

Abstract
The development of cancer is associated with disorders in the regulation of the cell cycle. The purpose of this review is to briefly summarize the known sequence of events that regulate cell cycle progression with an emphasis on the checkpoints and the mechanisms cell employ to insure DNA stability in the face of genotoxic stress. Key transitions in the cell cycle are regulated by the activities of various protein kinase complexes composed of cyclin and cyclin-dependent kinases (CDK) molecules. The cyclins are CDK binding partners which are required for kinase activity and their protein levels are intimately linked to the cell cycle stage. CDK activity can be regulated by other mechanisms, such as phosphorylation events, that may contribute to deregulation of cell cycle and the development of cancer. While fruits and vegetables are recommended for prevention of cancer, their active ingredients and mechanisms of action are less well understood. Here, we briefly present evidence that dietary agents identified from fruits and vegetables can act to modulate the effects of deregulated cell cycle checkpoints, and that this may contribute to the prevention of cancer. The agents include apigenin (celery, parsley), curcumin (turmeric), (−)-epigallocatechin-3-gallate (green tea), resveratrol (red grape, peanuts and berries), genistein (soybean), and silymarin (milk thistle). The teachings of Hippocrates are still true “let food be thy medicine and medicine be thy food”.
Keywords: Cell cycle, cyclins, cyclin dependent kinases, Cip1/p21, Kip1/p27, apigenin, curcumin, epigallocatechin-3-gallate, grape seed proanthocyanidins, genistein, resveratrol, silymarin
1. Introduction
Disruption of the normal regulation of cell-cycle progression and division are important events in the development of cancer. Complex networks of regulatory factors respond to the tumor microenvironment and stress signals, such as those resulting from damaged DNA, dictate whether cells proliferate or die. Life on earth copes with constant exposure to DNA-damaging agents, including solar radiation, polycyclic aromatic hydrocarbons and cigarette smoke, etc. The DNA contained in every mammalian cell is under constant attack by agents that can either directly damage one of its three billion bases or break the phosphodiester backbone on which the bases reside. The living cell has evolved such that it deals with both metabolic and external sources of DNA-damaging agents through elegant mechanisms that repair damage to the DNA. Cellular responses to DNA damage constitute one of the most important fields in cancer biology. Damage to cellular DNA can result in the development of cancer. This is evident from: (i) epidemiological studies [1], from animal models and from the observation that many human-cancer-susceptibility syndromes arise from mutations in genes involved in DNA-damage responses, (ii) DNA damage is used to cure cancer [2]. Most therapeutic modalities that are currently in use for the treatment of malignancies target the DNA, including radiation therapy and many chemo-therapeutic agents, and (iii) DNA damage is responsible for most of the side effects of therapy. Therefore, from the perspective of cancer, DNA damage causes the diseases, it is used to treat the disease, and is responsible for the toxicity of therapies for the disease.
Cells have developed several defensive mechanisms to cope with this constant attack on their DNA; however, the DNA-repair processes are not perfect. As there are various types of DNA lesion that can occur, a variety of different repair mechanisms exists. In addition to directly repairing DNA breaks or adducts, cells respond to DNA damage by halting cell-cycle progression or by undergoing programmed cell death, i.e., apoptosis. The term “cell-cycle checkpoints” refers to mechanisms by which the cell actively halts progression through the cell cycle until it can ensure that an earlier process, such as DNA replication or mitosis, is complete [3]. Normal cell cycle progression relies on the cell’s ability to translate extracellular signals, such as mitogenic stimuli and intact extracellular matrices, in order to efficiently replicate DNA and divide. Here, first we summarize briefly the components of cell cycle regulation in eukaryotic cells, and then present the current knowledge concerning the cell cycle regulatory effects of some dietary agents on the regulation of cell cycle in cancer cells from the perspective of the potential utilization of these agents for the prevention of cancer.
2. Overview of cell cycle progression
The cell cycle is the recurring sequence of events that includes the duplication of cell contents and subsequent cell division. For eukaryotic cells, the cell cycle has been defined as the interval between the completion of mitosis in a cell and the completion of mitosis by one or both of its daughter cells [4]. Traditionally the cell cycle in eukaryotic cell has been divided into four phases: Gap phase 1 (G1); DNA synthesis (S); Gap phase 2 (G2), during which the cell prepares itself for division; and mitosis (M) during which the chromosomes separate and the cell divides [5]. The rapid process in cellular mitosis includes chromosomal alignment in metaphase, segregation of sister chromosome in anaphase and subsequent division of cellular material leading to the next interphase [6]. The interphase consists of resting phase followed by cell growth and normal metabolic role to duplicate the genetic material in S-phase and further proof reading in the replication and prepare for mitosis that occurs in G2 phase. Strict regulation of this cell division cycle is crucial for duplication of genetic information with extremely high fidelity as well as to monitor correct segregation of this information during mitosis.
The progression of cell cycle from one phase to the next is regulated by sequential activation and inactivation of many “check points” that monitor the status of the cell as well as environmental cues as summarized in Figure 1 [7]. Checkpoints are defined operationally as a gene product or subset of gene products that when mutated confer independence on a cellular process that was previously dependent upon completion of another cellular process [6]. In order to ensure proper cell cycle progression, the cells go through countless internal checkpoints to verify proper completion of one step before proceeding onto the next step [8,9]. Among these, the phase change of the cycle is tightly regulated by cyclin-dependent kinase complexes (CDKs), which are activated when they become bound to regulatory proteins (cyclins) [10–12]. CDKs are protein kinases that require binding to cyclin subunits to become catalytically competent. Different members of the CDK family, in association with different cyclins, represent key switches at various points in the cell cycle. Cyclin-CDK complexes are regulated by phosphorylation and protein interaction events that tightly control the timing and extent of CDK activation. For example, in G1 phase, growth factors or other stimuli induce the production of cyclin D1, which upon association with CDK4 or CDK6 forms an active kinase. These kinases drive entry into the cell cycle by phosphorylating the retinoblastoma protein (pRb), and pRb causes the release of bound E2F transcription factors and expression of E2F [13], thereby allowing progression from G1 to S phase. The specific phase transition activated by E2F, including the transcription of specific genes of cyclin and kinase needed as well as the other enzymes, required DNA synthesis in S-phase, such as thymidine kinase and dihydrofolate reductase. This process is negatively regulated by cyclin kinase inhibitors (CDKI) and is also under surveillance at number of checkpoints.
Figure-1.
