mW microwave power, 6.32 × 104 receiver gain, 40.96 msec time constant, 9.76 GHz microwave frequency, sweep width = 200 G, field set = 3480 G, modulation frequency = 100 kHz, scan time = 42 sec; number of scans, 9.
To further explore the nature of the DEPMPO signals, the effects of the radical scavengers ethanol and formate were explored. In the presence of 5% ethanol, a specific hydroxyl radical scavenger, the DEPMPO/•OH adduct signal decreased by 50% whereas that for DEPMPO/•SG decreased by 30% (not shown). Ethanol would be expected to compete with DEPMPO for HO•, so the decline in the DEPMPO/•OH signal supports the generation of HO• in these cells. The generation of HO• in Cr(VI)-exposed cells is consistent with the known Fenton-like chemistry of the reactive Cr intermediates such as Cr(V) and the generation of HO• in vitro by human microsomal enzymes (Borthiry et al., 2007). The decline in DEPMPO/•SG in the presence of ethanol suggests that some of the GS• may result from reaction of GSH with HO•.
In the presence of 0.1M formate, the DEPMPO/•OH and DEPMPO/•SG signals were decreased by 68 and 44%, respectively, and the signal for the carbon dioxide radical anion (CO2•−) of DEPMPO was observed (not shown). Both HO• and GS• are known to oxidize formate to CO2•− (Karoui et al., 1996). These formate results therefore further support the generation of HO• and GS• in Cr(VI)-exposed cells.
4. Discussion
The redox states of Trx1 and Trx2 have been used to differentially assess the impacts of other oxidants on the thiol redox status of the cytosolic and mitochondrial compartments (Halvey et al., 2005; Hansen et al., 2006a). For the studies reported here, both insoluble and soluble Cr(VI) cause a dose- and time-dependent oxidation of Trx1 and Trx2 (Fig. 1–Fig. 3). From the perspective of conversion of all of the Trx to an oxidized form, Trx2 is more sensitive than Trx1. For example, 100% of Trx2 was converted to the oxidized form in cells exposed to 50 µM Cr(VI) for ≥30 min (Fig. 1), whereas 30 to 38% of Trx1 remained reduced after 30 min with 50 or 100 µM Cr(VI) (Fig. 2D). This is similar to reports for t-butyl hydroperoxide or diamide which preferentially oxidize Trx2 relative to Trx1 (Chen et al., 2006; Hansen et al., 2006a). However, it is in contrast to acrolein for which Trx1 in human endothelial cells is more sensitive than is Trx2 (Szadkowski and Myers, 2007). With some oxidants (e.g. epidermal growth factor-induced ROS) (Halvey et al., 2005; Hansen et al., 2006a), Trx2 is not significantly oxidized. The reason(s) for the enhanced sensitivity of Trx2 to Cr(VI) are not known, but could indicate a more robust interaction of Cr(VI) with the mitochondrial thioredoxin system or a lesser ability of the mitochondrial system to restore or maintain Trx2 in its reduced state once it has been oxidized.
Trx2 has only one dithiol, that of the active site, so it is either reduced (active) or oxidized (inactive). In contrast, Trx1 has two dithiols, one of which is at the active site. Only the partially oxidized form of Trx1 was observed in Cr(VI)-exposed cells, regardless of Cr(VI) concentration or time of exposure (Fig. 2, Fig. 3). This suggests that only one of the two dithiols of Trx1 is sensitive to Cr(VI) exposure. This is in contrast to treatment of these cells with acrolein, which can oxidize both dithiols of Trx1 (Fig. 2, Fig. 3). The acrolein results also show that its ability to oxidize Trx in cells is not unique to the endothelial cells which were previously reported (Szadkowski and Myers, 2007). Since acrolein is a component of all types of smoke, its ability to oxidize Trx in these human bronchial epithelial cells could have important implications for toxicity to bronchial epithelial cells in vivo.
