

Abstract
CD4+ T cells directly participate in bacterial clearance through secretion of proinflammatory cytokines. Although viral clearance relies heavily on CD8+ T cell functions, we sought to determine whether human CD4+ T cells could also directly influence viral clearance through cytokine secretion. We found that IFN-γ and TNF-α, secreted by IL-12-polarized Th1 cells, displayed potent antiviral effects against a variety of viruses. IFN-γ and TNF-α acted directly to inhibit HCV replication in an in vitro replicon system, and neutralization of both cytokines was required to block the antiviral activity that was secreted by Th1 cells. IFN-γ and TNF-α also exerted antiviral effects against VSV infection, but in this case, functional type I interferon receptor activity was required. Thus, in cases of VSV infection, the combination of IFN-γ and TNF-α secreted by human Th1 cells acted indirectly through the IFN-α/β receptor. These results highlight the importance of CD4+ T cells in directly regulating antiviral responses through proinflammatory cytokines acting in both a direct and indirect manner.
Keywords: Human, T cell, Viral, Cytokines
Introduction
Adaptive immune responses play a critical role in the clearance of infectious diseases and in providing long-term resistance against re-infection. CD4+ and CD8+ T cells orchestrate inflammatory processes through both cytolytic and cytokine-mediated effector mechanisms. In response to bacterial infections, CD4+ Th1 cells promote the recruitment and activation of phagocytic cells, such as macrophages and neutrophils, into sites of infection through the secretion of the chemokines CXCL8/IL-8 and CCL3/MIP-1α and the cytokines interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α). IFN-γ and TNF-α act in concert to promote the production of reactive oxygen and nitrogen radicals from phagocytic cells, which effectively sterilizes the site of infection (1–3). Thus, CD4+ T cells participate directly in antibacterial immunity through the secretion of proinflammatory cytokines. In contrast, viral infections are considered to rely predominantly on CD8+ T cell responses (4). CD4+ T cells clearly play a supporting role during viral infections through cytokine secretion and by providing critical help for B cell antibody production (5–12). Additionally, there are some reports detailing a population of CD4+ cytotoxic T cells which can directly lyse infected targets by cell-cell contact (7, 13, 14). However, the ability of CD4+ T cells to directly inhibit viral replication and spread has not been thoroughly examined.
Viral infections initiate a cascade of innate and adaptive immune responses that are collectively regulated by cytokines. Type I interferon (IFN-α/β) is one of the first cytokines secreted by virally infected cells and from professional antigen presenting cells through the activation of various pattern recognition receptors such as Toll-like receptors and RIG-I (15). IFN-α/β exerts potent antiviral activities directly on infected cells by inducing the expression of interferon sensitive genes (ISGs), thereby inhibiting virus replication and spread (16). In addition to these innate activities, IFN-α/β also enhances effector functions of natural killer (NK) cells and CD8+ cytolytic T lymphocytes (CTL) (17–20). However, the role of IFN-α/β in regulating CD4+ effector functions has been controversial. Early reports suggested that IFN-α/β could promote Th1 development through activation of STAT4 in an IL-12-independent manner (21–28). However, recent studies have demonstrated that IFN-α/β completely lacks the ability to drive Th1 development in human CD4+ T cells because unlike IL-12, IFN-α/β does not induce the expression of the Th1-specific transcription factor T-bet (29–31). However, in these studies, IFN-α/β did not inhibit the ability of IL-12 to promote Th1 development as assessed only by IFN-γ secretion. As both IL-12 and IFN-α/β are secreted to high levels by dendritic cells in response to viral infections (32), it is possible that IFN-α/β synergizes with IL-12 to regulate other potential CD4+ effector cytokines that may play important roles in inhibiting viral infections.
In addition to IFN-α/β, several other proinflammatory cytokines have been shown to exert antiviral activity. For example, IFN-γ shares various antiviral activities with IFN-α/β, such as upregulation of class I MHC, inhibition of viral replication, and the induction of an overlapping set of ISGs (16). In addition, TNF-α and TNF-β (lymphotoxin) have also been shown to inhibit viral replication directly as well as indirectly through the induction of IFN-β within infected cells (33–39). CD4+ Th1 cells represent a significant source of IFN-γ and TNF-α, and it is possible that CD4+ T cells play a much more central role in the course of viral infections than has previously been attributed to this subset. Indeed, studies in mice have demonstrated a CD4+-dependent component to clearance of Sendai virus, influenza A virus, and γ-herpesvirus (40–44). In cases of γ-herpesvirus infections, CD4+ T cells were shown to inhibit reactivation from latency, and neutralization of IFN-γ could inhibit this activity. However, administration of IFN-γ was not sufficient to maintain latency, particularly within infected B cells (44, 45). Based on these observations, it is likely that CD4+ T cells play a significant role in the inhibition of viral replication through the action of a complex mixture of cytokines, the nature of which has not been investigated.
We therefore sought to answer two distinct questions. First, how do innate cytokines present during viral infections shape effector CD4+ T cell responses? Second, can cytokines secreted by effector CD4+ T cells directly impact viral infections? We found that IL-12 is primarily responsible for the generation of antiviral CD4+ T cell effector cytokine responses. IL-12 drives the secretion of IFN-γ and TNF-α, which induce potent antiviral responses against a number of viruses. Further, we found that this antiviral effect on VSV infection requires IFN-α/β receptor (IFNAR) expression on the target cell, indicating the presence of a novel cytokine relay network.
Materials and Methods
Human subjects
100–120 ml of peripheral blood was obtained from healthy adult volunteers by venipuncture. Informed consent was obtained from each donor, and all procedures related to this study were approved by the institutional Internal Review Board (University of Texas Southwestern Medical Center).
Cell lines
THP-1 cells, a human monocytic lymphoma line, CV-1 cells, a green monkey fibroblast line, and HeLa cells, a human cervical carcinoma line, were purchased from American Type Culture Collection (Manassas, VA). 2fTGH cells, a human fibroblast line, and 2fTGH-derived IFNAR2-deficient U5A cells were a generous gift of G. Stark (Cleveland Clinic) (46–48). A7 replicon cells, a human hepatoma line carrying a replicating hepatitis C virus genome, have been previously described (49).
Cytokines, antibodies, and reagents
Recombinant human IL-4 (rhIL-4), rhIL-12, rhIFN-γ, rhTNF-α, and rhLTα1β2, and the anti-human IL-4, anti-human IFN-γ receptor (IFNγR1), and anti-human LT antibodies were purchased from R&D Systems (Minneapolis, MN). rhIFN-αA and rhIFN-ω, the anti-human IFN-α/β receptor (IFNAR2) and anti-human IFN-ω antibodies, and polyclonal antisera against human IFN-α and IFN-β were purchased from PBL Laboratories (Piscataway, NJ). rhIFN-β1a was a generous gift of M. Racke (University of Ohio). rhIL-2 was a generous gift of M. Bennett (University of Texas Southwestern Medical Center). The anti-human CD3, anti-human CD28, anti-human TNF-α, and allophycocyanin (APC)-conjugated anti-human TNF-α antibodies were purchased from BioLegend (San Diego, CA). The phycoerythrin (PE)-conjugated anti-human CD4 and fluorescein isothiocyanate (FITC)-conjugated anti-human IFN-γ antibodies were purchased from Caltag Laboratories (Burlingame, CA). The FITC-conjugated anti-human CD45RA and PE-Cy7-conjugated anti-human IFN-γ antibodies were purchased from BD Pharmingen (San Diego, CA). The anti-NS5A antibody was a generous gift of J. Ye (University of Texas Southwestern Medical Center). The anti-human ISG56 antibody was a generous gift of G. Sen (Cleveland Clinic) (50). The anti-human GAPDH antibody was purchased from Abcam (Cambridge, MA).
Preparation of human T cells and T cell conditioned media
Naïve human CD4+ T cells were isolated from whole blood of healthy adult volunteers as previously described (31). Briefly, heparinized whole blood was subjected to density centrifugation using Lymphocyte Separation Media (Mediatech, Inc., Herndon, VA). Peripheral blood mononuclear cells isolated from buffy coats were stained with FITC-conjugated anti-human CD45RA and PE-conjugated anti-human CD4 antibodies, and CD45RA+ CD4+ cells were sorted on a MoFlo cell sorter (Dako Cytomation, Fort Collins, CO). Cells were activated at 2–2.5 × 106 cells/ml for three days in complete Iscove’s Modified Dubelcco’s Medium (Hyclone, Logan, UT) supplemented with 10% fetal bovine serum (Valley Biomedical, Inc., Winchester, VA) (cIMDM) on culture plates coated with 5 μg/ml anti-human CD3 + 5 μg/ml anti-human CD28 in the presence of 50 units/ml rhIL-2. Cytokines and neutralizing antibodies were added as indicated in the figures at the following concentrations: anti-human IFN-γ (4S.B3), 5 μg/ml; anti-human IL-4, 2 μg/ml; anti-human IL-12 (20C2), 5 μg/ml; anti-human IFNAR2; 2 μg/ml; rhIL-12, 10 ng/ml; rhIL-4, 10 ng/ml; rhIFN-αA, 1000 units/ml. On day three, cells were split into fresh media containing IL-2 and were rested to day 7. On day 7, cells were washed in fresh cIMDM and left unstimulated or restimulated for 24 hours on culture plates coated with 5 μg/ml anti-CD3. Conditioned media from these cells was harvested and assayed for antiviral activity by in vitro infection.
Vesicular stomatitis virus infections
Cells were washed and resuspended in cIMDM at 6 × 106 cells/ml (THP-1 cells) or 3 × 106 cells/ml (2fTGH and U5A cells). Cells were infected with recombinant vesicular stomatitis virus carrying the GFP transgene (VSV-GFP) (generous gift of M. Whitt, University of Tennessee) (51) at 0.05–0.8 plaque-forming units (pfu)/cell for 2 minutes at room temperature. Cells were then transferred into wells of a 96-well plate containing cytokines or T cell conditioned media and incubated for 16 hours at 37°C, 5% CO2. Following infection, cells were washed and fixed, and analysis for GFP expression was performed on a FACScan or FACSCalibur cytometer (Becton Dickinson, Franklin Lakes, NJ), and the data was processed using FlowJo software (TreeStar, Ashland, OR). For experiments in which anti-human IFNAR2 or anti-human IFNγR1 neutralizing antibodies were used, cells were incubated with 5 μg/ml anti-human IFNAR2 or 10 μg/ml anti-human IFNγR1 for 2 minutes at room temperature immediately prior to infection.
Quantitation of VSV-GFP infection by plaque assay
THP-1 cells were cultured for 24 hours at 37°C, 5% CO2 in cIMDM in the absence or presence of 100 units/ml rhIFN-αA or T cell conditioned media (10% v/v). Cells were washed and resuspended at 6 × 106 cells/ml in cIMDM. Cells were infected with VSV-GFP at 0.7 pfu/cell for 15 minutes at room temperature. Cells were then washed in cIMDM, transferred to wells of a 96-well plate, and incubated for 24 hours at 37°C, 5% CO2. Confluent CV-1 cells were infected with supernatants from infected THP-1 cells at dilutions from 101–107 for 45 minutes at 37°C, 5% CO2. CV-1 cells were then washed and overlaid with cDMEM containing 0.6% agarose and cultured for 24–72 hours at 37°C, 5% CO2. Cells were stained with crystal violet for quantitation of plaque formation.