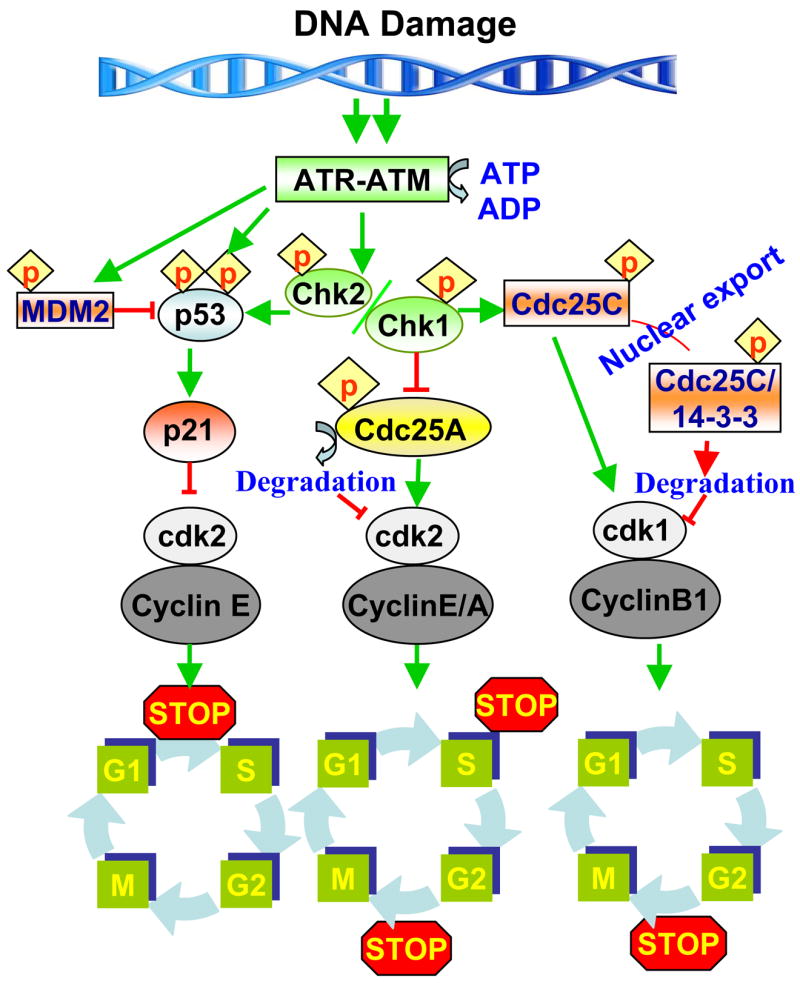
A schematic diagram illustrating cell cycle checkpoint pathways that are involved in the cell response to DNA damage. When cells incur DNA damage before entering into S phase, it is governed primarily by ATR. The ATR-activated p53 further activates p21 and leads to arrest of cells at the G1 phase. The ATM-activated check point kinase-1 (Chk1) arrests cells at the S phase by ubiquitin-dependent degradation of Cdc25A which inactivates the cdk2-cyclin E/A complex. In G2 arrest, Chk1 and Chk2 phosphorylate the dual specificity phosphatase Cdc25C, which creates a binding site for the 14-3-3 proteins. The 14-3-3/Cdc25C protein complexes are sequestered in the cytoplasm, thereby preventing Cdc25C from activating the Cdk1-Cdc2-Cyclin B1 complex and blocks entry into mitosis. The checkpoints are indicated by a stop sign.
The checkpoints occur predominantly at four stages of the cell cycle: in G1, at the G1/S transition, at the G2/M transition and at the metaphase/anaphase transition. The DNA damage checkpoint arrests cells in either the G1, S or G2 phase depending upon the cell cycle status of the cell at the time damage was incurred [14]. In addition to triggering arrest of cell cycle progression, checkpoints also can trigger the induction of necessary repair genes. Since CDK molecules regulate cell cycle progression, any of the genes involved in the regulation of kinase activity could be involved in the signal transduction machinery that leads to cell cycle arrest. The cell cycle arrest allows the repair of the genetic material thereby preventing secondary lesions and ensuring the appropriate progression into the next phase of the cycle.
2.1. Cyclins
Cyclins control various phases of the cell cycle through their ability to form a complex with a CDK partner. Their expression pattern dictates the point in the cell cycle at which they act. Some proteins are included in the ‘cyclin family’ due to their overall structural homology and conserved cyclin domains; however these ‘cyclins’ do not undergo cell cycle fluctuations and are involved in cellular processes other than cell cycle control. Thus, there are two types of cyclins: The cell-cycle related cyclins (Cyclins A, B, D and E) and the non-cell cycle related cyclins (e.g., cyclins H and C). Of the cell-cycle related cyclins, cyclins D and E play an important role in the transition from the G1 to S phase [15]. Cyclin D1 is part of a cell cycle control mode that is consistently deregulated in most human cancers. Studies of cyclin D1-null mice indicate, however, that it is dispensable in normal mouse development and is not required for cell growth in culture. It has been reported that ras-mediated tumorigenesis depends on signaling pathways that act preferentially through cyclin D1. Cyclin D1 expression and the activity of its associated kinase are up-regulated in keratinocytes in response to oncogenic ras. Furthermore, cyclin D1 deficiency results in a dramatic (up to 80%) reduction in the development of squamous tumors generated through either grafting or retroviral ras-transduced keratinocytes, phorbol ester treatment of ras transgenic mice, or a two-stage carcinogenesis [16].
Cyclin E plays a pivotal role in the regulation of G1-S transition and relates to malignant transformation of the cells. With its catalytic subunit CDK2, cyclin E is a key factor in the G1 checkpoint and promotes transition into S phase [17]. Cyclin E/CDK2 also plays a role in the initiation of DNA replication [18]. An oncogenic role for cyclin E has been suggested by studies of cyclin E-deficient cells which are resistant to transformation by myc alone or myc in combination with ras, a dominant negative p53, or E1A, suggesting that cyclin E is a key component in oncogenic signaling [19]. Induction of the p53 tumor suppressor gene after DNA damage inhibits the G1 cyclins/cyclin-dependent kinase activity via the p53 downstream mediator Cip1/p21 [20,21]. This inhibition causes cell cycle arrest to facilitate DNA repair [22,23]. Under normal conditions cyclin E is present at low level. Cylin E overexpressions were observed frequently in deeply invasive tumors, and can also be overexpressed in tumor tissue as biologically hyperactive low molecular weight isoforms which lack the normal N-terminus [24]. Constitutive over expression of cyclin E protein at all phases of the cell cycle is one of the features observed in breast cancer cell cycle and thought to result in premature DNA replication, genomic instability [25,26], and carcinogenesis [27]. In ovarian cancer patients, higher expression of cyclin E has been associated with low overall survival rates [28]. Although cyclin E overexpression has been linked as an independent poor prognostic to adverse outcomes in patients with gastric [29] and bladder carcinomas [30] but the prognosis is significant in non-small-cell lung carcinomas [31].
Cyclin A is associated with both CDK1 and CDK2, and has functions in both S phase and mitosis. Of these cyclin-cdk complexes, cyclin D-CDK4/6 activity drives cells through the early G1 phase of the cell cycle, whereas cyclin E-CDK2 and subsequently cyclin A-CDK2 activities are required for transition through the later G1 phase of the cell cycle past the restriction point up to which growth factor stimulation is mandatory. Cyclin A starts to accumulate during S phase and is abruptly disappear before metaphase. In cultured cells, cyclin A is synthesized and disappear after cyclin E but slightly earlier than cyclin B during G2 phase [32,33]. Consistent with its role in the control of DNA replication, cyclin A is synthesized at the onset of S phase and localizes to the sites of DNA replication [34,35]. Mostly two-types of cyclin A are known: an embryonic-specific cyclin A1 and a somatic cyclin A2. Conceptually, deregulation of cell cycle regulators such as cyclin A2 is likely to contribute to tumorigenesis. Cyclin A-CDK complex also contributes to tumorigenesis by phosphorylating other oncoproteins and tumor suppressors. The over expression of cyclin A alters the apoptotic function of p53 in breast cancer cells, which induces tumorigenic response [36].