Other studies have similarly shown that the two dithiols of Trx1 have differential susceptibility to oxidants. For example, the active site dithiol (C32/C35) is more sensitive to oxidation by diamide than is the second dithiol (C62/C69) (Watson et al., 2003). Similarly, while acrolein oxidizes both dithiols of Trx1, one of the dithiols is more sensitive to lower concentrations of acrolein (Szadkowski and Myers, 2007). Given the enhanced sensitivity of the active site to other oxidants, it seems likely that it is the active site of Trx1 that is oxidized following Cr(VI) exposure. This partially oxidized form would therefore be inactive. However, the C32/C35 active site disulfide is a substrate for reduction by TrxR (Watson et al., 2003), so it is possible that the redox status of Trx1 could be restored following oxidant removal. We did in fact observe this recovery of Trx1. In cells that were exposed to 25 or 50 µM Cr(VI) for 30 min, most of the Trx1 (≥85%) was restored to the reduced form after a 4 h Cr(VI)-free recovery period (not shown).
Both soluble and insoluble chromates are used industrially, and both Na2CrO4 (highly soluble) and ZnCrO4 (water insoluble) resulted in oxidation of Trx1 and Trx2 (Fig. 1–Fig. 3). When comparing the two, Na2CrO4 resulted in an earlier or more extensive oxidation than did ZnCrO4. For example, 50 µM Na2CrO4 for 30 min resulted in all of the Trx2 being oxidized (Fig. 1), whereas a mix of reduced and oxidized forms was seen for ZnCrO4 (Fig. 3). Similarly, 50 µM Na2CrO4 for 30 min resulted in about 45% of Trx1 oxidized, whereas with ZnCrO4 nearly all was still reduced (Fig. 3). Longer incubations were needed to result in significant Trx1 oxidation. This is not surprising as insoluble chromates would be expected to penetrate cells more slowly. Recent evidence for ZnCrO4 penetration into these cells comes from ESR studies which demonstrated a detectable Cr(V) signal after just 5 min exposure to ZnCrO4, although the signal was smaller than that seen with Na2CrO4 (Borthiry et al., 2008). Thus, at least a portion of the ZnCrO4 must enter cells fairly quickly, but the manner in which it enters the cells is not known. Among the insoluble chromates, ZnCrO4 proved the most soluble in complete medium (DMEM plus FBS) when followed over 7 days (Elias et al., 1991). In the Trx studies reported here, the cells were exposed to chromates in HBSS, however. The ZnCrO4 suspension was sonicated just before use to attempt to create as fine a dispersion as possible, and this may have helped facilitate cellular uptake. It is possible that the size or surface structure of the ZnCrO4 particles may make them more amenable to cellular components that facilitate dissolution or entry. With another insoluble chromate, PbCrO4, uptake into hamster embryo cells or human fibroblasts is facilitated by cell-enhanced extracellular dissolution of the particles (Elias et al., 1991; Xie et al., 2004). Such dissolution is not complete (Wise et al., 2002). Similarly, it is likely that only a portion of the insoluble ZnCrO4 was available to the cells during the time frame of the experiment. Nonetheless, the oxidation of Trx and the generation of Cr(V) in ZnCrO4-treated cells are consistent with the entry and reductive activation of at least a portion of the ZnCrO4.
Tessier and Pascal (Tessier and Pascal, 2006) estimated a daily occupational dose from welding fumes to be 0.4 to 1.6 mg Cr per m2 of lung airway. This estimate is based on presumed equal distribution over the entire alveolar surface area (approx. 100 m2). However, inhaled Cr is known to concentrate at bifurcations in the airways (Ishikawa et al., 1994b), so Cr doses at these sites are likely significantly greater than 0.4 to 1.6 mg per m2. Nonetheless, the levels of Cr used in these studies are similar to these estimated occupational exposures; 10 and 25 µM Cr correspond to 1.04 and 2.60 mg Cr per m2, respectively. Occupational exposures would therefore be at levels that would be expected to be sufficient to cause Trx oxidation.
There are several conceivable mechanisms by which Cr(VI) might mediate thioredoxin oxidation in cells. Some of these possibilities are: (a) direct oxidation by Cr(VI) or the reactive Cr intermediates such as Cr(V); (b) direct or indirect oxidation by Cr-generated ROS; (c) inhibition of TrxR by competition for electrons or other mechanisms; and (d) non-specific depletion/oxidation of total cellular thiols. The last possibility seems the least likely since Cr(VI) exposure did not significantly decrease GSH levels (Fig. 4). The other possibilities, based on the in vitro data with purified proteins, are shown in Fig. 8. Other in vitro studies have shown that Cr(VI) reduction can lead to ROS generation, so direct or indirect effects by ROS remain a possibility. Spin trapping studies were consistent with Cr(VI)-mediated ROS generation in these cells (Fig. 7). In vitro, the microsomal enzymes P450 reductase plus cytochrome b5 generate O2•− and HO• in the presence of low µM Cr(VI) (Borthiry et al., 2007). In order to detect ROS in cells by spin trapping (Fig. 7) higher concentrations of Cr(VI) were required relative to the in vitro experiments with purified enzymes (Borthiry et al., 2007). This is not surprising because of the large number of cell components that compete for trapping of the radicals by DEPMPO. In addition, the half-life of spin adducts is relatively short in biological systems because they can be converted to ESR silent species over time (Kosaka et al., 1992; Reinke et al., 1996). The fact that the spin adduct signals persisted over time in Cr(VI)-exposed cells suggests a continual generation of ROS, which could have contributed to Trx oxidation in cells.