A7 replicon cells and Western blotting
The generation and maintenance of the A7 replicon cell line has been previously described (49). A7 replicon cells were maintained in Dubelcco’s modified Eagle medium (Mediatech, Inc.) supplemented with 10% FBS (cDMEM) and 200 μg/ml G418 (Gemini Bio-products, Sacramento, CA). 24 hours prior to treatment, cells were washed with PBS and given cDMEM without antibiotic. The following day, media was removed, and cells were cultured in cDMEM containing cytokines or 5% (v/v) T cell conditioned media as indicated in the figures. Concentrations of cytokines were as follows: rhIFN-αA, 100 U/ml; rhIFN-γ, 5 ng/ml; rhTNF-α, 2.5 ng/ml. 48 hours later, cells were harvested and lysed in RIPA buffer, and proteins were separated by SDS-PAGE. Western blotting was performed using antibodies against HCV NS5A, human ISG56, or human GAPDH. For experiments in which neutralizing antibodies were used, cells were incubated with 5 μg/ml anti-hIFNAR2, 10 μg/ml anti-hIFNγR1, or 5 μg/ml anti-hTNF-α for 1 hour immediately prior to treatment with cytokine or T cell conditioned media and supplemented with the same antibodies 24 hours after the initiation of treatment.
Respiratory syncytial virus infections
HeLa cells were washed and resuspended in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (cDMEM) at 10 × 106 cells/ml. Cells were infected with recombinant respiratory syncytial virus carrying the GFP transgene (RSV-GFP) (generous gift of M. Peeples, Columbus Children’s Research Institute) (52) at 2–2.5 pfu/cell for 2 minutes at room temperature. Cells were then transferred into wells of a 96-well plate containing cytokines or T cell conditioned media and incubated for 72 hours at 37°C, 5% CO2. Following infection, cells were washed and fixed, and analysis for GFP expression was performed on a FACScan or FACSCalibur cytometer, and the data was processed using FlowJo software.
Intracellular staining for IFN-γ and TNF-α
Naïve human CD4+ T cells were differentiated for one or two consecutive weeks as described above. On day 7 or day 14, cells were washed and rested overnight in cIMDM. Cells were left unstimulated or were stimulated with 0.8 μg/ml phorbol 12-myristate 13-acetate (PMA) (A.G. Scientific, Inc., San Diego, CA) + 1 μM ionomycin (Sigma-Aldrich, St. Louis, MO) for 4 hours at 37°C, 5% CO2 in the absence or presence of 1 μg/ml Brefeldin A (Epicentre, Madison, WI). Intracellular staining was performed as previously described (53) using an APC-conjugated anti-human TNF-α antibody and either a FITC-conjugated or PE-Cy7-conjugated anti-human IFN-γ antibody. Cells were analyzed on a FACSCalibur or LSR II cytometer (Becton Dickinson), and the data was processed using FlowJo software.
Enzyme-linked immunosorbent assay (ELISA) for IFN-γ and TNF-α
Concentrations of human IFN-γ and TNF-α in T cell conditioned media were determined by ELISA using ELISA MAX kits (BioLegend) according to the manufacturer’s instructions.
Listeria innocua infections
THP-1 cells were washed and resuspended in antibiotic-free cIMDM at 2 × 106 cells/ml. Cells were activated with 0.8 μg/ml PMA for 48 hours at 37°C, 5% CO2 (54). Cells were washed, and cytokines or T cell conditioned media were then added for a further 48 hours at the concentrations indicated in the figures. Concentrations of cytokines were as follows: rhIFN-αA, 100 U/ml; rhIFN-γ, 10 ng/ml; rhTNF-α, 10 ng/ml. Cells were washed and infected with 3 colony-forming units (CFU)/cell Listeria innocua (generous gift of L. Hooper, University of Texas Southwestern Medical Center) for 45 minutes at 37°C, 5% CO2. Gentamicin (Sigma-Aldrich) was added at 50–100 μg/ml, and cytokines or T cell conditioned media were added at the concentrations indicated in the figures. Cells were incubated for 16 hours at 37°C, 5% CO2. Infected cells were lysed to release intracellular bacteria, and infection was assessed by plating on BHI agar plates.
Statistical analysis
Significance analysis was performed in Prism software (GraphPad Software, Inc., San Diego, CA) by one-way or two-way analysis of variance (ANOVA). Comparisons were considered significant at >95% confidence interval (p = <0.05).
Results
Human CD4+ T cells secrete an antiviral activity
Th1 cells are known to play a direct role in clearance of bacterial infections by secretion of IFN-γ. Since Th cells are known to secrete a variety of soluble mediators, we hypothesized that these cells may also play a role in viral pclearance by direct cytokine signaling to infected cells. To test this hypothesis, we established an in vitro infection model whereby THP-1 cells, a human monocyte line, were infected with vesicular stomatitis virus carrying a transgene for green fluorescent protein (VSV-GFP). The percentage of infected cells was monitored by flow cytometry (Fig. 1, A and B), whereas the relative secretion of live virus was quantified by plaque assay (Fig. 1E). With this model, we confirmed that VSV-GFP infection was blocked by treatment of infected cells with type I interferon (Fig. 1, A and B), and this effect was reversed by blocking the human type I interferon receptor (IFNAR) by a neutralizing antibody against the IFNAR2 subunit (Fig. 1A). IFN-α significantly reduced the percentage of infected cells, which correlated well with a significant decrease in secretion of live virus (Fig. 1. B and E).
FIGURE 1.

Soluble factors secreted by human CD4+ T cells inhibit VSV infection. A, THP-1 cells were infected for 16 hours with VSV-GFP. GFP expression was analyzed by flow cytometry. Representative FACS plots showing THP-1 cells left uninfected or infected with VSV-GFP in the presence or absence of 100 U/ml rhIFN-αA and/or a neutralizing anti-hIFNAR2 antibody. B, THP-1 cells were left uninfected (square) or were infected for 16 hours with VSV-GFP in the absence (triangle) or presence (circles) of increasing concentrations of rhIFN-αA. GFP expression was analyzed by flow cytometry. C, THP-1 cells were left uninfected (■) or were infected for 16 hours with VSV-GFP. Cells were treated at the time of infection with either media alone (▲) or with increasing doses of conditioned media from T cells polarized with the following conditions: Neutralized (anti-IFN-γ, anti-IL-4, anti-IL-12, and anti-IFNAR2), □; IL-12, (rhIL-12, anti-IFN-γ, anti-IL-4, and anti-IFNAR2) ;▵ IFN-αA, rhIFN-αA, anti-IFN-γ, anti-IL-4, and anti-IL-12) ∇; or IL-12 + IFN-αA, (rhIL-12 and rhIFN-αA in the presence of anti-IFN-γ and anti-IL-4) ○. GFP expression was analyzed by flow cytometry. *, p < 0.05 versus neutralized. D, THP-1 cells were infected for 16 hours with VSV-GFP in the absence or presence of T cell conditioned media from T cells which were left unstimulated (black bars) or were restimulated on day 7 for 24 hours with plate-bound anti-CD3 (hatched bars). GFP expression was analyzed by flow cytometry. E, THP-1 cells were cultured for 24 hours in the absence or presence of 100 U/ml rhIFN-αA or T cell conditioned media derived from cells polarized as indicated in the figure. Cells were then washed and then infected for an additional 24 hours with VSV-GFP, and viral replication was quantified by plaque assay.
We next examined the effect of CD4+ T cell-derived effector cytokines on VSV-GFP infection of THP-1 cells. In order to isolate the individual contributions of innate cues to the generation of antiviral effector responses, naïve (CD45RA+) human CD4+ T cells were differentiated with plate-bound anti-CD3 and anti-CD28 in the presence of cytokines or neutralizing antibodies for 7–14 days. These cells were then washed extensively in clean media and restimulated for 24 hours with plate-bound anti-CD3, and the conditioned media from these cells was harvested and used to treat VSV-GFP-infected THP-1 cells. Treatment of THP-1 cells with T cell conditioned media at the time of infection inhibited VSV-GFP infection as measured by GFP expression, and this effect was dose-dependent (Fig. 1C). Furthermore, conditioned media from resting CD4+ T cells did not inhibit VSV-GFP infection, indicating that the secretion of antiviral activity required secondary T cell activation (Fig. 1D, p < 0.05, anti-CD3-restimulated versus unstimulated, all conditions). Pre-treatment of THP-1 cells with T cell conditioned media for 24 hours prior to infection significantly inhibited VSV-GFP virus production from these cells as measured by plaque assay (Fig. 1E). We also noted that T cell conditioned media generated from T cells differentiated in the presence of IL-12 or a combination of IL-12 and IFN-α was consistently more effective at reducing VSV-GFP infection in the THP-1 cells than T cell conditioned media generated pfrom T cells differentiated in the presence of neutralizing antibodies or IFN-α alone (Fig. 1C, p < 0.05 versus neutralized). Conversely, T cell conditioned pmedia generated from T cells differentiated in the presence of IL-4 had no effect on VSV-GFP infection (Fig. 1E). Occasionally, we observed a slight difference in antiviral activity generated from Th cells differentiated in the presence of IL-12 alone versus IL-12 with IFN-α (Fig. 1C); however, this difference was not present in most experiments and was likely a result of donor variation.
In order to determine whether the activity secreted by human CD4+ T cells represented a general antiviral mechanism, we examined the ability of T cell conditioned media to inhibit infection with two other viruses: respiratory syncytial virus (RSV) and hepatitis C virus (HCV). HeLa cells were infected for 72 hours with RSV carrying a GFP transgene (RSV-GFP). Treatment of HeLa cells with T cell conditioned media at the time of infection significantly reduced RSV-GFP infection (Fig. 2A). In agreement with our previous results, conditioned media generated from T cells activated in the presence of IL-12, alone or in combination with IFN-α, contained greater antiviral activity against RSV-GFP than conditioned media from T cells activated under either neutralizing or IFN-α conditions (p < 0.05 versus neutralized).
FIGURE 2.