2.2. Cyclin dependent kinases (CDK)
CDKs are protein kinases that require binding to a cyclin subunit to become catalytically competent [37,38]. Different members of the CDK family, in association with different cyclins, switches throughout the cell cycle; other family members regulate transcription, differentiation, and nutrient uptake, as well as other cellular functions. CDKs are typically 300 amino acids in length and contain certain recognizable motifs. Even though CDK protein levels are constant throughout the cell cycle, the CDKs are only functional during distinct intervals within the cell cycle. Notably, to enter the S phase all cells must fulfill the same essential requirement: they must activate cyclin-dependent kinases. Four separate CDKs (CDK1, CDK2, CDK4, and CDK6) are responsible for controlling the various stages of the cell cycle [39]. At the G1/S transition phase, CDK4/6 and CDK2 govern the entry into S-phase. CDK2 continues to be active through S-phase with its decline in activity signaling exit from S-phase. CDK1 becomes active in G2 and its activity persists through mitosis [15]. The prototypic CDK, CDK1, associates with cyclins A and B, and acts at the G2/M interface. The progressive accumulation of A and B cyclins during the cell cycle and their abrupt degradation at the onset of anaphase, mediates entry and exit from mitosis, respectively.
CDKs in the G1 phase trigger DNA replication. In higher eukaryotes, the G1 CDKs including CDK2, combines with E-type cyclins (E1, E2) and cyclin A [37,38]. E-type cyclins are required for appropriate development of the mouse. Cyclin E levels are constantly high in the cells of early embryos, allowing CDK2 to initiates S phase as soon as M phase is over [38]. In most other cells, however, various mechanisms enforce the existence of the G1 phase by keeping CDK2 inactive until mitogenic signals intervene. Cyclin E expression is dependent on E2F transcription factors [40,41]. In mitotically resting cells, and in cells that have just emerged from M phase, E2F factors are bound to Rb or its family members, p107, and p130. Rb binding turns E2Fs into repressors or inactives transactivators [41].
2.3. CDK inhibitors (CDKI)
Primarily there are two families of CDK inhibitors (CDKI), each with multiple members. The first family is identified as the Cip1/p21 family of universal cyclin/CDK inhibitors which includes Cip1/p21, Kip1/p27 and Kip2/p57 [42,43]. This family of inhibitors binds both cyclin molecules, through conserved LFG residues in the cyclin box motif, and the CDK molecule simultaneously. External stimuli (i.e., environmental or nutrients) and internal signals (i.e., DNA damage) regulate the formation of cyclin-CDK complexes via CDKI, which include the members of Cip/Waf family (p21, p27, p57). The members of this family interacts with multiple cyclin-CDK complexes and members of the INK4 family (p15, p16, p18, p19), which specifically inhibit cyclin-D-CDK complexes. The members of the Cip1/p21 family are able to bind all cyclin/CDK complexes in vitro, but they have greater affinity for G1 cyclin/CDK complexes in vivo [44]. The members of the second family of inhibitors, the INK4 family, show specificity for CDK4 and CDK6 due to their specificity in binding with CDK4 or CDK6. The members of this family are: p16INK4A, p15INK4B, and p19INK4D [45]. The relative concentrations of each of the two families of inhibitors determine their distribution among the various cyclin/CDK complexes and ultimately affect G1 progression. The Cip1/p21 has the ability to inhibit CDK kinase activity and subsequently inhibit DNA replication, and also partly responsible for the G1 growth arrest phenotype, through cyclin/CDK kinase inhibition, that is observed after p53 upregulation [46,47]. The G1 phase arrest allows sufficient time for damaged cells to repair any damaged DNA before passage through R and entrance into S-phase. This inhibitory effect of Cip1/p21 acts as a surveillance mechanism for the cell to maintain genomic integrity. The Kip1/p27 also plays a role in the regulation of the restriction point. The function of Kip1/p27 is regulated by the presence or absence of mitogens and the fluctuations in the Kip1/p27 protein levels occur mainly as a consequence of translational and posttranslational modifications of the protein rather than changes in the mRNA levels [48,49]. CDK kinase activity is considered to play a major role in cancer progression. The functions of the members of the CDK family are regulated primarily by CDKI proteins and are commonly upregulated in response to antiproliferative signals [50]. The members of the INK4 family specifically block G1 progression by inhibiting the association of CDK4/6 with cyclin D [51]. P16INK4A, via its mechanism of inhibition through either CDK4 or CDK6 binding, is linked to cyclin D and Rb. A cyclin-CDK complex hyperphosphorylates Rb, leading to its release from E2F [52–54]. The free transcription factor E2F then activates the genes responsible for cellular proliferation by progression through G1 phase. Impairment of a growth-stimulatory signaling pathway (e.g., erbB1, raf, MAPK) has been shown to stimulate the expression of CDKIs. An activated CDKI binds to and subsequently inhibits cyclin-CDK activity, which interferes with hyperphosphorylation of Rb by keeping it in the hypophosphorylated form and bound to E2F, thereby blocking the proliferation of cells and inducing cell growth arrest [53–56].
3. Cell-cycle and cancer: checkpoints
Cells are constantly subject to mutation of their DNA, which is detrimental to the cells but only rarely results in the production of cells that can escape the normal constraints and flourish as pathologic tumors. Cells respond to DNA damage by halting cell cycle progression and/or by undergoing programmed cell death or apoptosis. Replication errors and DNA damage incurred by radiation or chemical agents constantly challenge the genetic integrity of a cell. The complex regulatory system, discussed in part above, detects these aberrations and stalls the cells in the gap phases (G1 or G2) until the damage is repaired, or triggers the pathways that lead to programmed cell death. Failure of the quality control check points or a loss of balance of the regulatory molecules plays a major role in the development of cancer. One of the key players in these pathways is the transcription factor p53, which is the most frequently mutated tumor suppressor gene and loss of p53 expression or function is associated with an increased risk of cancer in humans [46]. Some of the mechanisms and/or checkpoints at which cells halt their cycle in order to verify normal metabolic processes are listed below, and summarized in Figure 1.
3.1. The G1 and G1/S checkpoint
In the presence of DNA damage, the G1/S checkpoint prevents replication of damaged DNA through several distinct signal transduction pathways. Of the many regulatory checkpoints of the cell cycle, the acquisition of abnormalities at the G1/S checkpoint appears to be the most crucial step in the genesis and progression of cancer [12,13]. Upon DNA damage, the activated Chk1 phosphorylates Cdc25A, triggering its ubiquitination and degradation by the proteasome pathway, which is required for G1/S transition [14,57]. Ubiquitous degradation of Cdc25A results in the failure of Cdk2 activation and prevents Cdc45 from loading onto chromatin. Since Cdc45 is essential for recruitment of DNA polymerase a, this prevents the development of a new origin of replication. This unloading of Cdc45 plays a role in the initial cell cycle arrest at the G1/S boundary. Inactivation of the pRb and p53 pathways at the G1/S transition is a fundamental requirement for the genesis of most human cancers. The cell cycle protein p53, which has been termed ‘the guardian of the genome’ is one of the most important cell cycle proteins modulated by regulation of checkpoints at G1. Transcriptional responses by p53 are then required for maintaining the G1/S arrest. The expression and activity of p53 is regulated by post-transcriptional modification, such as phosphorylation, sumoylation, neddylation and acetylation. Phosphorylation of p53 on Ser15 by ATM or ATR and on Ser20 by Chk1 accumulates p53 protein in the nucleus by inhibiting its nuclear export and degradation. Under normal circumstances, p53 is dormant until activated by DNA damage or other genomic aberrations. The key transcriptional target of p53 is the Cip1/p21, an inhibitor of cyclin dependent kinases, which silences the G1/S promoting cyclin E/CDK2 kinases and thereby inhibits the G1/S transition [13,58]. This allows DNA repair or the induction of various pro-apoptotic factors (Puma, Bax, Noxa), oxidative stress response genes, and the feedback regulator, Mdm2. p53 is a short-lived protein that is stabilized and transcriptionally activated by ATM-mediated phosphorylation. Another G1-checkpoint function served by p53 through the activation of Cip1/p21, which binds to the cyclin D-CDK4 complex and prevents it from phosphorylating Rb, thereby suppressing the Rb/E2F pathway. Thus, the G1 checkpoint signals target two independent and critical tumor suppressor pathways, goverened by p53 and pRb, which are most commonly deregulated in human cancers [59,60].