Fig. 8.
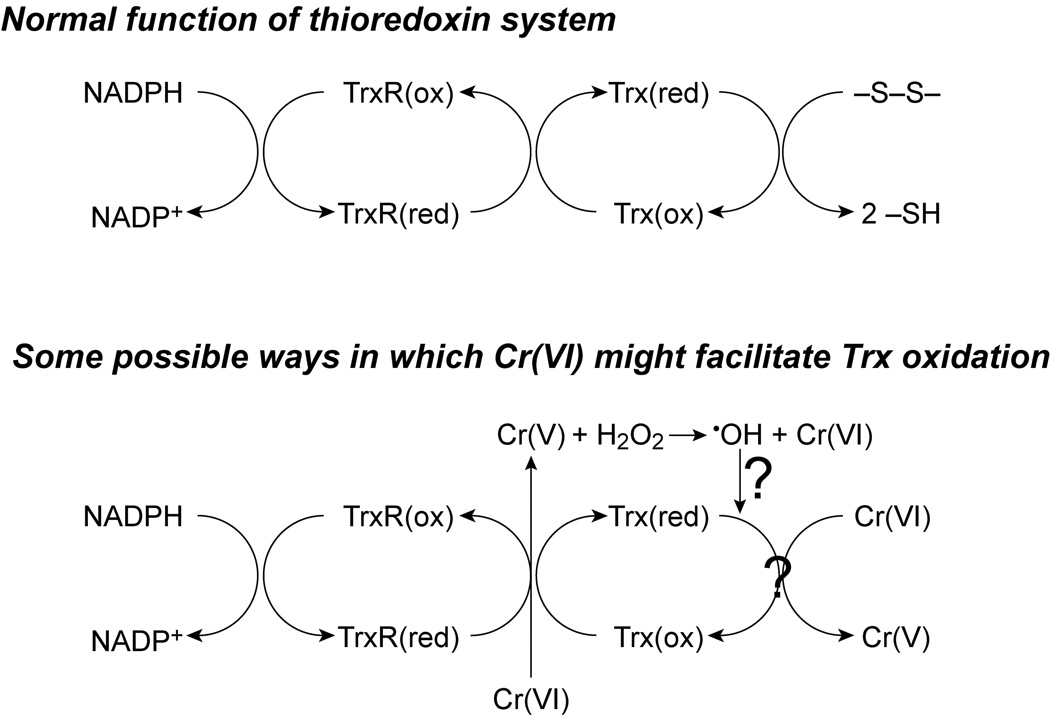
General scheme for possible ways in which Cr(VI) could result in Trx oxidation. The direct interactions of TrxR and Trx are based on the in vitro studies with purified proteins. The potential for Cr(V)-mediated ROS formation is based on other studies, and may not be directly linked to Cr(V) generation by the thioredoxin system as shown. The diagram is not meant to be inclusive of all possible interactions or Cr species.