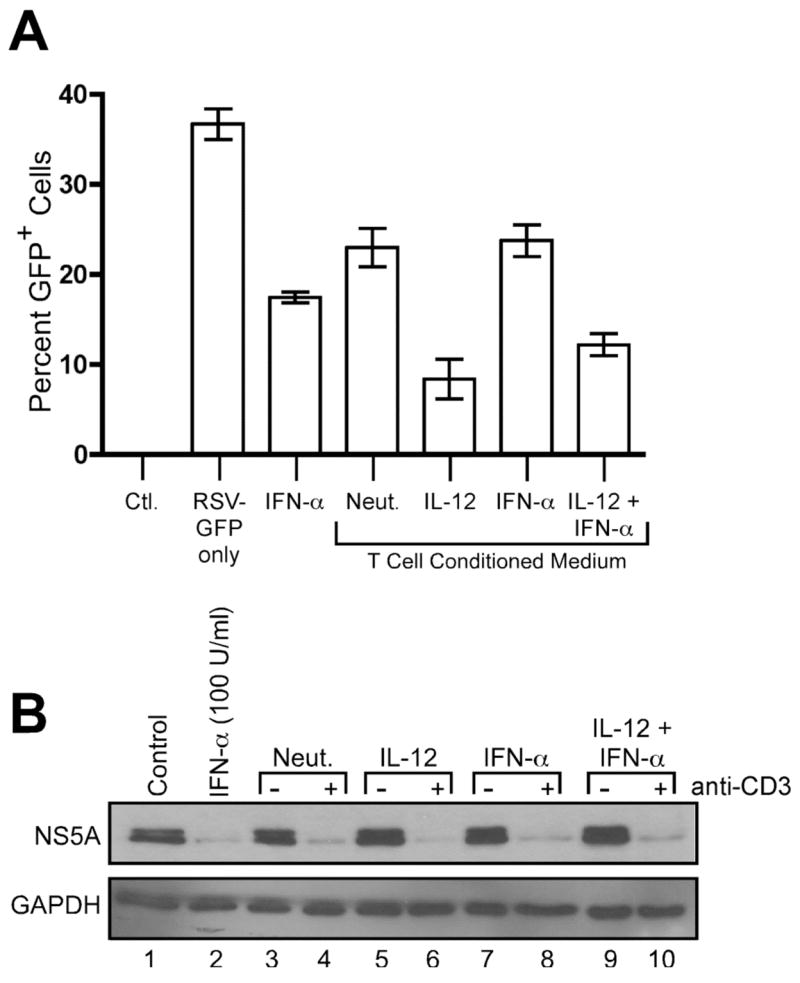
Inhibition of RSV and HCV replication factors secreted by human CD4+ T cells. A, HeLa cells were infected for 72 hours with RSV-GFP in the absence or presence of 100 U/ml rhIFN-αA or 10% (v/v) T cell conditioned media as indicated. GFP expression was analyzed by flow cytometry. Data are expressed as mean +/− SEM of three replicates. B, A7 replicon cells were incubated for 24 hours in the absence (lane 1) or presence of 100 U/ml rhIFN-αA (lane 2) or 5% (v/v) T cell conditioned media (lanes 3–10) as indicated. Cell lysates were prepared, and Western blotting was performed using antibodies against HCV NS5A and human GAPDH.
Inhibition of hepatitis C virus (HCV) infection by T cell conditioned media was examined using the A7 HCV replicon cell line. These cells carry a full-length, replicating HCV genome and express HCV proteins (49). Addition of 5% (v/v) T cell conditioned media to these cells reduced HCV NS5A protein synthesis, and this antiviral activity also required restimulation of the T cells by anti-CD3 crosslinking (Fig. 2B). At these concentrations, we did not observe obvious differences between conditioned media from different T cell conditions, which suggested that NS5A expression was particularly sensitive to very low levels of antiviral factors secreted by human CD4+ T cells. Taken together, these results demonstrate for the first time that effector cytokines secreted by human CD4+ T cells can directly inhibit viral infection in target cells.
IFN-γ and TNF-α secreted by Th cells demonstrate antiviral activity
The greatest antiviral activity against VSV and RSV was observed in conditioned media generated from T cells differentiated in the presence of IL-12, suggesting that the secreted factor was a Th1 cytokine. In accordance with previously published work (31), we verified that secretion of IFN-γ and TNF-α from human Th cells depended on IL-12, and IFN-α/β neither enhanced nor inhibited this effect (Fig. 3). Here, priming with IL-12 significantly enhanced the percentage of IFN-γ secreting cells (Fig. 3A), and the IFN-γ secreting cells were found to also secrete TNF-α (Fig. 3, A and B). Further, approximately 90% of cells were found to secrete TNF-α regardless of whether the cells were primed with neutralizing conditions or with IL-12 (Fig. 3, A and B). However, cells differentiated in the presence of IL-12 produced significantly higher concentrations of IFN-γ (16–18 fold) and TNF-α (3–5 fold) as compared to cells polarized under neutralizing conditions (Fig. 3, C and D).
FIGURE 3.
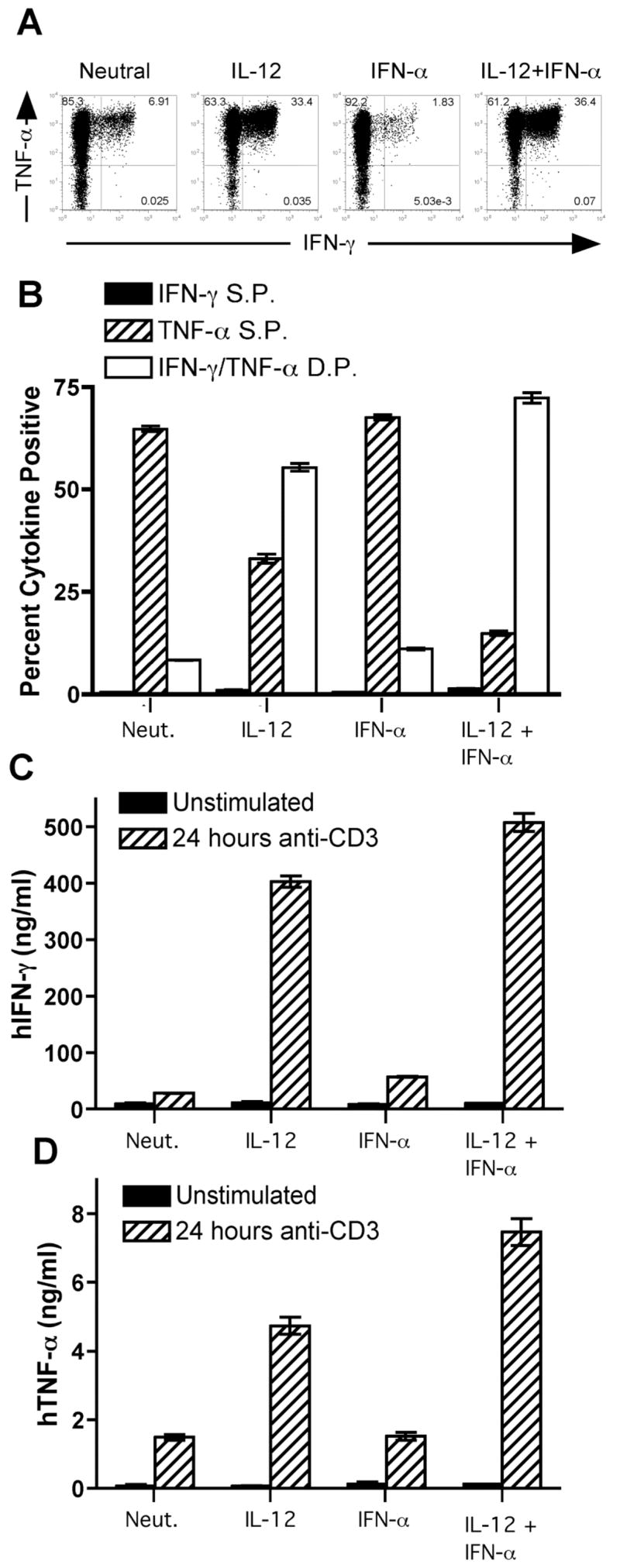
IL-12 promotes IFN-γ and TNF-α secretion by human CD4+ T cells. (A – D) Naïve (CD45RA+ CD4+) T cells were sorted from peripheral blood of healthy donors and activated for 7 (A, C – D) or 14 (B) days with plate-bound anti-CD3 and anti-CD28 in the presence of cytokines and neutralizing antibodies as indicated. (A – B) Cells were restimulated for 4 hours with PMA and ionomycin, and flow cytometry was performed using antibodies against human IFN-γ and human TNF-α. Black bars, percent IFN-γ single positive cells; hatched bars, percent TNF-α single positive cells; open bars, percent IFN-γ/TNF-α double positive cells. Data are expressed as mean +/− SEM of three replicates. C and D, Cells were washed and left unstimulated (open bars) or restimulated for 24 hours with plate-bound anti-CD3 (black bars). Supernatants were harvested, and ELISAs were performed for human IFN-γ (C) or human TNF-α (D). Data are expressed as mean +/− SEM of three replicates.
IFN-γ and TNF-α are proinflammatory cytokines that markedly inhibit intracellular bacterial infections. These cytokines act in concert to promote the oxidative burst within phagocytic cells. In order to confirm that the T cell conditioned media contained functionally relevant levels of these proinflammatory cytokines, we tested these supernatants for their ability to control Listeria infection within the THP-1 monocyte cell line. THP-1 cells were differentiated to a macrophage state with PMA and cultured in the presence or absence of recombinant cytokines (Fig. 4A) or T cell conditioned media (Fig. 4B) for 48 hours. The cells were subsequently infected with L. innocua, again in the presence or absence of recombinant cytokines or T cell conditioned media. As expected, bacterial replication was markedly inhibited by combined treatment with recombinant IFN-γ and TNF-α (Fig. 4A). Recombinant IFN-α also inhibited L. innocua infection (Fig. 4A), as has been previously reported (55). Additionally, treatment with T cell conditioned media significantly inhibited L. innocua infection, and conditioned media from cells differentiated in the presence of IL-12 displayed the greatest antibacterial activity in this assay (Fig. 4B). Thus, T cell conditioned media contain relevant levels of IFN-γ and TNF-α sufficient to inhibit intracellular bacterial replication. These data further indicate that the THP-1 monocyte cell line is sensitive to both IFN-γ and TNF-α signaling.
FIGURE 4.
