

Abstract
Schizophrenia and autism are thought to result from the interaction between a susceptibility genotype and environmental risk factors. The offspring of women who experience infection while pregnant have an increased risk for these disorders. Maternal immune activation (MIA) in pregnant rodents produces offspring with abnormalities in behavior, histology, and gene expression that are reminiscent of schizophrenia and autism, making MIA a useful model of the disorders. However, the mechanism by which MIA causes long-term behavioral deficits in the offspring is unknown. Here we show that the cytokine interleukin-6 (IL-6) is critical for mediating the behavioral and transcriptional changes in the offspring. A single maternal injection of IL-6 on day 12.5 of mouse pregnancy causes prepulse inhibition (PPI) and latent inhibition (LI) deficits in the adult offspring. Moreover, coadministration of an anti-IL-6 antibody in the poly(I:C) model of MIA prevents the PPI, LI, and exploratory and social deficits caused by poly(I:C) and normalizes the associated changes in gene expression in the brains of adult offspring. Finally, MIA in IL-6 knock-out mice does not result in several of the behavioral changes seen in the offspring of wild-type mice after MIA. The identification of IL-6 as a key intermediary should aid in the molecular dissection of the pathways whereby MIA alters fetal brain development, which can shed new light on the pathophysiological mechanisms that predispose to schizophrenia and autism.
Keywords: schizophrenia, autism, cytokine, poly(I:C), maternal immune activation, IL-6, influenza
Introduction
Birth in winter or spring months is an accepted risk factor for schizophrenia, and the preponderance of evidence suggests that the prevalence of influenza in winter months is responsible (Tochigi et al., 2004). Over 25 studies have analyzed schizophrenia incidence after influenza epidemics, and the majority have found an increased incidence among exposed offspring. More recently, Brown and colleagues (Brown and Susser, 2002; Brown et al., 2004; Brown, 2006) examined the medical records of >12,000 pregnant women and found that second-trimester respiratory infection increases the risk for schizophrenia in the offspring threefold to sevenfold. Because of the high prevalence of influenza infection, they estimate that 14–21% of schizophrenia cases are caused by maternal infection. These findings are also supported by an association between elevated cytokines or anti-influenza antibodies in maternal serum and schizophrenia in the offspring (Brown et al., 2004). Maternal infection may also play a role in the pathogenesis of autism (Patterson, 2002). These links are even more remarkable considering that the epidemiological studies are unable to screen for susceptibility genotype. Because of the strong genetic component in autism and schizophrenia, it is likely that only genetically susceptible individuals who were exposed to maternal infection would develop the disorder, suggesting that the risk associated with maternal infection may be considerably more than threefold to sevenfold in susceptible individuals.
Several lines of evidence indicate that the maternal immune response, rather than direct infection of the fetus, is responsible for the increased incidence of schizophrenia and autism in the offspring of mothers who suffer infections during pregnancy (Patterson, 2005). First, human influenza infection is usually confined to the respiratory tract. Moreover, in a mouse model, influenza infection during pregnancy produces behavioral deficits in the adult offspring (Shi et al., 2003), but we detected no virus in the fetuses (Shi et al., 2005). Most importantly, injecting either the double-stranded RNA poly(I:C) or bacterial lipopolysaccharide (LPS), which both induce strong innate immune responses in the absence of infection, into pregnant rodents produces behavioral and histological abnormalities in the adult offspring similar to those seen in the offspring of infected mothers (Shi et al., 2003; Zuckerman et al., 2003; Fortier et al., 2004a; Zuckerman and Weiner, 2005; Meyer et al., 2006; Ozawa et al., 2006). After maternal immune activation (MIA) by influenza infection, LPS, or poly(I:C), cytokine levels are altered in the maternal serum as well as the amniotic fluid, placenta, and fetal brain (Fidel et al., 1994; Cai et al., 2000; Urakubo et al., 2001; Gayle et al., 2004; Paintlia et al., 2004; Gilmore et al., 2005; Ashdown et al., 2006; Beloosesky et al., 2006; Meyer et al., 2006; Xu et al., 2006). Because cytokines drive the innate immune response, they are logical candidates for disruption of fetal brain development.
We have examined several proinflammatory cytokines as potential mediators of the effects of MIA on fetal brain development. Using the behavior of adult offspring as the readout, we first tested whether injection of single cytokines in the uninfected mother can mimic the effects of maternal infection on the offspring. To then test the involvement of endogenous cytokines, we asked whether anti-cytokine antibodies can block the effects of maternal poly(I:C) injection on the offspring and whether MIA affects the behavior of offspring of cytokine knock-out (KO) mice. The data identify interleukin-6 (IL-6) as a key mediator of the effects of MIA on fetal brain development.
Materials and Methods
Generation of animals
Female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) were obtained from our in-house breeding facility and were housed in ventilated cages under standard laboratory conditions. Mice were mated overnight, and the presence of a vaginal plug marked that day as embryonic day 0 (E0). Pregnant females were not disturbed, except for weekly cage cleaning, until E12.5 when they were weighed and pseudo-randomly assigned to one of seven groups. Each group initially contained at least five pregnant females.
Cytokine injection
Mice were given intraperitoneal injections of either 5 μg of carrier protein-free recombinant mouse IL-6 or 5 μg of carrier-free recombinant mouse interferon γ (IFNγ) (R & D Systems, Minneapolis, MN) freshly dissolved in 200 μl of 0.9% saline. Control mice were given injections of 200 μl of vehicle. Injections in the microgram range in adult mice cause acute behavioral effects (Swiergiel et al., 1997); our pilot experiments suggested that maternal doses in this range have a significant effect on adult offspring.
Cytokine blocking
Four groups of mice were given intraperitoneal injections of 20 mg/kg poly(I:C) (potassium salt; Sigma, St. Louis, MO) freshly dissolved in saline, because previous experiments found this dose to be most effective with intraperitoneal delivery (Shi et al., 2003). The manufacturer supplies poly(I:C) at 10% of the total weight of the salt, and dosage was based on the weight of poly(I:C) itself. Some of the mice were given coinjections of 100 μg of cytokine-neutralizing antibody targeted against IL-6 (anti-IL-6; rat IgG1), IFNγ (anti-IFNγ; rat IgG2a), or IL-1β (anti-IL-1β; rat IgG1) (R & D Systems), freshly dissolved in sterile saline. The total injection volume was 200 μl. The half-life of rat IgG1 and IgG2a in pregnant mice is similar to that of mouse IgG, with an α-phase lasting ∼24 h and a β-phase lasting >200 h, with 2–3% maternofetal transmission (Medesan et al., 1998). The antibody dose was calculated to be approximately a 10-fold molar excess of the maximal cytokine levels observed in response to poly(I:C). Control mice were given injections of either 200 μl of saline or 100 μg of anti-IL-6 freshly dissolved in 200 μl of saline.