Cr(VI) is capable of direct redox interactions with Trx1 and TrxR as demonstrated by the in vitro studies conducted here, i.e. Cr(VI) is reduced to Cr(V) and Trx1 is oxidized (Fig. 5, Fig. 6). Since TrxR has prominent Cr(VI)-reducing activity on its own, Cr(VI) has the potential to act as a competitive inhibitor of TrxR, i.e. the diversion of electrons to Cr(VI) could slow effective reduction of oxidized Trx. Over time, in the cells, the percent of oxidized Trx might therefore increase because the TrxR could no longer effectively retain the pool of Trx in the reduced state. Consistent with this possible mechanism is the observation that Trx oxidation in cells increased over time for each concentration of Cr(VI). While the addition of Trx1 did not enhance the Cr(VI)-reducing activity of TrxR, the data in Fig. 6 suggest some ability of Trx on its own to transfer electrons to Cr(VI). Since NADPH is not an efficient electron donor to Trx in the absence of TrxR, there was no mechanism to regenerate reduced Trx once it had reduced Cr(VI). Only the amount of Trx1 that was already reduced would have been able to reduce Cr(VI). Therefore, the ability of Trx1 to reduce Cr(VI) could be higher than what was observed in Fig. 6. Chemical reductants of Trx, such as dithiothreitol, can regenerate reduced Trx under these conditions and were considered as a possible experiment to support regeneration of reduced Trx in the absence of TrxR and NADPH. However, these chemical reductants of Trx are also potent chemical reductants of Cr(VI) themselves, which precluded their use in these experiments. If Cr(VI) were a robust direct chemical oxidant of Trx, we would have predicted a complete oxidation of purified Trx1 by the high concentrations of Cr(VI) used in vitro (Fig. 5), and a more rapid and complete oxidation of Trx in cells such as that caused by acrolein (Szadkowski and Myers, 2007). The in vitro data therefore argue that the interaction of Cr(VI) with TrxR is more robust than that with Trx1 and that this might contribute to the Trx oxidation observed in cells. However, some direct effects on Trx remain possible, as do other indirect mechanisms which might contribute to Trx oxidation in cells. The studies with the purified proteins were conducted using the cytosolic forms, Trx1 and TrxR1, because they are commercially available. Given that the mitochondrial forms (Trx2 and TrxR2) have active site domains that are similar to those of their cytosolic counterparts, it is possible that the mitochondrial forms may also have direct redox interactions with Cr(VI). This possibility needs to be tested experimentally.
The potential effects of some other metals on the thioredoxins have been explored in HeLa cells (Hansen et al., 2006b). Arsenic(III) or mercury(II) (10 or 100 µM for 4 h) caused oxidation of a portion of the Trx1 and very pronounced oxidation of Trx2. Their effects on purified Trx or TrxR were not explored, however. In contrast to arsenic and mercury, copper(II), iron(III), nickel(II) or zinc(II) did not cause oxidation of either thioredoxin (Hansen et al., 2006b). The redox activity of the metals does not therefore predict their effects on Trx. Cu(II), Fe(III), and Cr(VI) are all susceptible to reduction in cells and the reduced forms participate in Fenton-like reactions, but yet only Cr has an effect on Trx. Given the very low solubility of Fe(III) at physiological pH, one might argue that its bioavailability may have been low. However, both 10 and 100 µM Fe(III) caused pronounced declines in GSH levels (Hansen et al., 2006b), supporting that a significant portion was bioavailable and potentially redox active. Fe(III) is therefore the converse of what we observed for Cr(VI), i.e. Fe(III) does not affect Trx but can lead to GSH oxidation, whereas Cr(VI) oxidized Trx but did not significantly change GSH. The detection of GS• in Cr(VI)-exposed cells (Fig. 7) implies some oxidation of GSH. However, the cells were able to maintain their GSH redox status (Fig. 4), whereas they cannot maintain the reduced state of the thioredoxins (Fig. 1–Fig. 3). The amount of DEPMPO/•SG detected in Cr(VI)-exposed cells (Fig. 7) is only a very small percentage of total GSH. Simulation of the spectra in Fig. 7 indicated 75–113 pmol GSH per five million cells. Using an estimated cell volume of 1.4 pL (May and Qu, 2005), the intracellular DEPMPO/•SG is estimated at 10.7–16 µM. This is well below the typical mM levels of total GSH in cells (Meister and Anderson, 1983; Morrow et al., 2006). The relatively low levels of GS• spin adducts are therefore consistent with the essentially normal levels and redox status of GSH in Cr(VI)-exposed cells (Fig. 4).