3.2. The S-phase checkpoint
The intra-S-phase checkpoint network functions to avoid the duplication of damaged or broken DNA, which would be further propagated in mitosis eventually lead to genomic instability. This checkpoint is regulated by two distinct pathways, namely ATM/ATR–Chk1–Cdc25A and ATM–Nbs1–SMC1 [61]. Depending on the type of DNA damage, ATM or ATR phosphorylates Chk1, which in turn phosphorylates Cdc25A on its several serine residues, and then maintain an appropriate abundance of Cdc25A. In response to genotoxic stress, the activity of Chk1 and Chk2 is enhanced leading to downregulation of Cdc25A, which subsequently causes inactivation of cyclin E–Cdk2 [2]. The other type of intra-S-checkpoint reflects the impact of ATM-mediated phosphorylations of Nbs1 on several sites, in particular Ser343, which is required for activation of the Nbs1–Mre11–Rad50 complex [62,63]. Depending on the phosphorylation state of Nbs1, the cohesin protein SMC1 is phosphorylated on Ser957 and Ser966 by ATM, which is required for the intra-S checkpoint. Other mediator proteins, such as 53BP1, BRCA1, FANCD2 and MDC1, are also involved in the intra-S checkpoint. The above mentioned two kinds of intra-S-checkpoints have been documented in response to both ionizing radiation and UV radiation [61, 64].
3.3. The G2/M checkpoint
The G2/M checkpoint prevents cells from entering into mitosis when they experience DNA damage during G2 or when they carry unrepaired DNA from G1 or S to progress into G2 [65,66]. The critical target of the G2/M checkpoint is the mitosis promoting activity, which is regulated through the inhibition of cyclin B/Cdc2 kinase by Chk1- or p38-mediated subcellular sequestration, degradation and inhibition of the Cdc25 family of phosphatases. In addition, other upstream regulator of Cdc25c and cyclin B/CDK1 are also targeted in DNA damage induced G2 arrest. The maintenance phase of G2/M partially relies on the transcriptional programs regulated by BRCA1 and p53. P53-dependent mechanisms are also important for the maintenance of G2 arrest. The critical targets of p53 at G2/M are the Cdk inhibitor p21, GADD45, which causes the dissociation of the Cdc2 and cyclin complex, and 14-3-3 sigma, which sequesters the cyclin B/Cdc2 complex in the cytoplasm [66,67]. In addition, p53 appears to repress the transcription of Cdc2 and cyclin B. Two isoforms of MAP kinase, p38 a and, also have been implicated in the G2/M checkpoint [2].
Previously, Cdc25B and Cdc25C were thought to be the major effectors of the G2/M checkpoint response. However, recent reports have revealed that both Cdc25B- and Cdc25C-deficient cells have a normal G2/M checkpoint, suggesting the crucial role of Cdc25A in targeting the G2/M checkpoint. P53-independent mechanism also is sufficient to sustain the G2 arrest in p53 mutant tumor cells. This has inspired efforts to develop strategies to interfere with G2 checkpoints as a potential approach to the sensitization of cancer cells to radiation or drug-induced DNA damage and, thus, cell death [68].
4. Molecular targets of dietary agents on cell cycle progression of cancer cells
Cancer is a complex disease, in which there is genetic variability among not only different types of cancer but also among different patients with the same type of cancer, and even among different cells within the same tumor. Tumors also represent the culmination of multiple genetic abnormalities. As a consequence, the targeting of a single molecular target for therapeutic purposes might not be sufficient to elicit the desired outcome. Different nutrients, specifically dietary botanicals, can play a role in the regulation of both normal and pathologic processes. An improved understanding of the regulatory role of these nutrients on cell cycle regulatory checkpoints may help in the prevention and treatment of various cancers. For more than a decade, there has been considerable interest in the use of naturally occurring botanicals for the prevention of disease including prevention of various cancers. Although several dietary agents or nutrients have been shown to affect the cell cycle regulation on treatment with cancer cells, we briefly summarize the role of some common dietary agents as an example and present evidence that dietary agents can interfere with the abnormal progression of cell cycle regulation of cancer cells. The agents which we discuss include grape seed proanthocyanidins (GSPs), green tea polyphenol, (−)-epigallocatechin-3-gallate (EGCG), resveratrol (red grapes, peanuts and berries), silymarin/silibinin (milk thistle), genistein (soybean), curcumin (turmeric) and apigenin (celery, parsley). A brief discussion includes their effects on cancer cells in vitro and in vivo studies for their multiple roles in the regulation of cell cycle proteins/checkpoints. Their sources and structures are summarized briefly in Figure 2. The cell cycle checkpoints that are known to be targeted by dietary agents are summarized in Figure 3.
Figure-3.

A simplified schematic representation of the various cell cycle phases, and the different cyclins and their kinases that control progression through the cycle. At the core of this control is the cyclin dependent kinase (Cdk) family of serine/threonine kinases, which regulate cell cycle progression through phosphorylation of proteins that function at specific phase of the cell cycle. Different Cdks act at different phases of the cell cycle and their activity is dependent on association with a member of the cyclin family of regulatory sub-units. Different dietary agents, such as EGCG, GSPs, silymarin, apigenin, resveratrol, genistein and curcumin act at different checkpoints as illustrated in the Figure. Some of them act on multiple checkpoints or targets. The arrows indicate activation and blocked signs indicate inhibitory effects.
4.1. Grape seed proanthocyanidins
Grape (Vitis vinifera) seeds are potent source of proanthocyanidins (GSPs), which are composed mainly of dimers, trimers and highly polymerized oligomers of monomeric catechins [69]. GSPs have been shown to have potent anti-carcinogenic properties in several in vitro and in vivo tumor models. They have been shown to control abnormal regulation of cell cycle progression in cancer cells and promote apoptotic cell death. Treatment of human epidermoid carcinoma A431 cells with GSPs results in inhibition of cell proliferation and the promotion of cytotoxic effects in a dose-dependent manner, which was associated with the arrest of cells in the G1 phase. It was observed that treatment of A431 cells with GSPs resulted in a marked reduction in the expression levels of CDK2, CDK4 and CDK6. Similarly, a marked reduction in the expression levels of cyclins D1, D2 and E was observed after GSPs treatment [70]. The Cip1/p21 and Kip1/p27 regulate the progression of cells in the Go/G1 phase of the cell cycle and induction of these proteins causes a blockade of the G1 to S transition, thereby resulting in a Go/G1 phase arrest of the cell cycle [71]. The loss of CDKI in human cancers leads to uncontrolled cell proliferation [72]. In this context, the in vitro experimental data revealed that treatment of human epidermoid carcinoma A431 cells with GSPs resulted in a dose-dependent increase in the protein levels of Cip1/p21 and Kip1/p27. These in vitro observations indicate that the GSP-induced enhancement of the levels of CDKI may have an important role in the GSP-induced G1-phase arrest of cell cycle progression in A431 cells, possibly through their inhibition of CDK kinase activity [70]. This event may lead to the apoptotic cell death of cancer cells. Apoptosis plays a crucial role in eliminating the mutated neoplastic and hyperproliferating neoplastic cells from the system and therefore is considered as a protective mechanism against the development of cancer [73].