IL-6 KO mice
IL-6 KO mice, strain B6.129S2-IL6tm1Kopf/J, backcrossed for 11 generations onto a C57 background, were obtained from The Jackson Laboratory and maintained in our facility by homozygous breeding. Mice were mated and were given injections of 20 mg/kg poly(I:C) or saline, using the procedures outlined above.
All pregnant mice were single housed after injection and returned to the same cage rack. Mice were left undisturbed, except for weekly cage cleaning, until the pups were weaned at 3 weeks of age. Offspring were housed in same-sex groups of two to five animals.
Measurement of maternal cytokine levels
Pregnant females were given injections of saline, poly(I:C), poly(I:C) plus anti-IL-6, or poly(I:C) plus anti-IFNγ, as above. Three hours after injection, mice were killed with an overdose of sodium pentobarbital (Nembutal), and blood was collected via cardiac puncture into Eppindorf tubes and allowed to clot at room temperature for 1 h. Blood was centrifuged at 8000 × g for 10 min at 4°C, and the serum was aliquoted and stored at −80°C until use. ELISAs for IL-6, IFNγ, and IL-1β (R & D Systems) were preformed according to the manufacturer's instructions.
For immunoprecipitation of anti-cytokine antibodies, a biotinylated, anti-rat antibody that was preabsorbed with mouse serum to prevent binding to mouse antibodies (Vector Laboratories, Burlingame, CA) was conjugated to streptavidin magnetic beads (NEB, Ipswich, MA) overnight at 4°C. After washing the beads thoroughly with PBS, mouse serum was diluted 1:20 in PBS and incubated with the beads for 4 h at room temperature. The beads were removed magnetically, and the resulting serum was used directly for ELISA.
Cytokine detection array kits were purchased from RayBioTech (Norcross, GA), and the manufacturer's instructions were followed. Briefly, antibody-spotted membranes were treated with blocking solution, incubated overnight at 4°C with 50 μl of mouse serum to be tested, washed with wash buffer, and probed with biotinylated anti-cytokine antibodies, and binding was detected using streptavidin–HRP chemiluminescence.
Behavioral testing
Latent inhibition.
The protocol was modeled after Zuckerman and Weiner (2005). Each group of mice was randomly subdivided into two groups, pre-exposed (PE) and not pre-exposed (NPE). Mice were placed in a box (Coulborn Instruments, Allentown, PA) with a speaker mounted on the back wall and an infrared motion detector on the ceiling. PE mice were presented with 40 tones (2000 Hz, 30 s duration) separated by 30 ± 40 s to randomize the intertone interval. NPE mice were placed in the same enclosure for an equivalent amount of time. Immediately after pre-exposure, all mice were given three pairing trials of the 30 s tone immediately followed by a 1 s, 0.3 mA footshock delivered through the floor. Pairing trials were separated by 180 s. The next day, the mice were returned to the same enclosure for 8 min to measure context freezing (measured as described below). The following day, mice were again returned to the enclosure and, after a 180 s acclimation period, were presented with an 8 min tone presentation. Freezing during the tone presentation was measured by the sensors and defined as a period of ≥4 s during which movement was not detected. Data are presented as percentage of the time spent freezing during tone presentation, and latent inhibition (LI) is defined as the difference in the amount of freezing in response to the tone in PE mice compared with NPE mice.
Pilot experiments suggested that PE mice demonstrate a larger range of time spent freezing than NPE mice, so the groups were split unevenly [control-saline, 7 NPE mice and 20 PE mice; control-anti-IL-6, 7 NPE mice and 15 PE mice; poly(I:C), 6 NPE mice and 13 PE mice; poly(IC) plus anti-IL-6, 10 NPE mice and 29 PE mice; poly(IC) plus anti-IFNγ, 8 NPE mice and 12 PE mice; IL-6, 7 NPE mice and 10 PE mice; IFNγ, 3 NPE mice and 11 PE mice], resulting in small numbers of NPE animals in some groups. Initially, the NPE animals belonging to different groups were treated as separate groups, but ANOVA revealed no significant differences between the groups (F(6,41) = 0.9241, p ≫ 0.05) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Fear conditioning was therefore similar in all groups, and the NPE groups were merged for greater statistical power.
Prepulse inhibition.
The prepulse inhibition (PPI) apparatus (San Diego Instruments, San Diego, CA) consisted of a sound-insulated chamber with a speaker mounted on the ceiling. The subject was restrained in a Plexiglas cylinder inside the chamber, and a pliezo-electric sensor was mounted beneath the restraining device to measure the startle response. After a 5 min acclimation period, the subject was presented with six 120 dB pulses of white noise. The subject was then presented with 14 blocks of four different trial types in a pseudo-random order. Trial types included P5P, in which a prepulse of 5 dB above background (67 dB) precedes the startle stimulus by 100 ms; P15P, in which the prepulse is 15 dB above background; startle stimulus alone; and no stimulus. Trials were averaged for each individual, and PPI was defined as PPI(X) = (startle alone − PXP)/(startle alone), where X = 5 or 15.
Open field.
Mice were placed in a 50 × 50 cm white Plexiglas box brightly lit by fluorescent room lighting and six 60 W incandescent bulbs 4–6 feet above the box. Activity was recorded by a ceiling-mounted video camera and analyzed using Ethovision software (Noldus, Leesburg, VA). The software allows display of the paths taken by the mice, and it measures the total distance moved and the number of entries into the center of the arena (central 17 cm square) in a 10 min session.
Social interaction.
The testing apparatus consisted of a 60 × 40 cm Plexiglas box divided into three chambers as described previously (Sankoorikal et al., 2006). Mice could move between chambers through a small opening (6 × 6 cm) in the dividers. Plastic cylinders in each of the two side chambers contained the probe mice, and numerous 1 cm holes in the cylinders enabled test and probe mice to contact each other. Mice to be tested were placed in the center chamber, and an overhead camera recorded their movements. Mice were allowed 5 min to explore the box, after which an unfamiliar, same-sex probe mouse from the same experimental group was placed in one of two restraining cylinders. The Ethovision program measured time spent in each of the three chambers, and social preference was defined as (% time spent in the social chamber) − (% time spent in the opposite chamber).