In summary, this report demonstrates that both insoluble and soluble forms of chromate cause a dose- and time-dependent oxidation of Trx2 and Trx1 in human bronchial epithelial cells. With Cr(VI) treatments that oxidized all of the Trx2, some of the Trx1 was still in the reduced state, suggesting that Trx2 is more susceptible to complete oxidation. Only one of the dithiols, presumably the active site, of Trx1 was oxidized by Cr(VI). This is in contrast to the aldehyde acrolein which can oxidize both dithiols. Cr(VI)-mediated Trx oxidation is not the result of a general oxidation of cellular thiols. With purified proteins in vitro, Cr(VI) also results in Trx oxidation. Purified TrxR has pronounced Cr(VI) reducing activity, so it is possible that competition for electron flow from TrxR could impair the ability to reduce Trx. The in vitro data also suggest that Cr(VI) may also directly oxidize Trx. Since maintaining Trx1 and Trx2 largely in their reduced states is critical for their function, the ability of Cr(VI) to cause Trx oxidation in cells could contribute to its cytotoxic effects, and could have important implications for cell survival, redox-sensitive cell signaling, and the ability of cells to tolerate other oxidant insults. The impact of Cr(VI) on each of the thioredoxins and TrxR therefore warrant further study.
Acknowledgments
This research was supported by grant ES012707 from the National Institute of Environmental Health Sciences (NIEHS), NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
The ESR facilities of the Department of Biophysics are supported by National Biomedical ESR Center Grant EB001980 from the NIH.
We are grateful to: Dr. Kasem Nithipatikom and Marilyn Isbell for kindly providing the HPLC instruments and expertise; to Rachel Forbes for technical assistance with the GSH analysis; to Dr. David H. Petering for kindly supplying the Na253CrO4; and to Dr. Tak Yee Aw for providing the details of the glutathione analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arner ESJ, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Becker N, Chang-Claude J, Frentzel-Beyme R. Risk of cancer for arc welders in the Federal Republic of Germany: results of a second follow up (1983–8) Br. J. Ind. Med. 1991;48:675–683. doi: 10.1136/oem.48.10.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthiry GR, Antholine WE, Kalyanaraman B, Myers JM, Myers CR. Reduction of hexavalent chromium by human cytochrome b5: Generation of hydroxyl radical and superoxide. Free Radic. Biol. Med. 2007;42:738–755. doi: 10.1016/j.freeradbiomed.2006.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthiry GR, Antholine WE, Myers JM, Myers CR. Reductive activation of hexavalent chromium by human lung epithelial cells: generation of Cr(V) and Cr(V)-thiol species. J. Inorg. Biochem. 2008 doi: 10.1016/j.jinorgbio.2007.12.030. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner B, Beyersmann D. Modification of the erythrocyte anion carrier by chromate. Xenobiotica. 1985;15:735–741. doi: 10.3109/00498258509047435. [DOI] [PubMed] [Google Scholar]
- Cantoni O, Costa M. Analysis of the induction of alkali sensitive sites in the DNA by chromate and other agents that induce single strand breaks. Carcinogenesis. 1984;5:1207–1209. doi: 10.1093/carcin/5.9.1207. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cai J, Jones DP. Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett. 2006;580:6596–6602. doi: 10.1016/j.febslet.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie NT, Cantoni O, Evans RM, Meyn RE, Costa M. Use of mammalian DNA repair-deficient mutants to assess the effects of toxic metal compounds on DNA. Biochem. Pharmacol. 1984;33:1661–1670. doi: 10.1016/0006-2952(84)90289-2. [DOI] [PubMed] [Google Scholar]
- Costa M, Zhitkovich A, Toniolo P, Taioli E, Popov T, Lukanova A. Monitoring human lymphocytic DNA-protein cross-links as biomarkers of biologically active doses of chromate. Environ. Health Perspect. 1996;104:917–919. doi: 10.1289/ehp.96104s5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps F, Moulin JJ, Wild P, Labriffe H, Haguenoer JM. Mortality study among workers producing chromate pigments in France. Int. Arch. Occup. Environ. Health. 1995;67:147–152. doi: 10.1007/BF00626345. [DOI] [PubMed] [Google Scholar]