Although in vitro cell culture models are useful in obtaining mechanistic insights, the observations made using the in vitro systems need to be verified in vivo animal models to establish the relevance of the cellular findings. In an in vivo study, we found that administration of GSPs by oral gavage inhibits the growth of A431 tumor-xenografts in athymic nude mice. The mechanism of inhibition of the growth of tumors by GSPs was further confirmed by the analysis of mRNA levels of proliferating cell nuclear antigen (PCNA) and cyclin D1, as markers of tumor cell proliferation. A reduction in the mRNA expression of cyclin D1 and PCNA was observed in the tumor-xenograft samples of GSP-treated mice as compared to the tumor-xenograft samples of control mice that were not given GSPs by gavage. Further, the inhibition of the growth of tumor xenograft in athymic nude mice by GSPs was associated with the induction of apoptotic cell death of tumor cells [74]. Similar observation were noted when the prostate cancer cells, DU145 and LNCaP, were treated with grape seed extract. These studies indicate that treatment of prostate cancer cells with GSPs results in inhibition of proliferation, induction of apoptosis, G1 phase arrest, increases in Cip1/p21 and decreases in CDK4, CDK2 and cyclin E [75].
4.2. EGCG/green tea polyphenol
EGCG has been identified as a major and most effective constituent of green tea. Therefore most of the in vitro and in vivo studies of the effects of green tea have been conducted using EGCG. EGCG has been shown to induce apoptosis and cell cycle arrest in many cancer cells without affecting normal cells [76]. Treatment of various cancer cells (prostate, lung and skin) with EGCG altered the pattern of cell cycle proteins; specifically the inhibition of CDKs. EGCG also enhances the expression of CDKI proteins, such as Cip1/p21 and Kip1/p27 while reducing the expression of cyclin D1 and the phosphorylation of retinoblastoma protein. EGCG causes cell cycle arrest and promotes apoptosis via a dose- and time-dependent upregulation of Cip1/p21, Kip1/p27, and p16/INK4A and down-regulation of proteins such as cyclin D1, cyclin E, CDK2, and CDK4 [77]. EGCG caused growth arrest at G1 stage of cell cycle through regulation of cyclin D1, CDK4, CDK6, Cip1/p21 and Kip1/p27, and induced apoptosis through generation of reactive oxygen species and activation of caspase-3 and caspase-9 [78]. A comprehensive effect of EGCG has been described on various cell signaling targets in vitro and in vivo systems, which shows the multiple targets of EGCG against malignancies [79, 80].
4.3. Resveratrol
Several reports indicate that resveratrol, a polyphenol found at high concentrations in grapes and red wine, inhibits proliferation of cancer cells by inhibiting cell-cycle progression at different stages of the cell cycle [81–84]. Kuwajerwala et al. [85] have reported that treatment of prostate LNCaP cells with resveratrol induced the cells to enter into S phase, but subsequent progression through S phase was limited by the inhibitory effect of resveratrol on DNA synthesis. This unique ability of resveratrol may be responsible for its apoptotic and antiproliferative effects. Benitez et al. observed that treatment of LNCaP and PC-3 cells with resveratrol induced apoptosis and that this was associated with the reduced levels of expression of cyclins D1 and E and CDK4, as well as a reduction in cyclin D1/CDK4 kinase activity [86]. Resveratrol also reduced proliferation and induced apoptosis in human epidermoid carcinoma A431 cells in a dose- and time-dependent manner. Resveratrol-induced apoptosis in A431 cells was associated with a reduced level of expression of cyclins D1, D2 and E2; a reduction in the levels of CDK2, CDK4 and CDK6; and enhanced levels of Cip1/p21 and Kip1/p27 proteins [82]. Wolter et al. has shown the down-regulation of the cyclin D1/CDK4 complex by resveratrol in colon cancer cell lines [87].
4.4. Genistein/Apigenin
Genistein has been found to induce apoptosis and G2 arrest and inhibited proliferation in a variety of cancer cell lines, regardless of p53 status [88]. The dietary flavonoid apigenin, which is abundantly present in fruits and vegetables, induces G2/M phase arrest in two p53-mutant cancer cell lines, HT-29 and MG63, and simultaneously enhances the levels of Cip1/p21, a CDK inhibitory protein [89]. Oral administration of apigenin by gavage has been found to inhibit the growth of prostate tumor xenograft in athymic nude mice through the down-modulation of cyclins D1, D2 and E; CDK2, CDK4, and CDK6, and enhancement of the levels of Cip1/p21 and Kip1/p27 proteins [90]. Treatment of PC-3 and LNCaP cells with apigenin caused a marked reduction in the levels of cyclin D1 protein and decreases in CDK2, CDK4 and CDK6, which leads to G0/G1 phase arrest of the cell cycle and induction of apoptosis [91]. Apigenin induced G2/M phase cell cycle arrest and reduced the levels of cyclin A, cyclin B, phosphorylated forms of cdc2 and cdc25 in pancreatic cancer cell lines [92]. It was shown that the apoptosis induced by apigenin in Hep G2 cells was possibly mediated through the p53-dependent pathway and the induction of Cip1/p21 expression, which was probably associated with the cell cycle arrest in G2/M phase [93]. Treatment with apigenin resulted in growth-inhibition and G2/M phase arrest in two p53-mutant cancer cell lines, HT-29 and MG63. These effects were associated with a marked increase in the protein expression of Cip1/p21. These results suggest that there is a p53-independent pathway for apigenin in p53-mutant cell lines, which induces Cip1/p21 expression and growth-inhibition, and that apigenin may be a useful chemopreventive agent not only in wild-type p53 status, but also in cancer with mutant p53 [94].
4.5. Silymarin/silibinin
Treatment of prostate cancer cells with silibinin, an active constituent of silymarin, a plant flavonoid from milk thistle, induced apoptosis, which was associated with G1 phase arrest, inhibition of cyclin dependent kinases (CDK2, CDK4 and CDK6), and a reduction in the levels of cyclin D1 [Reviewed in 95]. In the same set of experiments, silibinin was found to increase the levels of CDKI (Cip1/p21 and Kip1/p27) proteins, suggesting their mechanistic involvement. Dietary agents also can synergize with chemotherapeutic drugs, thereby reducing the toxicity of these drugs. Silibinin has been found to synergize the growth-inhibitory effect of doxorubicin on prostate carcinoma DU145 cells, and this was associated with a significant G2/M arrest. The underlying mechanism of G2/M arrest indicated an inhibitory effect on Cdc25c, Cdc2 and cyclin B1 protein expression and Cdc2/p34 kinase activity [95]. Treatment of human malignant melanoma cells, A375-S2, with silymarin results in increased G(2)/M phase arrest, possibly providing a prolonged time for DNA repair. Consequently, silymarin protected A375-S2 cell against UV-induced apoptosis was partially through SIRT1 pathway and modulation of the cell cycle distribution [96]. Extensive research within the last decade has shown that silymarin can suppress the proliferation of a variety of tumor cells (e.g., prostate, breast, ovary, colon, lung, bladder), and this is accomplished through cell cycle arrest at the G1/S-phase, induction of cyclin-dependent kinase inhibitors (such as p15, Cip1/p21 and Kip1/p27), down-regulation of anti-apoptotic gene products (e.g., Bcl-2 and Bcl-xL), inhibition of cell-survival kinases (AKT, PKC and MAPK) and inhibition of inflammatory transcription factors (e.g., NF-kappaB). Silymarin can also down-regulate gene products involved in the proliferation of tumor cells (cyclin D1, EGFR, COX-2, TGF-beta, IGF-IR), invasion (MMP-9), angiogenesis and metastasis [97].