DNA microarray analysis
Mice were killed by cervical dislocation, and brains removed quickly. The olfactory bulb was peeled back and removed, and a 2 mm coronal section of cortex was removed from the front of the brain with a clean razor blade. This section was placed in a 1.5 ml tube and frozen in liquid nitrogen. Total RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, Ca). RNA quality was assessed using the Agilent (Palo Alto, CA) Bioanalyzer. Reverse transcription, in vitro transcription, and fragmentation were performed according to the manufacturer's recommendations (Affymetrix, Sunnyvale, CA). Samples were hybridized onto MOE430A mouse Affymetrix GeneChips that contained >22,000 probe sets using the Affymetrix hybridization station. To avoid microarray batch variation, only microarrays from a single lot were used. Microarrays were considered for use only if the average 3′:5′ ratio for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and actin did not exceed 1:1.2. Segmentation of scanned microarray images was performed by Microarray Analysis Suite 5.0. Determination of expression levels and scaling were performed using Robust Multi-array Average (Irizarry et al., 2003a,b). For scale linearity, the data were log2 transformed, and differential expression was established using the average log2 ratio (ALR) between the studied cohorts (|ALR| = 1 corresponds to a twofold increase or decrease, |ALR| = 0.585 represents a 50% change, and |ALR| = 0.263 depicts a 20% expression alteration).
Statistical analysis
All data are shown as mean ± SEM. The statistical significance of differences between two groups was assessed using the Student's t test, and differences among multiple groups were assessed using one-way ANOVAs followed by Bonferroni post hoc tests.
Results
Effects of exogenous cytokines
Our pilot studies indicated that maternal administration of IL-6, but not IL-1α, tumor necrosis factor α (TNFα), or IFNγ, causes PPI deficits in the adult offspring. PPI is the inhibition of a startle response when the startling stimulus is immediately preceded by a smaller, nonstartling stimulus of the same modality and is a measure of sensory-motor gating, attention, and distractibility. PPI deficits are observed in several mental disorders, including schizophrenia (Wynn et al., 2004) and autism (Perry et al., 2006). Furthermore, PPI deficits in the offspring elicited by maternal influenza infection respond to antipsychotic and psychomimetic drugs (Shi et al., 2003), and the PPI deficit resulting from poly(I:C) MIA is present in adult but not juvenile rats (Zuckerman et al., 2003), mimicking the adult onset of schizophrenia. The changes seen in this very relevant behavior prompted further study of the effects of maternal IL-6 administration.
In a large follow-up study (for numbers of animals, see supplemental Table 1, available at www.jneurosci.org as supplemental material), we confirmed that the offspring of pregnant mice given injections of 5 μg of IL-6 on E12.5 display lower PPI than either control offspring (p < 0.01) or offspring of mothers given injections of 5 μg of IFNγ (p < 0.05) (Fig. 1a).
Figure 1.

The offspring of mice given injections of recombinant IL-6 exhibit abnormal behaviors. a, Offspring of mice given injections of IL-6 but not IFNγ have a PPI deficit at a prepulse intensity of 85 dB (F(2,79) = 4.369; p < 0.05). *p < 0.05 versus control. b, PE offspring of control mice show normal LI compared with NPE animals, as do PE offspring of IFNγ-injected mothers. Offspring of IL-6-injected mothers, in contrast, do not demonstrate significant LI (F(3,103) = 10.22; p < 0.0001). *p < 0.05 versus NPE; **p < 0.001 versus NPE.
Both control offspring and offspring of mice given injections of IFNγ display significant LI compared with NPE mice (p < 0.001 and p < 0.05, respectively). The offspring of IL-6-injected mice lack significant LI (p > 0.05 vs NPE) (Fig. 1b). LI refers to the inhibition of a conditioned response to a stimulus when an individual has been repeatedly exposed to the stimulus before pairing with the unconditioned response. LI is a measure of the ability to ignore irrelevant stimuli, and its disruption is considered to be pertinent for the cognitive deficits in schizophrenia (Weiner, 2003). LI is disrupted in schizophrenic subjects and in amphetamine-treated humans and rats, restored to normal levels in schizophrenics by neuroleptic drugs, and enhanced in normal humans and rats by antipsychotic drugs (Weiner, 2003). As with PPI, MIA in rats disrupts LI in adult, but not prepubertal, offspring (Zuckerman et al., 2003), and the disrupted LI of poly(I:C)-exposed offspring responds to antipsychotic drugs (Zuckerman and Weiner, 2005). Disruption of LI, enhanced amphetamine-induced locomotion (Zuckerman et al., 2003; Ozawa et al., 2006), and altered concentrations of dopamine and its metabolites (Ozawa et al., 2006) caused by poly(I:C) MIA are consistent with a subcortical dopamine dysfunction that is a central feature of many theories of schizophrenia (Weiner, 2003). Thus, a single injection of IL-6 on E12.5 causes deficits in two relevant behaviors (LI and PPI) in the adult offspring.
Effects of anti-IL-6 on abnormal behaviors evoked by MIA
To test the role of endogenous IL-6 during MIA, we coadministered an IL-6-neutralizing antibody with poly(I:C) on E12.5. To control for nonspecific effects of antibody administration, we confirmed that a single maternal injection of anti-IL-6 antibody alone does not cause behavioral changes in the offspring (PPI, LI, and open field; data not shown). Furthermore, maternal coadministration of poly(I:C) and neutralizing antibodies of the same, or different, Ig subclass (IL-1β/IgG1 or anti-IFNγ/IgG2a) produces offspring that behave similarly to the offspring of mice given injections of poly(I:C) alone, indicating the lack of nonspecific effects (see below). To ensure the efficacy of the antibodies, we measured cytokine levels in each treatment group by ELISA and cytokine array assay. We find that IL-6 and IL-1β are elevated in mice given injections of poly(I:C) and that anti-IL-6 binds 98% of the total serum IL-6 induced by poly(I:C), without significantly altering the levels of other cytokines (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
We tested two separate, large sets of offspring for several relevant behaviors as adults. Offspring of mice given injections of poly(I:C) lack LI, as do the offspring of mice given injections of poly(I:C) plus anti-IFNγ-neutralizing antibody (Fig. 2a). In contrast, mice given injections of poly(I:C) plus anti-IL-6 antibody show normal LI (p < 0.01 vs NPE). Offspring of poly(I:C)-treated mothers also exhibit a deficit in PPI (p < 0.001), whereas the PPI of offspring of mice given injections of poly(I:C) plus anti-IL-6 antibody is not significantly different from controls but is significantly higher than the PPI of offspring of mice given injections of poly(I:C) alone (p < 0.05) (Fig. 2b). Thus, a brief neutralization of IL-6 during a critical period of embryonic development prevents two important behavioral deficits caused by MIA.