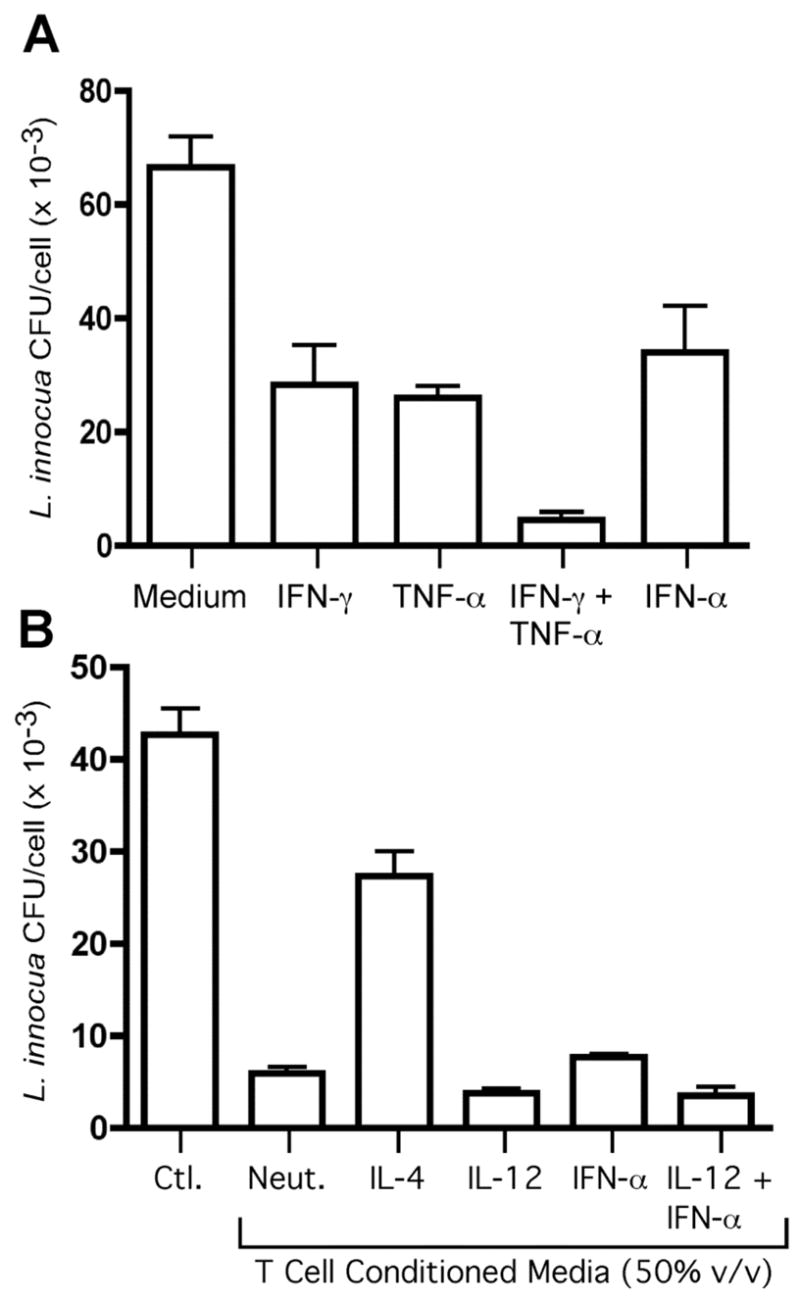
Inhibition of Listeria replication by human CD4+ T cells secreting IFN-γ and TNF-α. THP-1 cells were infected with Listeria innocua in the absence or presence of recombinant cytokines (A) or 50% (v/v) T cell conditioned media (B) for 16 hours. Cell monolayers were harvested, and L. innocua was quantified by colony-forming assay from cell lysates. Data are expressed as mean +/− SEM of three replicates.
As noted previously, T cell conditioned media inhibited HCV infection in A7 replicon cells (Fig. 2B). HCV suppresses antiviral signaling by type I interferon in infected host cells by a variety of mechanisms, including inhibition IFN-α/β synthesis through disruption of the RIG-I pathway (56–58). However, several reports have indicated that HCV is susceptible to the antiviral effects of IFN-γ (59–61). Therefore, we examined the role of IFN-γ and TNF-α secretion by Th cells in inhibition of HCV infection (Fig. 5). While recombinant TNF-α alone displayed no effect on HCV NS5A expression, recombinant IFN-γ alone inhibited HCV NS5A (Fig. 5A, compare lanes 2 and 3). Furthermore, addition of TNF-α marginally enhanced the antiviral effect of IFN-γ (Fig. 5A, compare lanes 3 and 5).
FIGURE 5.
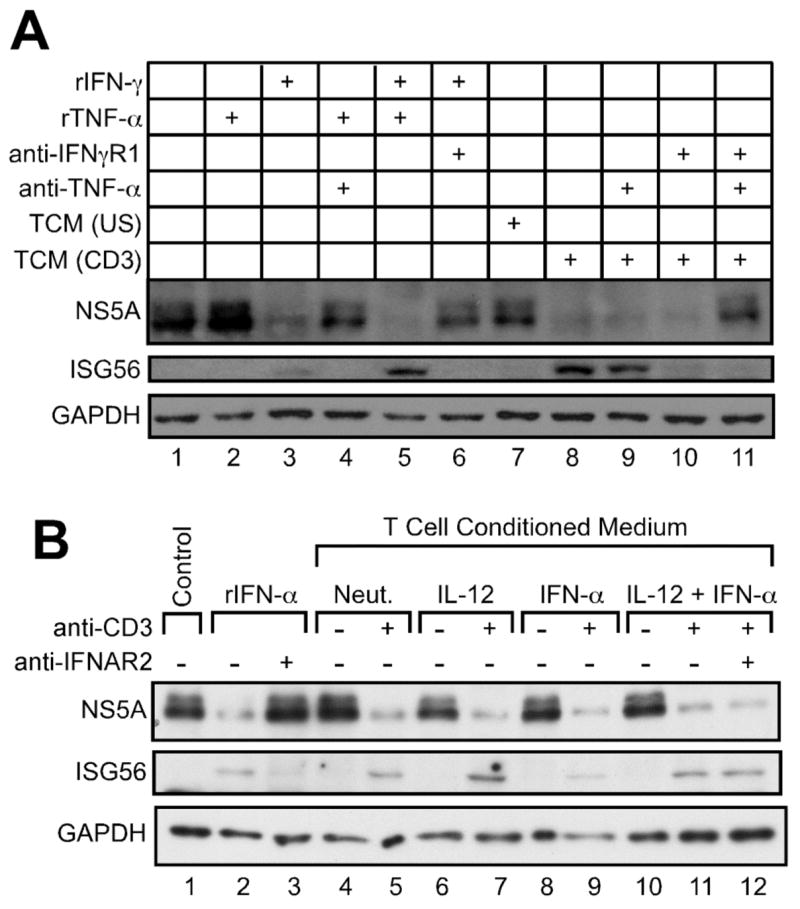
IFN-γ and TNF-α secreted by human CD4+ T cells exert antiviral activity against HCV infection. A, A7 replicon cells were incubated for 24 hours in the absence (lane 1) or presence (lanes 2–11) of cytokines, neutralizing anti-cytokine antibodies, and 5% (v/v) T cell conditioned media from IL-12 + IFN-α activated T cells as indicated. Cell lysates were prepared, and Western blotting was performed using antibodies against HCV NS5A, human ISG56, and human GAPDH. B, A7 replicon cells were incubated for 24 hours in the absence (lane 1) or presence of 100 U/ml rhIFN-αA (lanes 2–3) or 5% (v/v) T cell conditioned media (lanes 4–12), in the absence or presence of 5 μg/ml anti-hIFNAR2 antibody, as indicated. Cell lysates were prepared, and Western blotting was performed using antibodies against HCV NS5A, human ISG56, and human GAPDH.
As was previously observed, addition of T cell conditioned media to A7 HCV replicon cells reduced HCV NS5A protein synthesis (Fig. 5A). Neutralization of the R1 chain of the IFN-γ receptor (IFNγR1) on target cells, combined with neutralization of TNF-α in T cell conditioned media, reversed the previously observed antiviral activity of T cell conditioned media in this assay system (Fig. 5A, compare lanes 8 and 11), demonstrating an antiviral role for T cell-secreted IFN-γ and TNF-α in HCV infection. As expected, addition of neutralizing anti-hIFNAR2 antibody failed to reverse the antiviral effect of T cell conditioned media (Fig. 5B, compare lanes 11 and 12).
To elucidate a possible molecular mechanism for the antiviral effects of T cell conditioned media in the HCV replicon system, we examined the expression of ISG56, an interferon-stimulated gene known to inhibit HCV replication (62). Treatment of A7 replicon cells with recombinant IFN-α induced ISG56 expression, as expected (Fig. 5A, lane 2). Unexpectedly, recombinant IFN-γ also induced ISG56 expression in these cells, and addition of recombinant TNF-α enhanced this effect (Fig. 5A, lanes 3 and 5). Furthermore, ISG56 was induced by T cell conditioned media from T cells restimulated with anti-CD3 (Fig. 5, A and B), and this effect was reversed by blockade of IFN-γ and TNF-α signaling (Fig. 5A, lanes 8 and 11) but not by neutralization of IFNAR2 (Fig. 5B, lane 12).
IFN-γ and TNF-α signal through a cytokine relay network involving the type I interferon receptor
As IFN-γ and TNF-α were found to potently inhibit HCV gene expression, we wished to determine whether these two proinflammatory cytokines were also responsible for the antiviral activity of T cell conditioned media in VSV infection. Recombinant TNF-α alone showed little antiviral activity up to 50 ng/ml, while recombinant IFN-γ alone had a modest and dose-dependent effect on VSV infection (Fig. 6A). However, the combination of IFN-γ and TNF-α displayed a very potent and synergistic antiviral activity, comparable to the activity of 100 U/ml rhIFN-αA in this assay (Fig. 6A). Th1 cells also secrete lymphotoxin (LT), a member of the TNF superfamily (63), and some recent reports have demonstrated that LT secreted by NK cells has noncytopathic antiviral properties (38, 39). However, LT failed to demonstrate antiviral activity, either alone or in combination with IFN-γ (data not shown). Thus, VSV infection is sensitive to the combined effects of IFN-γ and TNF-α.
FIGURE 6.
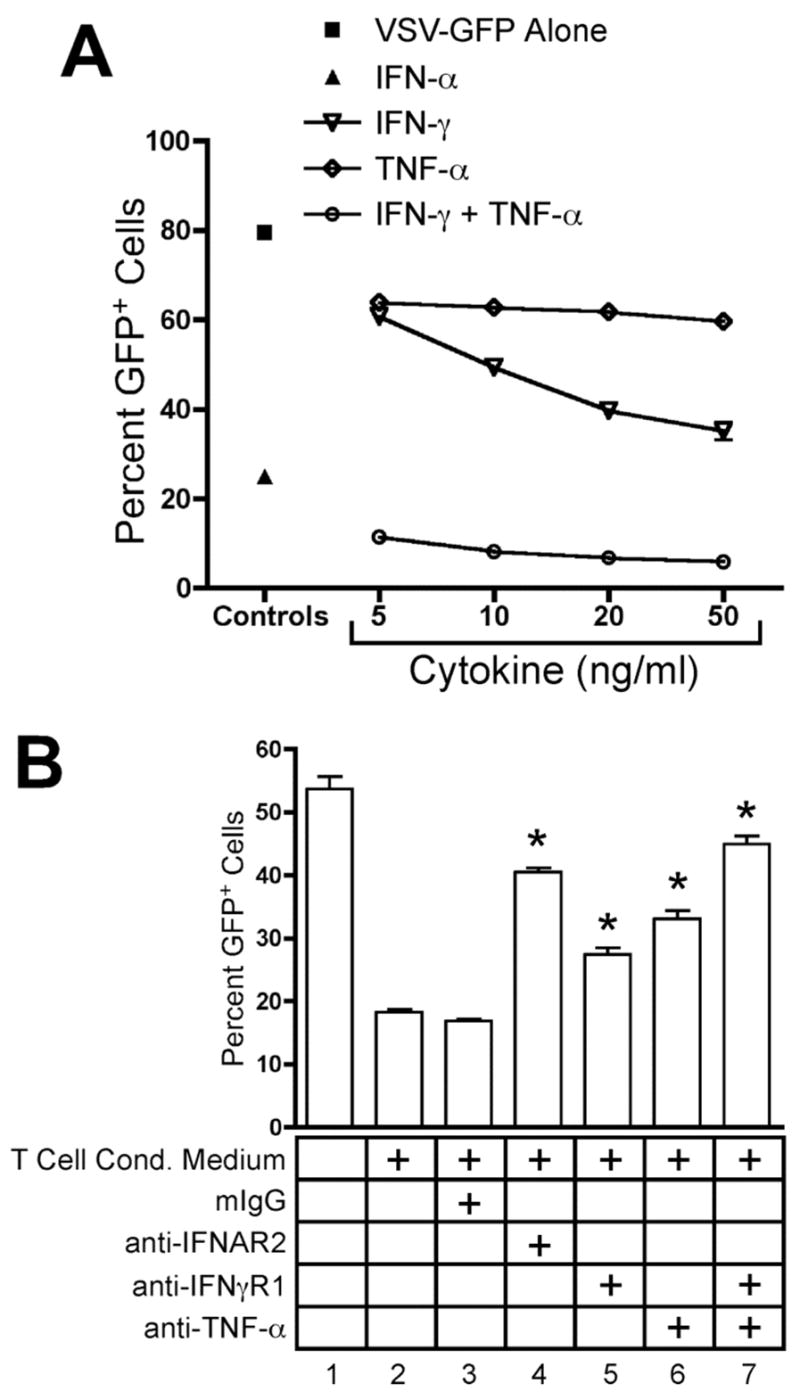
IFN-γ and TNF-α secreted by CD4+ T cells inhibit VSV infection. THP-1 cells were infected for 16 hours with VSV-GFP. GFP expression was analyzed by flow cytometry. Data are expressed as mean +/− SEM of three replicates. (A) THP-1 cells were infected in the absence (■) or presence of 100 U/ml rhIFN-αA (▴) or increasing concentrations of rhIFN-γ (
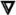 ), rhTNF-α (◇), or rhIFN-γ + rhTNF-α (○) as indicated. (B) THP-1 cells were infected in the absence (1) or presence (2–7) of 10% (v/v) T cell conditioned media from IL-12 + IFN-α activated T cells in the absence (2) or presence of 5 μg/ml mouse IgG1 isotype control antibody (3), 5 μg/ml anti-hIFNAR2 (4), 10 μg/ml anti-hIFNγR1 (5), 5 μg/ml anti-hTNF-α (6), or a combination of anti-hIFNγR1 and anti-hTNF-α (7).