4.6. Curcumin
Curcumin (Curcuma longa) is a common spice that is used commonly in the preparation of food in most Indian house-holds and in several Asian countries. Curcumin inhibits cell cycle progression of immortalized human umbilical vein endothelial cells by up-regulating the cyclin-dependent kinase inhibitors, Cip1/p21, Kip1/p27 and p53 [98]. In neuroblastoma cells, both curcumin and resveratrol upregulate p53 expression and induce nuclear translocation of p53, followed by induction of Cip1/p21 and Bax expression [99]. Treatment of Lovo cells and HCT-116 cells with curcumin resulted in an accumulation of the cells in the G2/M phase and prevented cells from entering the next cell cycle [100–102]. Curcumin inhibited the growth of glioma U251 cells in a dose-dependent manner, with the low dose of curcumin inducing G2/M cell cycle arrest. The high dose of curcumin not only enhanced G2/M cell cycle arrest, but also induced S phase arrest. Curcumin induces the expression of p53 and up-regulates the levels of Cip1/p21 and ING4 in glioma U251 cells [103]. Aggarwal et al. [104] have demonstrated a dose-and time-dependent down-regulation of expression of cyclin E by curcumin that correlates with a reduction in the proliferation of human prostate and breast cancer cells. The suppression of cyclin E expression was not cell-type dependent as down-regulation occurred in estrogen-positive and -negative breast cancer cells, androgen-dependent and -independent prostate cancer cells, leukemia and lymphoma cells, head and neck carcinoma cells, and lung cancer cells. This study indicated that curcumin enhanced the expression of tumor cyclin-dependent kinase inhibitors Cip1/p21 and Kip1/p27 as well as tumor suppressor protein p53 but suppressed the expression of Rb protein. Curcumin also promoted the accumulation of the cells in G1 phase of the cell cycle.
Collectively, it is apparent that dietary agents are important regulators of cellular proliferation and specific modulators of cell cycle-associated proteins. The present studies provide evidence that dietary agents have the ability to control the regulation of cell cycle progression in cancer cells through employing various molecular targets, as summarized in Figure 4; and may have the capability to inhibit the progression of cancers of many organs, if used appropriately and in a systematic manner. These findings reaffirm what Hippocrates said twenty-five centuries ago, “let food be thy medicine and medicine be thy food”.
Figure 4.

Molecular targets of cell cycle regulation in cancer cells in vitro and in vivo by dietary agents. Upward arrows (↑) indicate enhancement, and downward arrows (↓) indicate a reduction in the levels, or inhibition of the activity of the target molecules.
Table 1.
Dietary agents, their source and molecular structures.
| Dietary agents | Source | Botanical Name | Structure | Picture of the sources |
|---|---|---|---|---|
| EGCG | Green tea | Camellia sinensis |
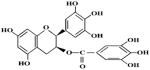
|

|
| Silymarin/silibinin | Milk thistle | Silybum marianum L. |
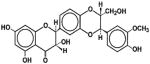
|

|
| Resveratrol | Grapes | Vitis vinifera |
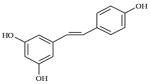
|

|
| Genistein | Soyabean | Glycine max |
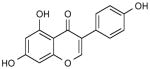
|

|
| Apigenin | Celery, Parsley & vegetables | Apium graveolens, Petroselinum crispum |
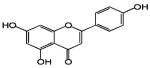
|

|
| Curcumin | Turmeric | Curcuma longa |
|

|
Acknowledgments
The work reported from the author’s laboratory was supported by the funds from National Institutes of Health (CA104428, AT002536) and Veteran Affairs Merit Review Award (SKK). The content of this article does not necessarily reflect the views or policies of the funding sources. The authors apologize for not discussing and citing numerous important publications because of the limitations of space and of the number of references.
References
- 1.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- 2.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 3.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 4.Baserga R, Wiebel F. The cell cycle of mammalian cells. Int Rev Exp Pathol. 1969;7:1–30. [PubMed] [Google Scholar]
- 5.Norbury C, Nurse P. Animal cell cycles and their control. Ann Rev Biochem. 1992;61:441–470. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- 6.McDonald ER, El-Deiry WS. Cell cycle control as a basis for cancer drug development. Int J Oncol. 2000;16:871–886. [PubMed] [Google Scholar]
- 7.Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature. 1970;225:159–164. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- 8.Murray A. Cell cycle checkpoints. Curr Opin Cell Biol. 1994;6:872–876. doi: 10.1016/0955-0674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 9.Murray AW. Coordinating cell cycle events. Cold Spring Harbor Symposia on Quantitative Biology. 1991;56:399–408. doi: 10.1101/sqb.1991.056.01.047. [DOI] [PubMed] [Google Scholar]
- 10.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 11.Michalides RJ. Cell cycle regulators: mechanisms and their role in aetiology, prognosis, and treatment of cancer. J Clin Pathol. 1999;52:555–568. doi: 10.1136/jcp.52.8.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto AE, Andre S, Laranjeira C, Soares J. Correlations of cell cycle regulators (p53, p21, pRb and mdm2) and c-erbB-2 with biological markers of proliferation and overall survival in breast cancer. Pathology. 2005;37:45–50. doi: 10.1080/00313020400011250. [DOI] [PubMed] [Google Scholar]
- 13.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 14.Mailand N, Falck J, Lukas C, Syljuâsen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 15.Sherr C. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 16.Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, Tennant RW, Weinberg RA, Yuspa SH, Conti CJ. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–2474. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 18.Krude T, Jackman M, Pines J, Laskey R. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell. 1997;88:109–119. doi: 10.1016/s0092-8674(00)81863-2. [DOI] [PubMed] [Google Scholar]
- 19.Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- 20.Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- 21.Gartel AL, Serfas MS, Tyner AL. p21-negative regulator of the cell cycle. Proc Soc Exp Biol Med. 1996;213:138–149. doi: 10.3181/00379727-213-44046. [DOI] [PubMed] [Google Scholar]
- 22.Pellegata NS, Antoniono RJ, Redpath JL, Stanbridge EJ. DNA damage and p53-mediated cell cycle arrest: a reevaluation. Proc Natl Acad Sci USA. 1996;93:15209–15214. doi: 10.1073/pnas.93.26.15209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fotedar R, Fitzgerald P, Rousselle T, Cannella D, Doree M, Messier H, Fotedar A. p21 contains independent binding sites for cyclin and cdk2: both sites are required to inhibit cdk2 kinase activity. Oncogene. 1996;12:2155–2164. [PubMed] [Google Scholar]
- 24.Porter DZ, Zhang N, Danes C, McGahren MJ, Harwell RM, Faruk S, Keyomarsi K. Tumor-specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol. 2001;21:6254–6269. doi: 10.1128/MCB.21.18.6254-6269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 26.Akli S, Zheng PJ, Multani AS, Wingate HF, Pathak S, Zhang N, Tucker SL, Chang S, Keyomarsi K. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res. 2004;64:3198–3208. doi: 10.1158/0008-5472.can-03-3672. [DOI] [PubMed] [Google Scholar]