Figure 2.

Abnormal behavior in MIA offspring is prevented by maternal treatment with anti-IL-6 antibody. a, Offspring of mice treated with poly(I:C) lack LI. Coinjection of anti-IL-6 with poly(I:C) restores significant LI, whereas coinjection of anti-IFNγ does not (F(4,132) = 7.566; p < 0.0001). **p < 0.001 versus NPE; *p < 0.01 versus NPE. b, Compared with controls, the offspring of mice treated with poly(I:C) show a PPI deficit at a prepulse level of 85 dB. Coinjection with anti-IL-6 prevents this deficit. The PPI of offspring of mice given coinjections of poly(I:C) and anti-IFNγ or anti-IL-1β are not significantly different from control or poly(I:C) (F(4,270) = 4.195; p < 0.005). *p < 0.001 versus control; #p < 0.05 versus poly(I:C). c, d, In the open-field test, offspring of mice treated with poly(I:C) make fewer entries than controls into the center (c) and travel less total distance (d). Offspring of mice given coinjections of anti-IL-6 enter the center as often as control mice [F(3,123) = 3.703; p < 0.05; *p < 0.05 vs control; #p < 0.05 vs poly(I:C)] and move a similar total distance [F(3,123) = 6.666; p < 0.0005; *p < 0.01 vs control; #p < 0.001 vs poly(I:C)]. Offspring of mice given coinjections of poly(I:C) and anti-IFNγ are not significantly different from controls or poly(I:C). e, Tracks recorded during the open-field session demonstrate increased thigmotaxis in offspring of poly(I:C)-treated mice compared with offspring of mice given coinjections of an IL-6 antibody. f, In the social interaction test, control mice show a strong preference for the social chamber [defined as (percentage of time in social chamber) − (percentage of time in opposite chamber)], whereas the offspring of poly(I:C)-treated mice show no such preference. Again, the deficit is corrected by maternal administration of IL-6 antibody (F(3,50) = 4.244; p < 0.01). *p < 0.05 versus control; #p < 0.05 versus poly(I:C).
Heightened anxiety and deficits in social interaction are hallmarks of schizophrenia and autism. Reluctance to enter the center portion of a well-lit open field is usually taken as a measure of heightened anxiety under mildly stressful conditions (Shi et al., 2003). Compared with controls, the offspring of poly(I:C)-injected mice exhibit a deficit in open-field exploration, as measured by entries into the center of the field (p < 0.05) or by total distance traversed (p < 0.01) (Fig. 2c,d). Neutralization of IL-6 prevents these deficits (p < 0.05 and 0.001, respectively). In the social interaction assay, control mice spend more time in the social chamber of the apparatus compared with the nonsocial chamber, whereas offspring of poly(I:C)-injected mice do not show a preference for the social chamber (p < 0.05) (Fig. 2f). Once again, neutralization of IL-6 prevents this deficit (p < 0.05).
Effects of MIA in IL-6 KO mice
We also used genetically altered mice to test the requirement for IL-6 in mediating MIA. The adult offspring of pregnant IL-6 KO mice given injections of poly(I:C) on E12.5 display robust PPI, similar to that found in adult offspring of saline-injected IL-6 KO mice (control, 73.0 ± 5.9%; poly(I:C), 81.2 ± 2.0%; p = 0.15). Moreover, the groups are similarly active in the open-field test (center entries: control, 12.7 ± 1.3; poly(I:C), 10.2 ± 1.9; p = 0.62; distance traveled: control, 3360 ± 201.8 cm; poly(I:C), 3202 ± 243.5 cm; p = 0.30), and both groups display a similar strong preference for the social chamber in the three-chamber social interaction test (control, 43.5 ± 4.6%; poly(I:C), 33.2 ± 6.7%; p = 0.22). In the LI paradigm, NPE offspring of both control- and poly(I:C)-injected females do not display any significant conditioning behavior (freezing; data not shown), making LI testing impossible in IL-6 KO mice. The abnormal conditioning behavior of the IL-6 KO mice suggests that IL-6 is important in brain development and/or function, as indicated by other behavioral studies (Armario et al., 1998; Butterweck et al., 2003; Braida et al., 2004). The key point in the present context is, however, that maternal poly(I:C) treatment has no effect on PPI, social interaction, or open-field behavior of the adult offspring, further demonstrating the importance of IL-6 in causing behavioral deficits in the offspring of MIA mice.
Effect of anti-IL-6 on changes in gene expression evoked by MIA
Another measure of how MIA affects fetal brain development is the alteration in gene expression in the brains of the offspring (Fatemi et al., 2005). Rather than focusing on the identification of individual genes, we used the microarray analysis to assess the overall changes to the transcriptome caused by IL-6 and MIA. mRNA was extracted from frontal cortex, which roughly corresponds to the human prefrontal cortex, an area that shows molecular, functional, and microanatomical alterations in human cognitive disorders (Lewis and Levitt, 2002). Tissue from five adult animals from control, poly(I:C), and poly(I:C) plus anti-IL-6 groups was individually processed and hybridized. Using a cutoff value of p < 0.01, 61 significant changes in gene expression are identified in the offspring of the poly(I:C)-treated mice compared with the saline-treated control animals. Fifty-five of these 61 genes (90%) do not show a statistically significant expression difference when comparing poly(I:C) plus anti-IL-6-treated versus saline-treated animals. Using a two-way (samples and genes) unsupervised hierarchical clustering of the gene expression intensities, the three experimental groups separate into distinct clusters (Fig. 3). Remarkably, four of the five mice in the poly(I:C) plus anti-IL-6 group cluster with the control group, rather than with the poly(I:C) group. A detailed analysis of the microarray results, including confirmation of changes by PCR and a detailed discussion of the genes identified, will be the subject of a future publication. The point of the analysis presented here is the demonstration that maternal anti-IL-6 treatment prevents the gene expression, as well as behavioral, changes caused by poly(I:C) MIA.