), rhTNF-α (◇), or rhIFN-γ + rhTNF-α (○) as indicated. (B) THP-1 cells were infected in the absence (1) or presence (2–7) of 10% (v/v) T cell conditioned media from IL-12 + IFN-α activated T cells in the absence (2) or presence of 5 μg/ml mouse IgG1 isotype control antibody (3), 5 μg/ml anti-hIFNAR2 (4), 10 μg/ml anti-hIFNγR1 (5), 5 μg/ml anti-hTNF-α (6), or a combination of anti-hIFNγR1 and anti-hTNF-α (7).
As demonstrated above, T cell conditioned media markedly inhibited VSV infection, and this activity was partially inhibited by blocking either the IFNγR1 or TNF-α (Fig. 6B). Further, neutralization of both cytokines resulted in much greater reversal (Fig. 6B, condition 7, p < 0.05 versus T cell conditioned media alone). As a control, we also preincubated target cells with neutralizing anti-IFNAR2 before VSV-GFP infection in the presence of T cell conditioned media. Surprisingly, neutralization of IFNAR2 reversed the antiviral effect of T cell conditioned media as effectively as blockade of IFN-γ and TNF-α (Fig. 6B, condition 4, p < 0.05 versus T cell conditioned media alone).
We could find no previous reports demonstrating secretion of type I interferon by CD4+ T cells. Thus, we were surprised to find that neutralization of the type I interferon receptor on target cells prevented the antiviral activity of T cell conditioned media against VSV-GFP. We therefore further pursued the role of type I interferon signaling in the observed antiviral activity secreted by Th cells. We found that the antiviral effect of recombinant IFN-γ and TNF-α could be reversed by neutralization of IFNAR2, indicating that this effect is dependent upon type I interferon signaling (Fig. 7A, p < 0.05, no antibody versus anti-IFNAR2). As noted previously, pre-treatment of THP-1 cells with neutralizing anti-IFNAR2 also abolished the antiviral effect of T cell conditioned media, indicating that the secreted activity requires this receptor (Fig. 7B, p < 0.05, no antibody versus anti-IFNAR2, all conditions). These data suggest the existence of a previously undescribed cytokine relay network whereby IFN-γ and TNF-α synergize to induce type I interferon signaling, which promotes viral clearance.
FIGURE 7.
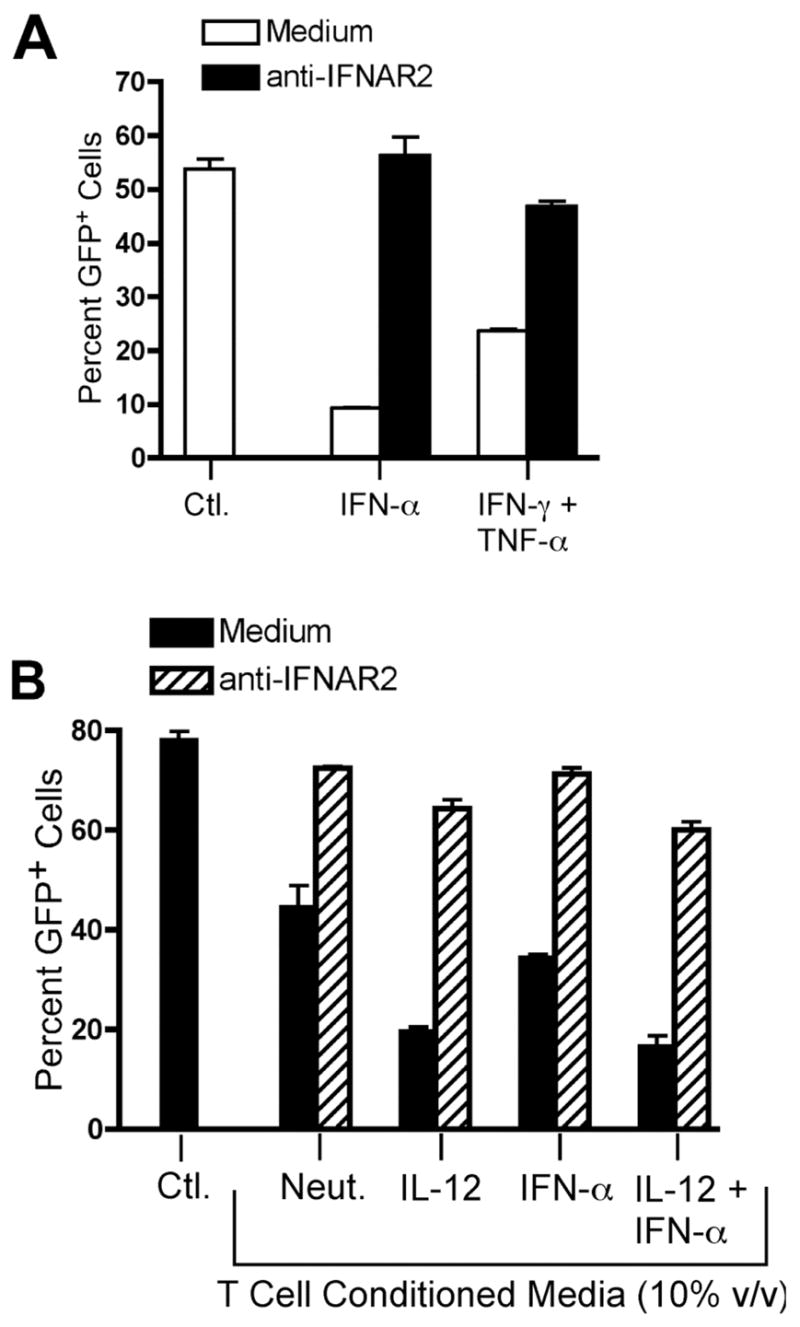
IFN-γ- and TNF-α-mediated antiviral activity requires availability of the IFNAR. THP-1 cells were infected for 16 hours with VSV-GFP. GFP expression was analyzed by flow cytometry. Data are expressed as mean +/− SEM of three replicates. A, THP-1 cells were infected in the absence or presence of 100 U/ml rhIFN-αA or a combination of 2.5 ng/ml rhIFN-γ and 2.5 ng/ml rhTNF-α in the absence (open bars) or presence (black bars) of 5 μg/ml anti-hIFNAR2 antibody. *, p < 0.05, two-way ANOVA. B, THP-1 cells were infected in the absence or presence of 10% (v/v) T cell conditioned media as indicated in the absence (black bars) or presence (hatched bars) of 5 μg/ml anti-hIFNAR2 antibody.
Either the Th cells or the THP-1 target cells could have been a source of type I interferon. In either case, neutralization of soluble type I interferon would reverse the antiviral activity. We therefore used neutralizing antibodies to examine the identity of the type I interferon involved in the observed antiviral activity. As noted previously, pre-treatment of THP-1 target cells with anti-IFNAR2 reversed the antiviral activity of T cell conditioned media (Fig. 8A, condition 4, p < 0.05 versus T cell conditioned media alone). However, addition of neutralizing anti-IFN-α, anti-IFN-β, or anti-IFN-ω antibodies to VSV-GFP infections failed to reverse the antiviral activity of T cell conditioned media, demonstrating that neither CD4+ T cells nor infected THP-1 cells secrete type I interferons (Fig. 8A, conditions 5–8). Addition of each antibody was sufficient to block 10–100 U/ml of its corresponding type I interferon activity in this assay (Fig. 8B), demonstrating that these antibodies possess the capacity to neutralize each specific type I interferon.
FIGURE 8.