- Dillon CT, Lay PA, Bonin AM, Cholewa M, Legge GJF, Collins TJ, Kostka KL. Permeability, cytotoxicity, and genotoxicity of chromium(V) and chromium(VI) complexes in V79 Chinese hamster lung cells. Chem. Res. Toxicol. 1998;11:119–129. doi: 10.1021/tx9701541. [DOI] [PubMed] [Google Scholar]
- Duling DR. Simulation of multiple isotropic spin-trap EPR spectra. J. Magn. Reson. B. 1994;104:105–110. doi: 10.1006/jmrb.1994.1062. [DOI] [PubMed] [Google Scholar]
- Elias Z, Poirot O, Baruthio F, Daniere MC. Role of solubilized chromium in the induction of morphological transformation of Syrian hamster embryo (SHE) cells by particulate chromium(VI) compounds. Carcinogenesis. 1991;12:1811–1816. doi: 10.1093/carcin/12.10.1811. [DOI] [PubMed] [Google Scholar]
- EPA. Integrated Risk Information System, Office of Health and Environmental Assessment. Chromium. Washington, D.C.: U.S. Environmental Protection Agency; 1999. [Google Scholar]
- Fariss MW, Reed DJ. High-performance liquid chromatography of thiols and disulfides: dinitrophenol derivatives. Meth. Enzymol. 1987;143:101–109. doi: 10.1016/0076-6879(87)43018-8. [DOI] [PubMed] [Google Scholar]
- Franchini I, Magnani F, Mutti A. Mortality experience among chromplating workers. Scand. J. Work Environ. Health. 1983;9:247–252. doi: 10.5271/sjweh.2413. [DOI] [PubMed] [Google Scholar]
- Frejaville C, Karoui H, Tuccio B, Le Moigne F, Culcasi M, Pietri S, Lauricella R, Tordo P. 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide: a new efficient phosphorylated nitrone for the in vitro and in vivo spin trapping of oxygen-centered radicals. J. Med. Chem. 1995;38:258–265. doi: 10.1021/jm00002a007. [DOI] [PubMed] [Google Scholar]
- Gadd GM, White C. Microbial treatment of metal pollution — a working biotechnology? TIBTECH. 1993 August;11 doi: 10.1016/0167-7799(93)90158-6. [DOI] [PubMed] [Google Scholar]
- Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem. J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006a;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic. Biol. Med. 2006b;40:138–145. doi: 10.1016/j.freeradbiomed.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Hayes RB. Carcinogenic effects of chromium. In: Langard S, editor. Biological and Environmental Aspects of Chromium. vol. 5. Amsterdam: Elsevier Biomedical Press; 1982. pp. 221–239. [Google Scholar]
- Ishikawa Y, Nakagawa K, Satoh Y, Kitagawa T, Sugano H, Hirano T, Tsuchiya E. Characteristics of chromate workers' cancers, chromium lung deposition and precancerous bronchial lesions: an autopsy study. Br. J. Cancer. 1994a;70:160–166. doi: 10.1038/bjc.1994.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Nakagawa K, Satoh Y, Kitagawa T, Sugano H, Hirano T, Tsuchiya E. "Hot spots" of chromium accumulation at bifurcations of chromate workers' bronchi. Cancer Res. 1994b;54:2343–2346. [PubMed] [Google Scholar]
- Jannetto PJ, Antholine WE, Myers CR. Cytochrome b5 plays a key role in human microsomal chromium(VI) reduction. 2001;159:119–133. doi: 10.1016/s0300-483x(00)00378-4. [DOI] [PubMed] [Google Scholar]
- Jennette KW. Microsomal reduction of the carcinogen chromate produces chromium(V) J. Am. Chem. Soc. 1982;104:874–875. [Google Scholar]
- Karoui H, Hogg N, Fréjaville C, Tordo P, Kalyanaraman B. Characterization of sulfur-centered radical intermediates formed during the oxidation of thiols and sulfite by peroxynitrite. J. Biol. Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- Kosaka H, Katsuki Y, Shiga T. Spin trapping study on the kinetics of Fe2+ autoxidation: formation of spin adducts and their destruction by superoxide. Arch. Biochem. Biophys. 1992;293:401–408. doi: 10.1016/0003-9861(92)90412-p. [DOI] [PubMed] [Google Scholar]
- Langard S. Role of chemical species and exposure characteristics in cancer among persons occupationally exposed to chromium compounds. Scand. J. Work Environ. Health. 1993;19 Suppl. 1:81–89. [PubMed] [Google Scholar]