Figure 3.
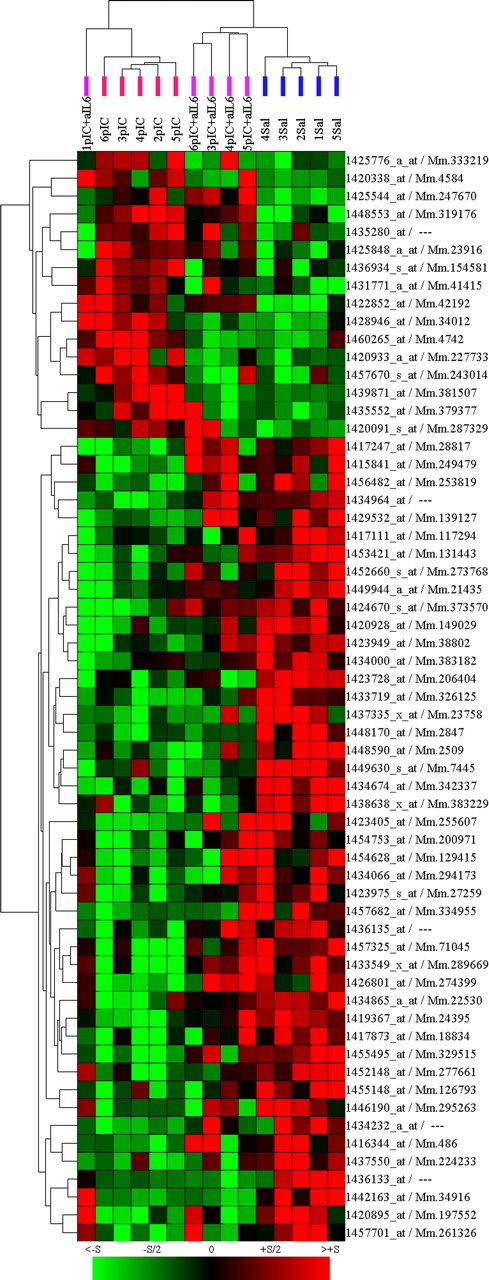
Unsupervised clustering of microarray data show anti-IL-6 treatment rescues transcriptome changes in MIA offspring. Sixty-one genes show significant (p < 0.01) expression differences between the adult offspring of poly(I:C)-treated and saline-injected mice. Two-dimensional, unsupervised clustering of these genes (x-axis genes, y-axis samples) reveals control (Sal; blue bars), poly(I:C) (pIC; red bars), and poly(I:C) plus anti-IL-6 (pIC+aIL6; purple bars) animals cluster according to treatment, with only one pIC+aIL6 outlier. Significantly, the pIC+aIL6 animals cluster with saline-injected controls, rather than with pIC offspring. Each column represents expression values from a single animal. Genes are annotated by Affymetrix probe set/Unigene identifiers. Color intensity represents the magnitude of the gene expression change compared with the overall average intensity (green, decreased; red, increased; black, unchanged).
Discussion
Maternal infection is an environmental risk factor for both schizophrenia and autism (Brown and Susser, 2002; Patterson, 2002). The lack of evidence for direct infection of the fetus (Shi et al., 2005) (but see Aronsson et al., 2002), and the fact that multiple pathogens cause similar results in humans [e.g., influenza (Brown, 2006), herpes (Babulas et al., 2006), rubella (Chess, 1977)], indicates that MIA, in general, is detrimental to the developing brain. The data presented here confirms previous reports (see Introduction) that MIA causes behavioral and gene-expression changes in the offspring of pregnant mice. When IL-6 is eliminated from the maternal immune response using genetic methods or with blocking antibodies, however, the behavioral deficits associated with MIA are not present in the adult offspring. Antibodies to IL-1β or IFNγ did not prevent behavioral deficits, suggesting that the anti-IL-6 effect is specific. Furthermore, maternal exposure to IL-6, in the absence of poly(I:C) or infection, is sufficient to cause two key deficits in the adult offspring, PPI and LI disruption. Thus, IL-6 is central to the process by which MIA causes long-term behavioral alterations in the offspring.
We also show that blocking IL-6 eliminates virtually all of the transcriptional changes caused by MIA. The microarray data presented here are essentially numerical data that estimate the extent to which treatment with the anti-IL-6 antibody is able to normalize changes in expression. Our sole aim in including the numerical data here is to demonstrate that blocking IL-6 prevents >90% of the changes seen in offspring of poly(I:C)-injected females, showing that gene expression changes, as well as behavioral changes, are normalized by eliminating IL-6 from the maternal immune response.
Poly(I:C) signals through Toll-like receptor 3 via a nuclear factor κB-dependent mechanism (Alexopoulou et al., 2001). Although usually cited for its ability to induce interferons (Toth et al., 1990; Katafuchi et al., 2003; Voss et al., 2006), poly(I:C) is also a strong inducer of IL-1, IL-6, and TNF-α (Fortier et al., 2004b; Traynor et al., 2004; Gilmore et al., 2005). Our pilot data suggesting that IL-1α, TNF-α, and IFNγ do not cause behavioral changes in the offspring may be surprising, because these cytokines induce IL-6 in vivo (Gadient and Otten, 1997). However, high levels of maternal IL-6 are necessary for the behavioral changes we observe in the adult offspring, and it may be that these cytokines [as well as lower intraperitoneal doses of IL-6 or poly(I:C)] do not produce levels of IL-6 sufficient to have an effect on the fetus.