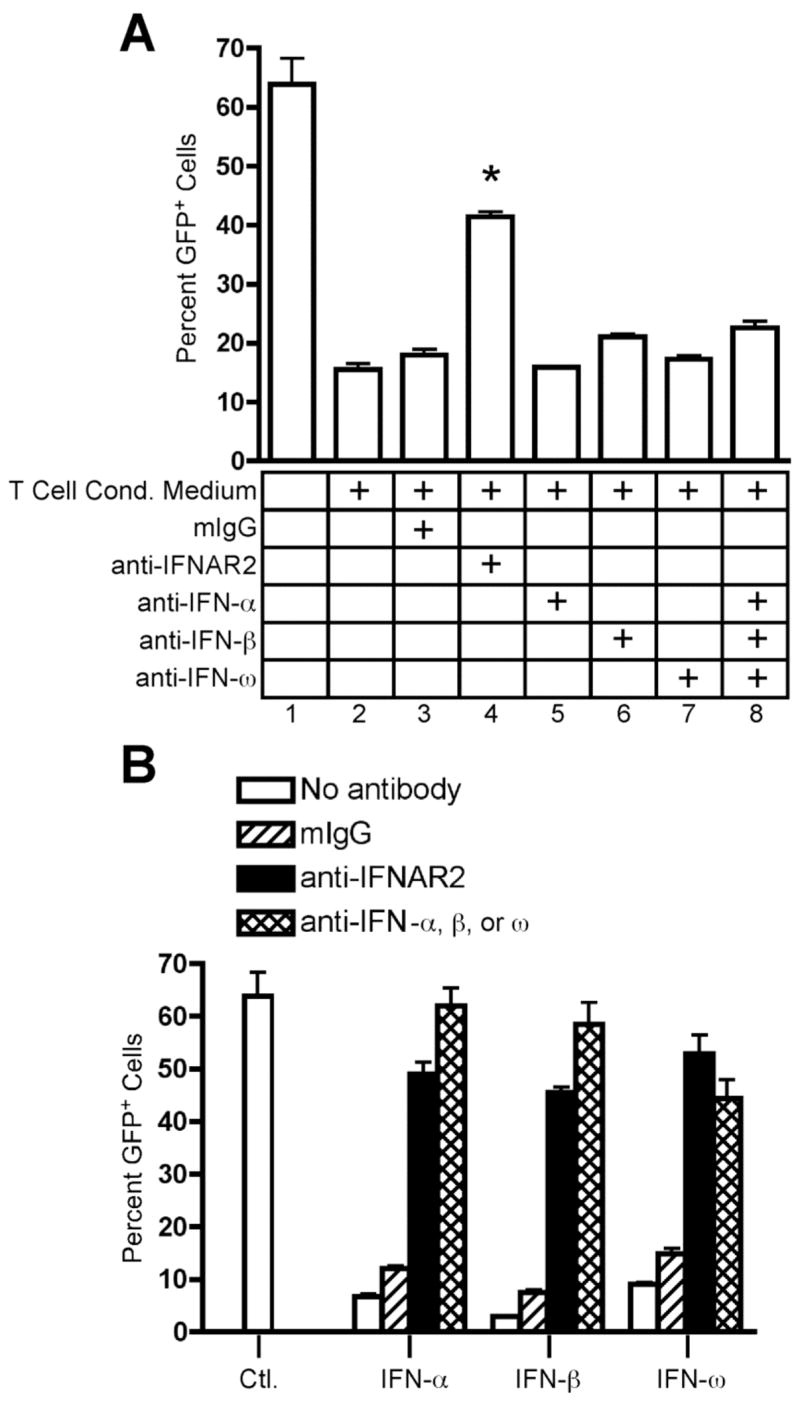
Neutralization of IFN-α, IFN-β, and IFN-ω fails to inhibit the antiviral activity of human CD4+ T cells. THP-1 cells were infected for 16 hours with VSV-GFP. GFP expression was analyzed by flow cytometry. Data are expressed as mean +/− SEM of three replicates. (A) THP-1 cells were infected in the absence (1) or presence (2–8) of 10% (v/v) T cell conditioned media from IL-12 + IFN-α activated T cells in the absence (2) or presence of 5 μg/ml mouse IgG1 isotype control antibody (3), 5 μg/ml anti-hIFNAR2 (4), 5 μg/ml anti-hIFN-α (5), 1700 NU/ml anti-hIFN-β (6), 5 μg/ml anti-hIFN-ω (7), or a combination of anti-hIFN-α, anti-hIFN-β, and anti-hIFN-ω (8). (B) THP-1 cells were infected in the absence or presence of 100 U/ml rhIFN-αA or 100 U/ml rhIFN-β or 10 U/ml rhIFN-ω in the absence (open bars) or presence of 5 μg/ml mouse IgG1 isotype control antibody (hatched bars), 5 μg/ml anti-hIFNAR2 antibody (black bars), or 5 μg/ml anti-hIFN-α antibody or 1700 NU/ml anti-hIFN-β antibody or 5 μg/ml anti-hIFN-ω antibody (double hatched bars) as indicated.
We also assayed human CD4+ T cells for secretion of IFN-α and IFN-β by ELISA. We found no detectable IFN-α or IFN-β protein in T cell conditioned media (data not shown). Additionally, we quantified IFN-β secretion from untreated and T cell conditioned media-treated uninfected and VSV-GFP-infected THP-1 cells, but we found no detectable secretion of IFN-β from these cells (data not shown). We further examined both human Th cells and THP-1 cells for induction of mRNA transcripts for IFN-α, IFN-β, IFN-ω, IFN-ε, and IFN-κ by quantitative real-time polymerase chain reaction (qPCR), but no transcripts were detected (data not shown). Taken together, these data demonstrated no detectable type I interferon production from either CD4+ T cells or THP-1 target cells.
Given the lack of detectable type I interferon production in this assay, it was possible that the anti-IFNAR2 antibody was inhibiting the previously observed antiviral activity through pathways not involving the human IFNAR. Therefore, we sought to further verify the role of type I interferon signaling in the observed antiviral activity. We made use of a genetically modified human fibroblast cell line, U5A, in which the gene for the human IFNAR2 subunit has been ablated (46–48). We compared VSV-GFP infection in these cells to the parent cell line, 2fTGH, which expresses an intact IFNAR. In agreement with our results in THP-1 cells, treatment of VSV-GFP-infected wild-type 2fTGH cells with a combination of recombinant IFN-γ and TNF-α at the time of infection significantly reduced viral infection. This antiviral activity was reversed in the IFNAR2-deficient U5A cells (Fig. 9A, p < 0.05 versus 2fTGH), confirming that the antiviral activity of IFN-γ and TNF-α is dependent upon type I IFN signaling. Furthermore, infection of wild-type 2fTGH cells with VSV-GFP could be inhibited by treatment at the time of infection with recombinant human IFN-αA or T cell conditioned media. However, the antiviral effects of both IFN-αA and T cell conditioned media were severely attenuated in the IFNAR2-deficient U5A cells (Fig. 9B, p < 0.05 versus 2fTGH). These results confirm the involvement of type I interferon signaling in VSV-GFP inhibition by effector cytokines secreted by human CD4+ T cells.
FIGURE 9.
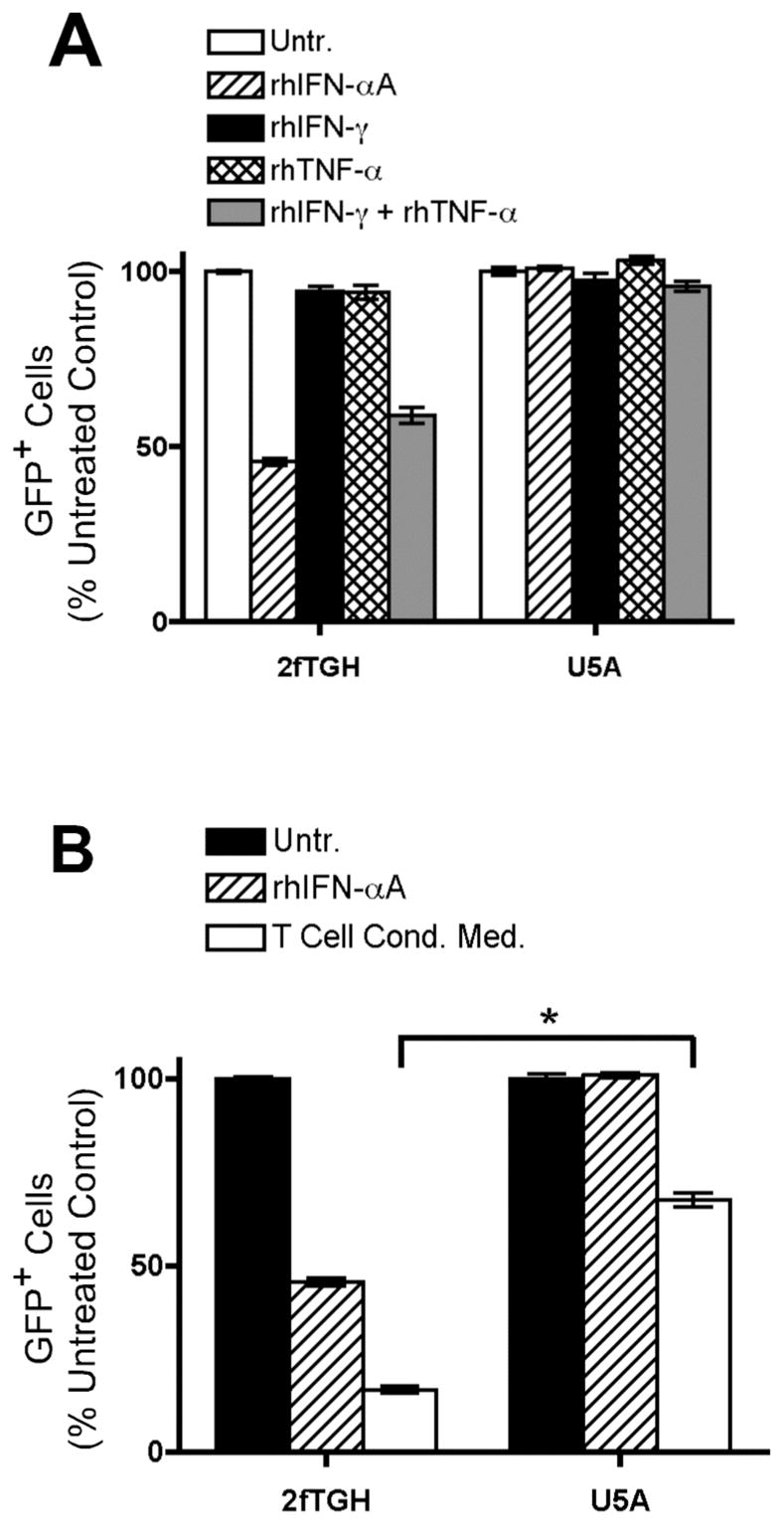
IFN-γ and TNF-α signal through a cytokine relay network involving the type I interferon receptor. Wild-type 2fTGH cells and hIFNAR2-deficient U5A cells were infected for 16 hours with VSV-GFP. GFP expression was analyzed by flow cytometry. Data are expressed as mean +/− SEM of three replicates. A, 2fTGH and U5A cells were infected in the absence (open bars) or presence of 100 U/ml rhIFN-αA (hatched bars), 2.5 ng/ml rhIFN-γ (black bars), 2.5 ng/ml rhTNF-α (double hatched bars), or a combination of 2.5 ng/ml rhIFN-γ and 2.5 ng/ml rhTNF-α (gray bars). B, 2fTGH and U5A cells were infected in the absence (black bars) or presence of 100 U/ml rhIFN-αA (hatched bars) or 10% (v/v) T cell conditioned media generated from IL-12 + IFN-α activated hCD4+ T cells (white bars). *, p < 0.05, one-way ANOVA.
Discussion
In the present study, we have demonstrated that secretion of IFN-γ and TNF-α represents a direct, cytokine-mediated antiviral activity of human CD4+ T cells. Elevated secretion of these cytokines was directed by IL-12; we found no significant contribution, positive or negative, of IFN-α/β. A combination of IFN-γ and TNF-α produced by Th1 cells promotes antiviral responses by two distinct mechanisms. First, IFN-γ and TNF-α can transmit an antiviral signal via a type I interferon-independent pathway, as in the case of HCV infection. In this case, the antiviral activity could be mediated by direct effects of IFN-γ and TNF-α or through the induction of another, non-IFN-α/β cytokine. Alternatively, the activity can be mediated through a cytokine relay network, as in the case of VSV infection, in which type I interferon signaling is required for the antiviral effect.