- 27.Bortner DM, Rosenberg MP. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol. 1997;17:453–459. doi: 10.1128/mcb.17.1.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sui L, Dong Y, Ohno M, Sugimoto K, Tai Y, Hando T, Tokuda M. Implication of malignancy and prognosis of p27(kip1), Cyclin E, and Cdk2 expression in epithelial ovarian tumors. Gynecol Oncol. 2001;83:56–63. doi: 10.1006/gyno.2001.6308. [DOI] [PubMed] [Google Scholar]
- 29.Jang SJ, Park YW, Park MH, Lee JD, Lee YY, Jung TJ, Kim IS, Choi IY, Ki M, Choi BY, Ahn MJ. Expression of cell-cycle regulators, cyclin E and p21WAF1/CIP1, potential prognostic markers for gastric cancer. Eur J Surg Oncol. 1999;25:157–163. doi: 10.1053/ejso.1998.0619. [DOI] [PubMed] [Google Scholar]
- 30.Del Pizzo JJ, Borkowski A, Jacobs SC, Kyprianou N. Loss of cell cycle regulators p27(Kip1) and cyclin E in transitional cell carcinoma of the bladder correlates with tumor grade and patient survival. Am J Pathol. 1999;155:1129–1136. doi: 10.1016/S0002-9440(10)65216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukuse T, Hirata T, Naiki H, Hitomi S, Wada H. Prognostic significance of cyclin E overexpression in resected non-small cell lung cancer. Cancer Res. 2000;60:242–244. [PubMed] [Google Scholar]
- 32.Pines J, Hunter T. Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature. 1990;346:760–763. doi: 10.1038/346760a0. [DOI] [PubMed] [Google Scholar]
- 33.Erlandsson F, Linnman C, Ekholm S, Bengtsson E, Zetterberg A. A detailed analysis of cyclin A accumulation at the G(1)/S border in normal and transformed cells. Exp Cell Res. 2000;259:86–95. doi: 10.1006/excr.2000.4889. [DOI] [PubMed] [Google Scholar]
- 34.Sobczak-Thepot J, Harper F, Florentin Y, Zindy F, Brechot C, Puvion E. Localization of cyclin A at the sites of cellular DNA replication. Exp Cell Res. 1993;206:43–48. doi: 10.1006/excr.1993.1118. [DOI] [PubMed] [Google Scholar]
- 35.Cardoso MC, Leonhardt H, Nadal-Ginard B. Reversal of terminal differentiation and control of DNA replication: cyclin A and Cdk2 specifically localize at subnuclear sites of DNA replication. Cell. 1993;74:979–992. doi: 10.1016/0092-8674(93)90721-2. [DOI] [PubMed] [Google Scholar]
- 36.Bukholm IR, Husdal A, Nesland JM, Langerød A, Bukholm G. Overexpression of cyclin A overrides the effect of p53 alterations in breast cancer patients with long follow-up time. Breast Cancer Res Treat. 2003;80:199–206. doi: 10.1023/A:1024527220362. [DOI] [PubMed] [Google Scholar]
- 37.Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 38.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 39.Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 40.Sears RC, Nevins JR. Signaling networks that link cell proliferation and cell fate. J Biol Chem. 2002;277:11617–11620. doi: 10.1074/jbc.R100063200. [DOI] [PubMed] [Google Scholar]
- 41.Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol. 2002;14:684–691. doi: 10.1016/s0955-0674(02)00388-5. [DOI] [PubMed] [Google Scholar]
- 42.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 43.Lee MH, Reynisdóttir I, Massagué J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995;9:639–49. doi: 10.1101/gad.9.6.639. [DOI] [PubMed] [Google Scholar]
- 44.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, Fox MP, Wei N. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y, Wiman KG, Mercer WE, Kastan MB, Kohn KW, Elledge SJ, Kinzler KW, Vogelstein B. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 47.Duliv V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 48.Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- 49.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 50.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-CDK protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 51.Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- 52.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 53.Hunter T, Pines J. Cyclins and cancer II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y, Martindale JL, Gorospe M, Holbrook NJ. Regulation of p21WAF1/CIP1 expression through mitogen-activated protein kinase signaling pathway. Cancer Res. 1996;56:31–35. [PubMed] [Google Scholar]
- 55.Blagosklonny MV, Prabhu NS, El-Deiry WS. Defects in p21WAF1/CIP1, Rb, and c-myc signaling in phorbol ester-resistant cancer cells. Cancer Res. 1997;57:320–325. [PubMed] [Google Scholar]
- 56.Graña X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- 57.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 58.Wahl GM, Carr AM. The evaluation of diverse biological response to DNA damage: insights from yeast and p53. Nature Cell Biol. 2001;3:E277–E286. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- 59.Michael D, Oren M. The p53 and mdm2 families in cancer. Curr Opin Genet Dev. 2002;12:53–59. doi: 10.1016/s0959-437x(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 60.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 61.Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30:290–294. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 62.Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 63.Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, Shay JW, Ziv Y, Shiloh Y, Lee EY. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
- 64.Pichierri P, Rosselli F. The DNA crosslink induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004;23:1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu B, Kim S-T, Lim D-S, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing radiation. Mol Cell Biol. 2002;22:1049–1059. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 67.Taylor WR, Stark GR. Regulation of G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 68.Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nature Rev Cancer. 2004;4:216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
- 69.Prieur C, Rigaud J, Cheynier V, Moutounet M. Oligomeric and polymeric procyanidins from grape seeds. Phytochemistry. 1994;36:781–789. [Google Scholar]
- 70.Meeran SM, Katiyar SK. Grape seed proanthocyanidins promote apoptosis in human epidermoid carcinoma A431 cells through alterations in Cdki-Cdk-cyclin cascade, and caspase-3 activation via loss of mitochondrial membrane potential. Exp Dermatol. 2007;16:405–415. doi: 10.1111/j.1600-0625.2007.00542.x. [DOI] [PubMed] [Google Scholar]
- 71.Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of cdks, their cyclin activators, and CIP and INK4 inhibitors. J Mol Biol. 1999;287:821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 72.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 73.Hickman JA. Apoptosis induced by anticancer drugs. Cancer Metastasis Rev. 1992;11:121–139. doi: 10.1007/BF00048059. [DOI] [PubMed] [Google Scholar]
- 74.Meeran SM, Katiyar SK. Proanthocyanidins inhibit mitogenic and survival-signaling in vitro and tumor growth in vivo. Front Biosci. 2008;13:887–897. doi: 10.2741/2729. [DOI] [PubMed] [Google Scholar]