- Levy LS, Venitt S. Carcinogenicity and mutagenicity of chromium compounds: the association between bronchial metaplasia and neoplasia. Carcinogenesis. 1986;7:831–835. doi: 10.1093/carcin/7.5.831. [DOI] [PubMed] [Google Scholar]
- May JM, Qu ZC. Transport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis. Arch. Biochem. Biophys. 2005;434:178–186. doi: 10.1016/j.abb.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu. Rev. Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Mikalsen A, Alexander J, Ryberg D. Microsomal metabolism of hexavalent chromium. Inhibitory effect of oxygen and involvement of cytochrome P-450. Chem.-Biol. Interact. 1989;69:175–192. doi: 10.1016/0009-2797(89)90076-8. [DOI] [PubMed] [Google Scholar]
- Morrow CS, Peklak-Scott C, Bishwokarma B, Kute TE, Smitherman PK, Townsend AJ. Multidrug resistance protein 1 (MRP1, ABCC1) mediates resistance to mitoxantrone via glutathione-dependent drug efflux. Mol. Pharmacol. 2006;69:1499–1505. doi: 10.1124/mol.105.017988. [DOI] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Cloning and sequence of cymA, a gene encoding a tetraheme cytochrome c required for reduction of iron(III), fumarate, and nitrate by Shewanella putrefaciens MR-1. J. Bacteriol. 1997;179:1143–1152. doi: 10.1128/jb.179.4.1143-1152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Iron stimulates the rate of reduction of hexavalent chromium by human microsomes. Carcinogenesis. 1998;19:1029–1038. doi: 10.1093/carcin/19.6.1029. [DOI] [PubMed] [Google Scholar]
- Myers CR, Myers JM, Carstens BP, Antholine WE. Reduction of chromium(VI) to chromium(V) by human microsomal enzymes: effects of iron and quinones. Toxic Subst. Mech. 2000;19:25–51. [Google Scholar]
- Nakagawa K, Matsubara T, Kinoshita I, Tsuchiya E, Sugano H, Hirano T. Surveillance study of a group of chromate workers — early detection and high incidence of lung cancer (in Japanese) Lung Cancer. 1984;24:301–310. [Google Scholar]
- Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;283:G1352–G1359. doi: 10.1152/ajpgi.00183.2002. [DOI] [PubMed] [Google Scholar]
- Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- Pias EK, Aw TY. Early redox imbalance mediates hydroperoxide-induced apoptosis in mitotic competent undifferentiated PC-12 cells. Cell Death Differ. 2002;9:1007–1016. doi: 10.1038/sj.cdd.4401064. [DOI] [PubMed] [Google Scholar]
- Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu. Rev. Biophys. Biomol. Struct. 2001;30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- Raithel H-J, Schaller K-H, Kraus T, Lehnert G. Biomonitoring of nickel and chromium in human pulmonary tissue. Int. Arch. Occup. Environ. Health. 1993;65:S197–S200. doi: 10.1007/BF00381340. [DOI] [PubMed] [Google Scholar]
- Reinke LA, Moore DR, McCay PB. Degradation of DMPO adducts from hydroxyl and 1-hydroxyethyl radicals by rat liver microsomes. Free Radic. Res. 1996;25:467–474. doi: 10.3109/10715769609149069. [DOI] [PubMed] [Google Scholar]
- Shi X, Chiu A, Chen CT, Halliwell B, Castranova V, Vallyathan V. Reduction of chromium(VI) and its relationship to carcinogenesis. J. Toxicol. Environ. Health. 1999a;Pt. B 2:87–104. doi: 10.1080/109374099281241. [DOI] [PubMed] [Google Scholar]
- Shi X, Dalal NS. The role of superoxide radical in chromium(VI)-generated hydroxyl radical: the Cr(VI) Haber-Weiss cycle. Arch. Biochem. Biophys. 1992;292:323–327. doi: 10.1016/0003-9861(92)90085-b. [DOI] [PubMed] [Google Scholar]
- Shi X, Ding M, Ye J, Wang S, Leonard SS, Zang L, Castranova V, Vallyathan V, Chiu A, Dalal N, Liu K. Cr(IV) causes activation of nuclear transcription factor-kB, DNA strand breaks and dG hydroxylation via free radical reactions. J. Inorg. Biochem. 1999b;75:37–44. doi: 10.1016/S0162-0134(99)00030-6. [DOI] [PubMed] [Google Scholar]