In agreement with our results, several other groups have shown that CD4+ T cells have the capacity to promote viral clearance in vivo in a “helper-independent” fashion. For instance, clearance of Sendai virus, gammaherpesvirus (γHV68), or influenza A virus can proceed in a CD4+ T cell-dependent fashion in the absence of B cells and CD8+ T cells (40–44). Additionally, memory Th cells generated against VSV in CTL-nonresponsive mice provide protection in an antibody-independent manner (64). In many cases, a deficiency in IFN-γ in vivo abolished the antiviral capacity of CD4+ T cells (42, 64, 65), and adoptive transfer of an antigen-specific Th1 clone conferred protection from γHV68 infection (45). However, the target of IFN-γ was undetermined in these studies. Therefore, it was possible that viral clearance could have been mediated by a population of innate cells, such as NK cells, which were activated in the presence of IFN-γ. Here, we definitively demonstrate for the first time that cytokines secreted by Th cells directly impact viral clearance from infected targets.
Furthermore, CD4+ T cell-mediated control of cytomegalovirus (CMV) in salivary glands requires IFN-γ, but, paradoxically, treatment of virally infected mice with recombinant IFN-γ failed to clear the virus (66). We have shown that both IFN-γ and TNF-α are required to achieve robust viral inhibition by Th1 cell-secreted factors. Therefore, in vivo treatment of CMV-infected animals with a combination of recombinant IFN-γ and TNF-α could promote viral clearance when neither cytokine alone possessed this activity.
Several groups have reported that TNF-α can induce secretion of IFN-β from target cells and that this IFN-β can synergize with IFN-γ for viral inhibition (33, 34, 36, 37, 67, 68). However, this effect relied upon pre-treatment of target cells with cytokines for 16–24 hours before in vitro infection. In contrast, we have demonstrated an antiviral activity of IFN-γ and TNF-α which does not require pre-treatment of target cells. Thus, secretion of these cytokines by CD4+ T cells at peripheral sites could have beneficial effects even after cells were already infected.
We found that the antiviral activity of T cell-secreted IFN-γ and TNF-α was independent of type I interferon signaling in the case of HCV infection. Surprisingly, this activity was completely dependent upon the presence of a functional IFNAR in the case of VSV infection. It is currently unclear whether this phenomenon is specific to VSV or represents a more general antiviral mechanism. However, we noted during the course of our experiments that Sendai virus, which blocks type I interferon signaling in infected cells, was also completely resistant to the antiviral effects of T cell conditioned media (K. A. H. and M. G., Jr., unpublished observations).
While the observed antiviral effect of IFN-γ and TNF-α is dependent upon signaling through the IFNAR in the case of VSV, we were unable to detect induction of known type I interferon genes in target cells. This further excludes induction of IFN-β by TNF-α as a mechanism for the observed antiviral effect. Many possible explanations exist for this novel antiviral effect of IFN-γ and TNF-α during VSV infection. For instance, IFN-γ and TNF-α may be inducing expression of a novel type I interferon gene in virally infected target cells. Several new type I interferon genes have been described in recent years (69–71); a more extensive search may reveal other, distantly related family members located within or even outside the IFN locus.
Alternatively, IFN-γ and TNF-α may synergize to directly activate IFNAR signaling via a mechanism such as receptor sharing in order to induce type I IFN-like effects in specialized situations. There are many known cases in which two or more unrelated receptors are activated by the same ligand. For instance, glial cell-derived neurotrophic factor (GDNF) signals through both the receptor tyrosine kinase RET and the Ig-domain-containing receptor NCAM (72). Alternately, a single receptor subunit can be shared among multiple distinct receptors, as in the case of the common gamma chain which is used for cytokine signaling (73). Consistent with our in vitro studies, it is interesting to note that Müller et. al. demonstrated that the antiviral effects of IFN-γ against VSV were impaired in murine cells lacking IFNAR expression (74). However, other IFN-γ signaling pathways were unaffected in cells from IFNAR−/− mice, and IFNγR−/− mice showed no defect in VSV clearance.
Many viruses encode intracellular or extracellular mechanisms to antagonize antiviral cytokine secretion and signaling by infected host cells. For instance, poxviruses encode soluble, secreted forms of the IFNAR, IFNγR, and TNFR which can neutralize host cytokines (75–77). A variety of viruses, including HCV, influenza A virus, and Sendai virus, also inhibit intracellular induction of type I interferon by blockade of the RIG-I pathway (57, 78–80). In such cases, exogenously delivered cytokines from Th cells could provide alternative pathways to overcome these blocks and promote pathogen clearance in a noncytopathic manner.
IFN-α is widely used to treat HCV infections, but many patients fail to respond to this therapy. HCV and other flaviviruses, such as West Nile Virus, inhibit IFNAR signal transduction in target cells through inactivation of downstream signaling intermediates (56, 58, 81). In accordance with previous reports, we demonstrated that IFN-γ possessed substantial antiviral activity against HCV (59–61). However, Frese et. al. found no role for TNF-α, either alone or in combination with IFN-γ, in inhibition of HCV replication (60). In contrast, we observed cooperation between IFN-γ and TNF-α in suppressing HCV NS5A protein expression. Furthermore, our data show that IFN-γ and TNF-α inhibit HCV infection by a type I IFN-independent mechanism. Therefore, Th1 responses generated during infections with these viruses could represent an important alternative mechanism for pathogen clearance when type I IFN is ineffective.
Acknowledgments
We thank Angela Mobley and the Flow Cytometry Core Facility at UT Southwestern Medical Center for assistance with flow cytometry. We gratefully acknowledge Drs. Lora Hooper, James Forman, and Nicolai van Oers as well as Dyan Fox, Hilario Ramos, and Jonathan Huber for helpful suggestions and for critically reviewing the manuscript.
Abbreviations in this paper
- IFNAR
IFN-α/β receptor
- LT
lymphotoxin
Footnotes
This study was supported by the following grants from the NIH/NIAID: AI060389 and AI40035 awarded to MG, and AI056222 awarded to JDF. AMD was supported by a training grant from the NIH/GM (GM00820317).
References
- 1.Brocke S, Hahn H. Heat-killed Listeria monocytogenes and L. monocytogenes soluble antigen induce clonable CD4+ T lymphocytes with protective and chemotactic activities in vivo. Infect Immun. 1991;59:4531–4539. doi: 10.1128/iai.59.12.4531-4539.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canessa A, Pistoia V, Roncella S, Merli A, Melioli G, Terragna A, Ferrarini M. An in vitro model for Toxoplasma infection in man: interaction between CD4+ monoclonal T cells and macrophages results in killing of trophozoites. J Immunol. 1988;140:3580–3588. [PubMed] [Google Scholar]
- 3.Geginat G, Lalic M, Kretschmar M, Goebel W, Hof H, Palm D, Bubert A. Th1 cells specific for a secreted protein of Listeria monocytogenes are protective in vivo. J Immunol. 1998;160:6046–6055. [PubMed] [Google Scholar]
- 4.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 5.Urbani S, Amadei B, Fisicaro P, Tola D, Orlandini A, Sacchelli L, Mori C, Missale G, Ferrari C. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology. 2006;44:126–139. doi: 10.1002/hep.21242. [DOI] [PubMed] [Google Scholar]
- 6.Scherle PA, Gerhard W. Functional analysis of influenza-specific helper T cell clones in vivo: T cells specific for internal viral proteins provide cognate help for B cell responses to hemagglutinin. J Exp Med. 1986;164:1114–1128. doi: 10.1084/jem.164.4.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol. 2006;177:2888–2898. doi: 10.4049/jimmunol.177.5.2888. [DOI] [PubMed] [Google Scholar]
- 8.Salkowitz JR, Sieg SF, Harding CV, Lederman MM. In vitro human memory CD8 T cell expansion in response to cytomegalovirus requires CD4+ T cell help. J Infect Dis. 2004;189:971–983. doi: 10.1086/382032. [DOI] [PubMed] [Google Scholar]
- 9.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 11.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 12.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasukawa M, Ohminami H, Yakushijin Y, Arai J, Hasegawa A, Ishida Y, Fujita S. Fas-independent cytotoxicity mediated by human CD4+ CTL directed against herpes simplex virus-infected cells. J Immunol. 1999;162:6100–6106. [PubMed] [Google Scholar]
- 14.Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, Harpur AG, Wilks AF, Yasukawa K, Taga T, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89–92. doi: 10.1126/science.8272872. [DOI] [PubMed] [Google Scholar]
- 15.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. TYPE I INTERFERONS (alpha/beta) IN IMMUNITY AND AUTOIMMUNITY. Annu Rev Immunol. 2005;23:307–335. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 16.Stark GR, I, Kerr M, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 17.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 18.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 19.Orange JS, Biron CA. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J Immunol. 1996;156:1138–1142. [PubMed] [Google Scholar]
- 20.Orange JS, Biron CA. Characterization of early IL-12, IFN-α/β, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156:4746–4756. [PubMed] [Google Scholar]
- 21.Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med. 1993;178:1655–1663. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wenner CA, Guler ML, Macatonia SE, O’Garra A, Murphy KM. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 23.Sareneva T, Matikainen S, Kurimoto M, Julkunen I. Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J Immunol. 1998;160:6032–6038. [PubMed] [Google Scholar]
- 24.Rogge L, D’Ambrosio D, Biffi M, Penna G, Minetti LJ, Presky DH, Adorini L, Sinigaglia F. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol. 1998;161:6567–6574. [PubMed] [Google Scholar]
- 25.Hibbert L, Pflanz S, De Waal Malefyt R, Kastelein RA. IL-27 and IFN-alpha signal via Stat1 and Stat3 and induce T-Bet and IL-12Rbeta2 in naive T cells. J Interferon Cytokine Res. 2003;23:513–522. doi: 10.1089/10799900360708632. [DOI] [PubMed] [Google Scholar]
- 26.Tyler DR, Persky ME, Matthews LA, Chan S, Farrar JD. Pre-assembly of STAT4 with the human IFN-alpha/beta receptor-2 subunit is mediated by the STAT4 N-domain. Molec Immunol. 2007;44:1864–1872. doi: 10.1016/j.molimm.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–69. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat Immunol. 2000;1:70–76. doi: 10.1038/76940. [DOI] [PubMed] [Google Scholar]
- 29.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. 2004;172:61–69. doi: 10.4049/jimmunol.172.1.61. [DOI] [PubMed] [Google Scholar]