Although it is remarkable that a single intraperitoneal injection of IL-6 is capable of altering the fetal brain, leading to abnormal adult behavior, the cytokine stimulation by poly(I:C) is also quite transient. Poly(I:C)-treated mice display sickness behavior (lethargy, hunched posture, hindlimb stiffness), beginning ∼30 min after injection and lasting ∼6 h (data not shown), and a biphasic temperature response consisting of 4–8 h of hyperthermia followed by 12–24 h of hypothermia (Traynor et al., 2004; Cunningham et al., 2007). However, the effects of poly(I:C) treatment are mild compared with experimental influenza infection, in which sickness behavior and hypothermia last for several days (Yang and Evans, 1961; Shi et al., 2003). Previous work with pregnant rats demonstrated that three injections of IL-6 over 5 d caused increased latency to find the platform in the Morris water maze in the adult offspring (Samuelsson et al., 2006). The total amount of IL-6 administered to the rats was similar to the total amount administered in our study, although the rats received the dose spread over 5 d. The offspring of the IL-6-injected rats also exhibited pyknotic cells similar to those reported in the offspring of rats given injections of poly(I:C) (Zuckerman et al., 2003). This pathology is not, however, observed in the offspring of maternal poly(I:C)-treated mice (Meyer et al., 2006) (and our data not shown). Early postnatal administration of epidermal growth factor or leukemia inhibitory factor over several days also cause PPI deficits, tested in adulthood (Futamura et al., 2003; Watanabe et al., 2004), but in none of these previous studies were the roles of endogenous cytokines examined.
Ideally, one would like to extend these cytokine-blocking experiments to the model of maternal infection with influenza virus (Shi et al., 2003), because this model more closely recapitulates the human data related to mental illness. However, when pregnant C57 females are infected with influenza virus and given an anti-IL-6 injection, they become more severely ill than mice given saline injections and either die or suffer miscarriage (data not shown). Similar results were obtained with influenza infection of IL-6 KO mice (data not shown). A previous study on experimental influenza infection in nonpregnant IL-6 KO mice reported changes in weight loss, body temperature, and anorexia but did not report effects on survival (Kozak et al., 1997). That study also only lasted for 5 d, and we do not see major differences until 6–7 d, when wild-type mice begin to recover and KO mice do not. The increased severity of infection in IL-6-compromised mice forced us to use the poly(I:C), pathogen-free MIA model for the IL-6 blocking experiments.
The observation that elimination of IL-6 in the MIA model almost completely abrogates abnormal behaviors and transcriptome changes in the offspring suggests that searches for other mediators of MIA should be directed upstream and downstream of IL-6, rather than in other signaling pathways. IL-6 is a pleiotropic cytokine that signals through heterodimerization of gp130 and IL-6 receptor (IL-6R) on the cell surface (Bauer et al., 2007). However, most cells express gp130, and the soluble form of the IL-6R that is present in blood allows trans signaling (McLoughlin et al., 2005), which enables many cells to respond to IL-6. In considering the location of IL-6 action in the MIA model, three major sites of signaling seem likely: the maternal immune system, the maternal/fetal interface (i.e., the placenta), and the fetal brain.
The most obvious possibility is that IL-6 acts directly on the fetal brain. IL-6 is known to play a role in brain development, learning, and memory and in the CNS response to disease and injury (Bauer et al., 2007). IL-6 is central to inflammation-induced working memory disruption (Sparkman et al., 2006) and plays an important role in long-term potentiation in normal rats (Balschun et al., 2004). During development, the STAT (signal transducers and activators of transcription) pathway, through which IL-6 signals, regulates the balance between neurogenesis and gliogenesis (He et al., 2005) and IL-6 triggers brain endothelial cells to divide and migrate (Yao et al., 2006). After injury, IL-6 can assume very different roles, triggering either neuronal survival or neuronal degeneration, through mechanisms that are not well understood (Gadient and Otten, 1994; Wagner, 1996; Harry et al., 2006). Regarding access to the fetus, radiolabeled IL-6 can enter the rat fetus during mid, but not late, gestation (Dahlgren et al., 2006), which correlates with human data showing influenza infection increases risk for schizophrenia only in the second trimester (Brown, 2006). Elevated levels of cytokine protein and mRNA (including IL-6) have been detected in embryonic serum and brain after MIA (Fidel et al., 1994; Cai et al., 2000; Urakubo et al., 2001; Gayle et al., 2004; Paintlia et al., 2004; Gilmore et al., 2005; Ashdown et al., 2006; Beloosesky et al., 2006; Meyer et al., 2006; Xu et al., 2006). IL-6 can regulate brain-derived neurotrophic factor (BDNF) expression (Murphy et al., 2000), and a decrease in BDNF is found in embryos and placentas 24 h after poly(I:C) administration (Gilmore et al., 2005). Thus, although the pleiotropic nature of IL-6 makes it difficult to predict the precise mechanism of action in the brain, many plausible pathways exist.
A second target of interest is the placenta, because IL-6 could alter the transfer of nutrients, hormones, or other key molecules to the fetus. IL-6 alters vascular permeability in the adult brain after bacterial challenge (Paul et al., 2003), and expression of genes responsible for the integrity of the placental barrier are decreased after MIA (Beall et al., 2005). This could have significant effects on transfer of potentially harmful proteins (i.e., antibodies) into the fetal environment, or could allow maternal immune cells to infiltrate the fetus.
IL-6 could also act on the maternal immune system, activating lymphocyte migration and cytotoxicity and degrading maternal tolerance of the fetus. Normal pregnancy is characterized by a shift in basal cytokine production and other adjustments to prevent rejection of the fetus (Sargent et al., 2006). Severe MIA causes loss of pregnancy in rodents, and depletion of uterine natural killer (uNK) cells prevents this loss, indicating that uNK cells mediate the effect (Arad et al., 2005). IL-6 could also enhance production of maternal antibodies, which could cross-react with the fetal brain, as has been proposed to occur in autism (Warren et al., 1990; Dalton et al., 2003; Vincent et al., 2003; Singer et al., 2006; Zimmerman et al., 2007). Future research on the mechanism of MIA effects on fetal brain development and on potential therapeutic approaches can therefore productively focus on the effects of IL-6 on the maternal–fetal unit.
Footnotes
This work was supported by the National Institute of Mental Health (K.M., P.H.P.) and the McKnight, Cure Autism Now, and Autism Speaks Foundations (P.H.P.). We thank Benjamin Deverman, Natalia Malkova, Limin Shi, Ali Khoshnan, and Lorena Sandoval for assistance and advice; Kathleen Hamilton for administrative support; and Amanda Miles for skillful technical assistance with the microarray experiments. Ben Deverman and Joanna Jankowsky also provided useful comments on this manuscript.