- Shi X, Dong Z, Dalal NS, Gannett PM. Chromate-mediated free radical generation from cysteine, penicillamine, hydrogen peroxide, and lipid hydroperoxides. Biochim. Biophys. Acta. 1994;1226:65–72. doi: 10.1016/0925-4439(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Shi XL, Dalal NS. One-electron reduction of chromate by NADPH-dependent glutathione reductase. J. Inorg. Biochem. 1990;40:1–12. doi: 10.1016/0162-0134(90)80034-u. [DOI] [PubMed] [Google Scholar]
- Soderdahl T, Enoksson M, Lundberg M, Holmgren A, Ottersen OP, Orrenius S, Bolcsfoldi G, Cotgreave IA. Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. FASEB J. 2003;17:124–126. doi: 10.1096/fj.02-0259fje. [DOI] [PubMed] [Google Scholar]
- Standeven AM, Wetterhahn KE. Is there a role for reactive oxygen species in the mechanism of chromium(VI) carcinogenesis? Chem. Res. Toxicol. 1991;4:616–625. doi: 10.1021/tx00024a003. [DOI] [PubMed] [Google Scholar]
- Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis. 1992;13:1319–1324. doi: 10.1093/carcin/13.8.1319. [DOI] [PubMed] [Google Scholar]
- Stern RM. Assessment of risk of lung cancer for welders. Arch. Environ. Health. 1983;38:148–155. doi: 10.1080/00039896.1983.10543996. [DOI] [PubMed] [Google Scholar]
- Sugden KD. Formation of modified cleavage termini from the reaction of chromium(V) with DNA. J. Inorg. Biochem. 1999;77:177–183. doi: 10.1016/s0162-0134(99)00189-0. [DOI] [PubMed] [Google Scholar]
- Sugden KD, Campo CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: a possible mechanism for chromate genotoxicity. Chem. Res. Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Fukuda K. Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch. Toxicol. 1990;64:169–176. doi: 10.1007/BF02010721. [DOI] [PubMed] [Google Scholar]
- Szadkowski A, Myers CR. Acrolein oxidizes the cytosolic and mitochondrial thioredoxins in human endothelial cells. Toxicology. 2007;243:164–176. doi: 10.1016/j.tox.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taioli E, Zhitkovich A, Kinney P, Udasin I, Toniolo P, Costa M. Increased DNA-protein crosslinks in lymphocytes of residents living in chromium-contaminated areas. Biol. Trace Elem. Res. 1995;50:175–180. doi: 10.1007/BF02785408. [DOI] [PubMed] [Google Scholar]
- Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, Zamzami N, Jan G, Kroemer G, Brenner C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene. 2007;26:2606–2620. doi: 10.1038/sj.onc.1210074. [DOI] [PubMed] [Google Scholar]
- Tessier DM, Pascal LE. Activation of MAP kinases by hexavalent chromium, manganese and nickel in human lung epithelial cells. Toxicol. Lett. 2006;167:114–121. doi: 10.1016/j.toxlet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Tordo P. Spin-trapping: recent developments and applications. In: Gilbert BC, Atherton NM, Davies MJ, editors. Electron Paramagnetic Resonance. Specialist Periodical Reports. vol. 16. Cambridge: The Royal Society of Chemistry; 1998. pp. 116–144. [Google Scholar]
- Tsapakos MJ, Hampton TH, Wetterhahn KE. Chromium(VI)-induced DNA lesions and chromium distribution on rat kidney, liver and lung. Cancer Res. 1983;43:5662–5667. [PubMed] [Google Scholar]
- Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- Whiting RF, Stich HF, Koropatnick DJ. DNA damage and DNA repair in cultured human cells exposed to chromate. Chem.-Biol. Interactions. 1979;26:267–280. doi: 10.1016/0009-2797(79)90030-9. [DOI] [PubMed] [Google Scholar]
- Wise JP, Sr, Wise SS, Little JE. The cytotoxicity and genotoxicity of particulate and soluble hexavalent chromium in human lung cells. Mutat. Res. 2002;517:221–229. doi: 10.1016/s1383-5718(02)00071-2. [DOI] [PubMed] [Google Scholar]
- Xie H, Holmes AL, Wise SS, Gordon N, Wise JP., Sr Lead chromate-induced chromosome damage requires extracellular dissolution to liberate chromium ions but does not require particle internalization or intracellular dissolution. Chem. Res. Toxicol. 2004;17:1362–1367. doi: 10.1021/tx0498509. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A, Voitkun V, Kluz T, Costa M. Utilization of DNA-protein cross-links as a biomarker of chromium exposure. Environ. Health Perspect. 1998;106 suppl. 4:969–974. doi: 10.1289/ehp.98106s4969. [DOI] [PMC free article] [PubMed] [Google Scholar]