- 30.Berenson LS, Gavrieli M, Farrar JD, Murphy TL, Murphy KM. Distinct Characteristics of Murine STAT4 Activation in Response to IL-12 and IFN-{alpha} J Immunol. 2006;177:5195–5203. doi: 10.4049/jimmunol.177.8.5195. [DOI] [PubMed] [Google Scholar]
- 31.Ramos HJ, Davis AM, George TC, Farrar JD. IFN-{alpha} Is Not Sufficient to Drive Th1 Development Due to Lack of Stable T-bet Expression. J Immunol. 2007;179:3792–3803. doi: 10.4049/jimmunol.179.6.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes TK, Kaspar TA, Coppenhaver DH. Synergy of antiviral actions of TNF and IFN-γ: evidence for a major role if TNF-induced IFN-β. Antiviral Res. 1988;10:1–9. doi: 10.1016/0166-3542(88)90010-1. [DOI] [PubMed] [Google Scholar]
- 34.Mestan J, Brockhaus M, Kirchner H, Jacobsen H. Antiviral activity of tumor necrosis factor. Synergism with interferons and induction of oligo-2′,5′-adenylate synthetase. J Gen Virol. 1988;69:3113–3120. doi: 10.1099/0022-1317-69-12-3113. [DOI] [PubMed] [Google Scholar]
- 35.Matikainen S, Sirén J, Tissari J, Veckman V, Pirhonen J, Severa M, Sun Q, Lin R, Meri S, Uzé G, Hiscott J, Julkunen I. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J Virol. 2006;80:3515–3522. doi: 10.1128/JVI.80.7.3515-3522.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mestan J, Digel W, Mittnacht S, Hillen H, Blohm D, Möller A, Jacobsen H, Kirchner H. Antiviral effects of recombinant tumor necrosis factor in vitro. Nature. 1986;323:816–819. doi: 10.1038/323816a0. [DOI] [PubMed] [Google Scholar]
- 37.Wong GHW, Goeddel DV. Tumor necrosis factor α and β inhibit virus replication and synergize with interferons. Nature. 1986;323:819–822. doi: 10.1038/323819a0. [DOI] [PubMed] [Google Scholar]
- 38.Banks TA, Rickert S, Benedict CA, Ma L, Ko M, Meier J, Ha W, Schneider K, Granger SW, Turovskaya O, Elewaut D, Otero D, French AR, Henry SC, Hamilton JD, Scheu S, Pfeffer K, Ware CF. A lymphotoxin-IFN-β axis essential for lymphocyte survival revealed during cytomegalovirus infection. J Immunol. 2005;174:7217–7225. doi: 10.4049/jimmunol.174.11.7217. [DOI] [PubMed] [Google Scholar]
- 39.Iversen AC, Norris PS, Ware CF, Benedict CA. Human NK cells inhibit cytomegalovirus replication through a noncytolytic mechanism involving lymphotoxin-dependent induction of IFN-β. J Immunol. 2005;175:7568–7574. doi: 10.4049/jimmunol.175.11.7568. [DOI] [PubMed] [Google Scholar]
- 40.Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibility complex-restricted CD8+ T cells. J Exp Med. 1991;174:875–880. doi: 10.1084/jem.174.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong W, Marshall D, Coleclough C, Woodland DL. CD4+ T cell priming accelerates the clearance of Sendai virus in mice, but has a negative effect on CD8+ T cell memory. J Immunol. 2000;164:3274–3282. doi: 10.4049/jimmunol.164.6.3274. [DOI] [PubMed] [Google Scholar]
- 42.Zhong W, Roberts AD, Woodland DL. Antibody-independent antiviral function if memory CD4+ T cells in vivo requires regulatory signals from CD8+ effector T cells. J Immunol. 2001;167:1379–1386. doi: 10.4049/jimmunol.167.3.1379. [DOI] [PubMed] [Google Scholar]
- 43.Topham DJ, Doherty PC. Clearance of an influenza A virus by CD4+ T cells is inefficient in the absence of B cells. J Virol. 1998;72:882–885. doi: 10.1128/jvi.72.1.882-885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sparks-Thissen RL, Braaten DC, Kreher S, Speck SH, Virgin HWI. An optimized CD4 T-cell response can control productive and latent gammaherpesvirus infection. J Virol. 2004;78:6827–6835. doi: 10.1128/JVI.78.13.6827-6835.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sparks-Thissen RL, Braaten DC, Hildner K, Murphy TL, Murphy KM, Virgin HWI. CD4 T cell control of acute and latent murine gammaherpesvirus infection requires IFNγ. Virology. 2005;338:201–208. doi: 10.1016/j.virol.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 46.Lutfalla G, Holland SJ, Cinato E, Monneron D, Reboul J, Rogers NC, Smith JM, Stark GR, Gardiner K, Mogensen KE, et al. Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. EMBO J. 1995;14:5100–5108. doi: 10.1002/j.1460-2075.1995.tb00192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pellegrini S, John J, Shearer M, Kerr IM, Stark GR. Use of a selectable marker regulated by alpha interferon to obtain mutations in the signaling pathway. Mol Cell Biol. 1989;9:4605–4612. doi: 10.1128/mcb.9.11.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol. 1995;15:1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foy E, Li K, Wang C, Sumpter R, Jr, Ikeda M, Lemon SM, Gale M., Jr Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 50.Sumpter RJ, Wang C, Foy E, Loo YM, Gale MJ. Viral evolution and interferon resistance of hepatitis C virus RNA replication in a cell culture model. J Virol. 2004;78:11591–11604. doi: 10.1128/JVI.78.21.11591-11604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuura Y, Tani H, Suzuki K, Kimura-Someya T, Suzuki R, Aizaki H, Ishii K, Moriishi K, Robison CS, Whitt MA, Muiyamura T. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology. 2001;286:263–275. doi: 10.1006/viro.2001.0971. [DOI] [PubMed] [Google Scholar]
- 52.Hallak LK, Collins PL, Knudson W, Peeples ME. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology. 2000;271:264–275. doi: 10.1006/viro.2000.0293. [DOI] [PubMed] [Google Scholar]
- 53.Persky ME, Murphy KM, Farrar JD. IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol. 2005;174:294–301. doi: 10.4049/jimmunol.174.1.294. [DOI] [PubMed] [Google Scholar]
- 54.Sukumaran SK, Shimada H, Prasadarao NV. Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun. 2003;71:5951–5961. doi: 10.1128/IAI.71.10.5951-5961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peck R. Gamma interferon induces monocyte killing of Listeria monocytogenes by an oxygen-dependent pathway; alpha- or beta-interferons by oxygen-independent pathways. J Leukoc Biol. 1989;46:434–440. doi: 10.1002/jlb.46.5.434. [DOI] [PubMed] [Google Scholar]
- 56.Heim MH, Moradpour D, Blum HE. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J Virol. 1999;73:8469–8475. doi: 10.1128/jvi.73.10.8469-8475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foy E, Li K, Sumpter RJ, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale MJ. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Basu A, Meyer K, Ray RB, Ray R. Hepatitis C virus core protein modulates the interferon-induced transacting factors of Jak/Stat signaling pathway but does not affect the activation of downstream IRF-1 or 561 gene. Virology. 2001;288:379–390. doi: 10.1006/viro.2001.1100. [DOI] [PubMed] [Google Scholar]
- 59.Frese M, Schwärzle V, Barth K, Krieger N, Lohmann V, Mihm S, Haller O, Bartenschlager R. Interferon-γ inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology. 2002;35:694–703. doi: 10.1053/jhep.2002.31770. [DOI] [PubMed] [Google Scholar]
- 60.Frese M, Barth K, Kaul A, Lohmann V, Schwärzle V, Bartenschlager R. Hepatitis C virus RNA replication is resistant to tumor necrosis factor-α. J Gen Virol. 2003;84:1253–1259. doi: 10.1099/vir.0.18997-0. [DOI] [PubMed] [Google Scholar]
- 61.Windisch MP, Frese M, Kaul A, Trippler M, Lohmann V, Bartenschlager R. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J Virol. 2005;79:13778–13793. doi: 10.1128/JVI.79.21.13778-13793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang C, Pflugheber J, Sumpter R, Jr, Sodora DL, Hui D, Sen GC, Gale M., Jr Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cherwinski HM, Schumacher JH, Brown KD, Mosmann TR. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J Exp Med. 1987;166:1229–1244. doi: 10.1084/jem.166.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Binder D, Kündig TM. Antiviral protection by CD8+ versus CD4+ T cells: CD8+ T cells correlating with cytotoxic activity in vitro are more efficient in antivaccinia virus protection than CD4-dependent IL. J Immunol. 1991;146:4301–4307. [PubMed] [Google Scholar]
- 65.Steed A, Buch T, Waisman A, Virgin HWI. Gamma interferon blocks gammaherpesvirus reactivation from latency in a cell type-specific manner. J Virol. 2007;81:6134–6140. doi: 10.1128/JVI.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucin P, Pavic I, Polic B, Jonjic S, Koszinowski UH. Gamma interferon-dependent clearance of cytomegalovirus infection in salivary glands. J Virol. 1992;66:1977–1984. doi: 10.1128/jvi.66.4.1977-1984.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lucin P, Jonjic S, Messerle M, Polic B, Hengel H, Koszinowski UH. Late phase inhibition of murine cytomegalovirus replication by synergistic action of interferon-gamma and tumor necrosis factor. J Gen Virol. 1994;75:101–110. doi: 10.1099/0022-1317-75-1-101. [DOI] [PubMed] [Google Scholar]
- 68.Mayer A, Gelderblom H, Kümel G, Jungwirth C. Interferon-γinduced assembly block in the replication cycle of adenovirus 2: augmentation by tumor necrosis factor-α. Virology. 1992;187:372–376. doi: 10.1016/0042-6822(92)90330-r. [DOI] [PubMed] [Google Scholar]
- 69.Hardy MP, Owczarek CM, Jermiin LS, Ejdebäck M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics. 2004;84:331–345. doi: 10.1016/j.ygeno.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 70.Adolf GR, Maurer-Fogy I, Kalsner I, Cantell K. Purification and characterization of natural human interferon ω1. J Biol Chem. 1990;265:9290–9295. [PubMed] [Google Scholar]
- 71.LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, Taylor K, Buergin M, Chinchilla D, Roshke V, Chen G, Ruben SM, Pitha PM, Coleman TA, Moore PA. Interferon-κ, a novel type I interferon expressed in human keratinocytes. J Biol Chem. 2001;276:39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 72.Ben-Shlomo I, Hsueh AJ. Three’s company: two or more unrelated receptors pair with the same ligand. Molec Endocrinol. 2005;19:1097–1109. doi: 10.1210/me.2004-0451. [DOI] [PubMed] [Google Scholar]
- 73.Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, Yamauchi A, Bloom ET, Mietz J, John S, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331–339. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 74.Müller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 75.Alcamí A, Khanna A, Paul NL, Smith GL. Vaccinia virus strains Lister, USSR and Evans express soluble and cell-surface tumor necrosis factor receptors. J Gen Virol. 1999;80:949–959. doi: 10.1099/0022-1317-80-4-949. [DOI] [PubMed] [Google Scholar]
- 76.Alcamí A, Smith GL. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J Virol. 1995;69:4633–4639. doi: 10.1128/jvi.69.8.4633-4639.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alcamí A, Symons JA, Smith GL. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol. 2000;74:11230–11239. doi: 10.1128/jvi.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cárdenas WB, Loo YM, Gale MJ, Hartman AL, Kimberlin CR, Martínez-Sobrido L, Saphire EO, Basler CF. Ebola virus VP35 binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mibayashi M, Martínez-Sobrido L, Loo YM, Cárdenas WB, Gale MJ, García-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kato A, Kiyotani K, Kubota T, Yoshida T, Tashiro M, Nagai Y. Importance of the anti-interferon capacity of Sendai virus C protein for pathogenicity in mice. J Virol. 2007;81:3264–3271. doi: 10.1128/JVI.02590-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale MJ. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]