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Arad M, Atzil S, Shakhar K, Adoni A, Ben-Eliyahu S. Poly I-C induces early embryo loss in f344 rats: a potential role for NK cells. Am J Reprod Immunol. 2005;54:49–53. doi: 10.1111/j.1600-0897.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- Armario A, Hernandez J, Bluethmann H, Hidalgo J. IL-6 deficiency leads to increased emotionality in mice: evidence in transgenic mice carrying a null mutation for IL-6. J Neuroimmunol. 1998;92:160–169. doi: 10.1016/s0165-5728(98)00199-4. [DOI] [PubMed] [Google Scholar]
- Aronsson F, Lannebo C, Paucar M, Brask J, Kristensson K, Karlsson H. Persistence of viral RNA in the brain of offspring to mice infected with influenza A/WSN/33 virus during pregnancy. J Neurovirol. 2002;8:353–357. doi: 10.1080/13550280290100480. [DOI] [PubMed] [Google Scholar]
- Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry. 2006;11:47–55. doi: 10.1038/sj.mp.4001748. [DOI] [PubMed] [Google Scholar]
- Babulas V, Factor-Litvak P, Goetz R, Schaefer CA, Brown AS. Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am J Psychiatry. 2006;163:927–929. doi: 10.1176/ajp.2006.163.5.927. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wetzel W, Del Rey A, Pitossi F, Schneider H, Zuschratter W, Besedovsky HO. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi: 10.1096/fj.04-1625fje. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Beall MH, Amidi F, Gayle DA, Wang S, Beloosesky R, Ross MG. Placental and fetal membrane Nephrin and Neph1 gene expression: response to inflammation. J Soc Gynecol Investig. 2005;12:298–302. doi: 10.1016/j.jsgi.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Beloosesky R, Gayle DA, Amidi F, Nunez SE, Babu J, Desai M, Ross MG. N-acetyl-cysteine suppresses amniotic fluid and placenta inflammatory cytokine responses to lipopolysaccharide in rats. Am J Obstet Gynecol. 2006;194:268–273. doi: 10.1016/j.ajog.2005.06.082. [DOI] [PubMed] [Google Scholar]
- Braida D, Sacerdote P, Panerai AE, Bianchi M, Aloisi AM, Iosue S, Sala M. Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav Brain Res. 2004;153:423–429. doi: 10.1016/j.bbr.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Susser ES. In utero infection and adult schizophrenia. Ment Retard Dev Disabil Res Rev. 2002;8:51–57. doi: 10.1002/mrdd.10004. [DOI] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- Butterweck V, Prinz S, Schwaninger M. The role of interleukin-6 in stress-induced hyperthermia and emotional behaviour in mice. Behav Brain Res. 2003;144:49–56. doi: 10.1016/s0166-4328(03)00059-7. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Chess S. Follow-up report on autism in congenital rubella. J Autism Child Schizophr. 1977;7:69–81. doi: 10.1007/BF01531116. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain Behav Immun. 2007;21:490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Dahlgren J, Samuelsson AM, Jansson T, Holmang A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60:147–151. doi: 10.1203/01.pdr.0000230026.74139.18. [DOI] [PubMed] [Google Scholar]
- Dalton P, Deacon R, Blamire A, Pike M, McKinlay I, Stein J, Styles P, Vincent A. Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol. 2003;53:533–537. doi: 10.1002/ana.10557. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse. 2005;57:91–99. doi: 10.1002/syn.20162. [DOI] [PubMed] [Google Scholar]
- Fidel PL, Jr, Romero R, Wolf N, Cutright J, Ramirez M, Araneda H, Cotton DB. Systemic and local cytokine profiles in endotoxin-induced preterm parturition in mice. Am J Obstet Gynecol. 1994;170:1467–1475. doi: 10.1016/s0002-9378(94)70180-6. [DOI] [PubMed] [Google Scholar]
- Fortier ME, Joober R, Luheshi GN, Boksa P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J Psychiatr Res. 2004a;38:335–345. doi: 10.1016/j.jpsychires.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Fortier ME, Kent S, Ashdown H, Poole S, Boksa P, Luheshi GN. The viral mimic, polyinosinic:polycytidylic acid, induces fever in rats via an interleukin-1-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2004b;287:R759–R766. doi: 10.1152/ajpregu.00293.2004. [DOI] [PubMed] [Google Scholar]
- Futamura T, Kakita A, Tohmi M, Sotoyama H, Takahashi H, Nawa H. Neonatal perturbation of neurotrophic signaling results in abnormal sensorimotor gating and social interaction in adults: implication for epidermal growth factor in cognitive development. Mol Psychiatry. 2003;8:19–29. doi: 10.1038/sj.mp.4001138. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten U. Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain Res. 1994;637:10–14. doi: 10.1016/0006-8993(94)91211-4. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten UH. Interleukin-6 (IL-6)—a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–390. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- Gayle DA, Beloosesky R, Desai M, Amidi F, Nunez SE, Ross MG. Maternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brain. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1024–R1029. doi: 10.1152/ajpregu.00664.2003. [DOI] [PubMed] [Google Scholar]
- Gilmore JH, Jarskog LF, Vadlamudi S. Maternal poly I:C exposure during pregnancy regulates TNF alpha, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J Neuroimmunol. 2005;159:106–112. doi: 10.1016/j.jneuroim.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Lawler C, Brunssen SH. Maternal infection and white matter toxicity. Neurotoxicology. 2006;27:658–670. doi: 10.1016/j.neuro.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, de Vellis J, Sun YE. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003a;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003b;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Katafuchi T, Kondo T, Yasaka T, Kubo K, Take S, Yoshimura M. Prolonged effects of polyriboinosinic:polyribocytidylic acid on spontaneous running wheel activity and brain interferon-alpha mRNA in rats: a model for immunologically induced fatigue. Neuroscience. 2003;120:837–845. doi: 10.1016/s0306-4522(03)00365-8. [DOI] [PubMed] [Google Scholar]