- 75.Agarwal C, Sharma Y, Agarwal R. Anticarcinogenic effect of a polyphenolic fraction isolated from grape seeds in human prostate carcinoma DU145 cells: modulation of mitogenic signaling and cell-cycle regulators and induction of G1 arrest and apoptosis. Mol Carcinog. 2000;28:129–38. doi: 10.1002/1098-2744(200007)28:3<129::aid-mc1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 76.Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst. 1997;89:1881–1886. doi: 10.1093/jnci/89.24.1881. [DOI] [PubMed] [Google Scholar]
- 77.Gupta S, Ahmad N, Nieminen AL, Mukhtar H. Growth inhibition, cell-cycle dysregulation, and induction of apoptosis by green tea constituent (−)-epigallocatechin-3-gallate in androgen-sensitive and androgen-insensitive human prostate carcinoma cells. Toxicol Appl Pharmacol. 2000;164:82–90. doi: 10.1006/taap.1999.8885. [DOI] [PubMed] [Google Scholar]
- 78.Shankar S, Suthakar G, Srivastava RK. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front Biosci. 2007;12:5039–5051. doi: 10.2741/2446. [DOI] [PubMed] [Google Scholar]
- 79.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006;66:2500–2505. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 80.Yusuf N, Irby C, Katiyar SK, Elmets CA. Photoprotective effects of green tea polyphenols. Photodermatol Photoimmunol Photomed. 2007;23:48–56. doi: 10.1111/j.1600-0781.2007.00262.x. [DOI] [PubMed] [Google Scholar]
- 81.Estrov Z, Shishodia S, Faderl S, Harris D, Van Q, Kantarjian HM, Talpaz M, Aggarwal BB. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood. 2003;102:987–995. doi: 10.1182/blood-2002-11-3550. [DOI] [PubMed] [Google Scholar]
- 82.Ahmad N, Adhami VM, Afaq F, Feyes DK, Mukhtar H. Resveratrol causes WAF-1/p21-mediated G(1)-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin Cancer Res. 2001;7:1466–1473. [PubMed] [Google Scholar]
- 83.Hsieh TC, Juan G, Darzynkiewicz Z, Wu JM. Resveratrol increases nitric oxide synthase, induces accumulation of p53 and p21(WAF1/CIP1), and suppresses cultured bovine pulmonary artery endothelial cell proliferation by perturbing progression through S and G2. Cancer Res. 1999;59:2596–2601. [PubMed] [Google Scholar]
- 84.Liang YC, Tsai SH, Chen L, Lin-Shiau SY, Lin JK. Resveratrol-induced G2 arrest through the inhibition of CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem Pharmacol. 2003;65:1053–1060. doi: 10.1016/s0006-2952(03)00011-x. [DOI] [PubMed] [Google Scholar]
- 85.Kuwajerwala N, Cifuentes E, Gautam S, Menon M, Barrack ER, Reddy GP. Resveratrol induces prostate cancer cell entry into s phase and inhibits DNA synthesis. Cancer Res. 2000;62:2488–2492. [PubMed] [Google Scholar]
- 86.Benitez DA, Pozo-Guisado E, Alvarez-Barrientos A, Fernandez-Salguero PM, Castellón EA. Mechanisms involved in resveratrol-induced apoptosis and cell cycle arrest in prostate cancer-derived cell lines. J Androl. 2007;28:282–293. doi: 10.2164/jandrol.106.000968. [DOI] [PubMed] [Google Scholar]
- 87.Wolter F, Akoglu B, Clausnitzer A, Stein J. Downregulation of the cyclin D1/Cdk4 complex occurs during resveratrol-induced cell cycle arrest in colon cancer cell lines. J Nutr. 2001;131:2197–2203. doi: 10.1093/jn/131.8.2197. [DOI] [PubMed] [Google Scholar]
- 88.Li M, Zhang Z, Hill DL, Chen X, Wang H, Zhang R. Genistein, a dietary isoflavone, down-regulates the MDM2 oncogene at both transcriptional and posttranslational levels. Cancer Res. 2005;65:8200–8208. doi: 10.1158/0008-5472.CAN-05-1302. [DOI] [PubMed] [Google Scholar]
- 89.Takagaki N, Sowa Y, Oki T, Nakanishi R, Yogosawa S, Sakai T. Apigenin induces cell cycle arrest and p21/WAF1 expression in a p53-independent pathway. Int J Oncol. 2005;26:185–189. [PubMed] [Google Scholar]
- 90.Shukla S, Gupta S. Molecular targets for apigenin-induced cell cycle arrest and apoptosis in prostate cancer cell xenograft. Mol Cancer Ther. 2006;5:843–852. doi: 10.1158/1535-7163.MCT-05-0370. [DOI] [PubMed] [Google Scholar]
- 91.Shukla S, Gupta S. Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma dephosphorylation in human prostate cancer cells. Cell Cycle. 2007;6:1102–1114. doi: 10.4161/cc.6.9.4146. [DOI] [PubMed] [Google Scholar]
- 92.Ujiki MB, Ding XZ, Salabat MR, Bentrem DJ, Golkar L, Milam B, Talamonti MS, Bell RH, Jr, Iwamura T, Adrian TE. Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer. 2006;5:76. doi: 10.1186/1476-4598-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chiang LC, Ng LT, Lin IC, Kuo PL, Lin CC. Anti-proliferative effect of apigenin and its apoptotic induction in human Hep G2 cells. Cancer Lett. 2006;237:207–214. doi: 10.1016/j.canlet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 94.Takagaki N, Sowa Y, Oki T, Nakanishi R, Yogosawa S, Sakai T. Apigenin induces cell cycle arrest and p21/WAF1 expression in a p53-independent pathway. Int J Oncol. 2005;26:185–189. [PubMed] [Google Scholar]
- 95.Deep G, Agarwal R. Chemopreventive efficacy of silymarin in skin and prostate cancer. Integr Cancer Ther. 2007;6:130–145. doi: 10.1177/1534735407301441. [DOI] [PubMed] [Google Scholar]
- 96.Li LH, Wu LJ, Tashiro SI, Onodera S, Uchiumi F, Ikejima T. Activation of the SIRT1 pathway and modulation of the cell cycle were involved in silymarin’s protection against UV-induced A375-S2 cell apoptosis. J Asian Nat Prod Res. 2007;9:245–252. doi: 10.1080/10286020600604260. [DOI] [PubMed] [Google Scholar]
- 97.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006;26:4457–4498. [PubMed] [Google Scholar]
- 98.Park MJ, Kim EH, Park IC, Lee HC, Woo SH, Lee JY, Hong YJ, Rhee CH, Choi SH, Shim BS, Lee SH, Hong SL. Curcumin inhibits cell cycle progression of immortalized human umbilical vein endothelial (ECV304) cells by up-regulating cyclin-dependent kinase inhibitor, p21WAF1/CIP1, p27KIP1 and p53. Int J Oncol. 2002;21:379–383. [PubMed] [Google Scholar]
- 99.Liontas A, Yeger H. Curcumin and resveratrol induce apoptosis and nuclear translocation and activation of p53 in human neuroblastoma. Anticancer Res. 2004;24:987–998. [PubMed] [Google Scholar]
- 100.Jaiswal AS, Marlow BP, Gupta N, Narayan S. Beta-catenin-mediated transactivation and cell–cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene. 2002;21:8414–8427. doi: 10.1038/sj.onc.1205947. [DOI] [PubMed] [Google Scholar]
- 101.Moragoda L, Jaszewski R, Majumdar AP. Curcumin induced modulation of cell cycle and apoptosis in gastric and colon cancer cells. Anticancer Res. 2001;21:873–878. [PubMed] [Google Scholar]
- 102.Chen H, Zhang ZS, Zhang YL, Zhou DY. Curcumin inhibits cell proliferation by interfering with the cell cycle and inducing apoptosis in colon carcinoma cells. Anticancer Res. 1999;19:3675–3680. [PubMed] [Google Scholar]
- 103.Liu E, Wu J, Cao W, Zhang J, Liu W, Jiang X, Zhang X. Curcumin induces G2/M cell cycle arrest in a p53-dependent manner and upregulates ING4 expression in human glioma. J Neurooncol. 2007 Jun 27; doi: 10.1007/s11060-007-9421-4. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 104.Aggarwal BB, Banerjee S, Bharadwaj U, Sung B, Shishodia S, Sethi G. Curcumin induces the degradation of cyclin E expression through ubiquitin-dependent pathway and up-regulates cyclin-dependent kinase inhibitors p21 and p27 in multiple human tumor cell lines. Biochem Pharmacol. 2007;73:1024–1032. doi: 10.1016/j.bcp.2006.12.010. [DOI] [PubMed] [Google Scholar]