- Kozak W, Poli V, Soszynski D, Conn CA, Leon LR, Kluger MJ. Sickness behavior in mice deficient in interleukin-6 during turpentine abscess and influenza pneumonitis. Am J Physiol. 1997;272:R621–R630. doi: 10.1152/ajpregu.1997.272.2.R621. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, Ernst M, Topley N, Jones SA. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci USA. 2005;102:9589–9594. doi: 10.1073/pnas.0501794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medesan C, Cianga P, Mummert M, Stanescu D, Ghetie V, Ward ES. Comparative studies of rat IgG to further delineate the Fc:FcRn interaction site. Eur J Immunol. 1998;28:2092–2100. doi: 10.1002/(SICI)1521-4141(199807)28:07<2092::AID-IMMU2092>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PG, Borthwick LA, Altares M, Gauldie J, Kaplan D, Richardson PM. Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons. Eur J Neurosci. 2000;12:1891–1899. doi: 10.1046/j.1460-9568.2000.00074.x. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh AK. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Curr Opin Neurobiol. 2002;12:115–118. doi: 10.1016/s0959-4388(02)00299-4. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal influenza infection leads to neuropathology and behavioral abnormalities in adult offspring. Neuropsychopharmacology. 2005;30:S9–S9. [Google Scholar]
- Paul R, Koedel U, Winkler F, Kieseier BC, Fontana A, Kopf M, Hartung HP, Pfister HW. Lack of IL-6 augments inflammatory response but decreases vascular permeability in bacterial meningitis. Brain. 2003;126:1873–1882. doi: 10.1093/brain/awg171. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2006;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Samuelsson AM, Jennische E, Hansson HA, Holmang A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1345–R1356. doi: 10.1152/ajpregu.00268.2005. [DOI] [PubMed] [Google Scholar]
- Sankoorikal GM, Kaercher KA, Boon CJ, Lee JK, Brodkin ES. A mouse model system for genetic analysis of sociability: C57BL/6J versus BALB/cJ inbred mouse strains. Biol Psychiatry. 2006;59:415–423. doi: 10.1016/j.biopsych.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Sargent IL, Borzychowski AM, Redman CW. NK cells and human pregnancy–an inflammatory view. Trends Immunol. 2006;27:399–404. doi: 10.1016/j.it.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int J Dev Neurosci. 2005;23:299–305. doi: 10.1016/j.ijdevneu.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Singer HS, Morris CM, Williams PN, Yoon DY, Hong JJ, Zimmerman AW. Antibrain antibodies in children with autism and their unaffected siblings. J Neuroimmunol. 2006;178:149–155. doi: 10.1016/j.jneuroim.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci. 2006;26:10709–10716. doi: 10.1523/JNEUROSCI.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiergiel AH, Smagin GN, Dunn AJ. Influenza virus infection of mice induces anorexia: comparison with endotoxin and interleukin-1 and the effects of indomethacin. Pharmacol Biochem Behav. 1997;57:389–396. doi: 10.1016/s0091-3057(96)00335-8. [DOI] [PubMed] [Google Scholar]
- Tochigi M, Okazaki Y, Kato N, Sasaki T. What causes seasonality of birth in schizophrenia? Neurosci Res. 2004;48:1–11. doi: 10.1016/j.neures.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Toth FD, Juhl C, Norskov-Lauritsen N, Mosborg Petersen P, Ebbesen P. Interferon production by cultured human trophoblast induced with double stranded polyribonucleotide. J Reprod Immunol. 1990;17:217–227. doi: 10.1016/0165-0378(90)90004-p. [DOI] [PubMed] [Google Scholar]
- Traynor TR, Majde JA, Bohnet SG, Krueger JM. Intratracheal double-stranded RNA plus interferon-gamma: a model for analysis of the acute phase response to respiratory viral infections. Life Sci. 2004;74:2563–2576. doi: 10.1016/j.lfs.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res. 2001;47:27–36. doi: 10.1016/s0920-9964(00)00032-3. [DOI] [PubMed] [Google Scholar]
- Vincent A, Dalton P, Clover L, Palace J, Lang B. Antibodies to neuronal targets in neurological and psychiatric diseases. Ann NY Acad Sci. 2003;992:48–55. doi: 10.1111/j.1749-6632.2003.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Voss T, Rummel C, Gerstberger R, Hubschle T, Roth J. Fever and circulating cytokines induced by double-stranded RNA in guinea pigs: dependence on the route of administration and effects of repeated injections. Acta Physiol (Oxf) 2006;187:379–389. doi: 10.1111/j.1748-1716.2006.01587.x. [DOI] [PubMed] [Google Scholar]
- Wagner JA. Is IL-6 both a cytokine and a neurotrophic factor? J Exp Med. 1996;183:2417–2419. doi: 10.1084/jem.183.6.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RP, Cole P, Odell JD, Pingree CB, Warren WL, White E, Yonk J, Singh VK. Detection of maternal antibodies in infantile autism. J Am Acad Child Adolesc Psychiatry. 1990;29:873–877. doi: 10.1097/00004583-199011000-00005. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Hashimoto S, Kakita A, Takahashi H, Ko J, Mizuno M, Someya T, Patterson PH, Nawa H. Neonatal impact of leukemia inhibitory factor on neurobehavioral development in rats. Neurosci Res. 2004;48:345–353. doi: 10.1016/j.neures.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Weiner I. The “two-headed” latent inhibition model of schizophrenia: modeling positive and negative symptoms and their treatment. Psychopharmacology (Berl) 2003;169:257–297. doi: 10.1007/s00213-002-1313-x. [DOI] [PubMed] [Google Scholar]
- Wynn JK, Dawson ME, Schell AM, McGee M, Salveson D, Green MF. Prepulse facilitation and prepulse inhibition in schizophrenia patients and their unaffected siblings. Biol Psychiatry. 2004;55:518–523. doi: 10.1016/j.biopsych.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Xu DX, Chen YH, Wang H, Zhao L, Wang JP, Wei W. Tumor necrosis factor alpha partially contributes to lipopolysaccharide-induced intra-uterine fetal growth restriction and skeletal development retardation in mice. Toxicol Lett. 2006;163:20–29. doi: 10.1016/j.toxlet.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Yang YT, Evans CA. Hypothermia in mice due to influenza virus infection. Proc Soc Exp Biol Med. 1961;108:776–780. doi: 10.3181/00379727-108-27064. [DOI] [PubMed] [Google Scholar]
- Yao JS, Zhai W, Young WL, Yang GY. Interleukin-6 triggers human cerebral endothelial cells proliferation and migration: the role for KDR and MMP-9. Biochem Biophys Res Commun. 2006;342:1396–1404. doi: 10.1016/j.bbrc.2006.02.100. [DOI] [PubMed] [Google Scholar]
- Zimmerman AW, Connors SL, Matteson KJ, Lee LC, Singer HS, Castaneda JA, Pearce DA. Maternal antibrain antibodies in autism. Brain Behav Immun. 2007;21:351–357. doi: 10.1016/j.bbi.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]