

Abstract
Diabetes mellitus (DM) affects approximately 170 million individuals worldwide and is expected to alter the lives of at least 366 million individuals within a future span of 25 years. Of even greater concern is the premise that these projections are underestimated since they assume obesity levels will remain constant. Type 1 insulin-dependent DM accounts for only 5-10 percent of all diabetics but represents a highly significant health concern, since this disorder begins early in life and leads to long-term complications. In contrast, Type 2 DM is recognized as the etiology of over 80 percent of all diabetics and is dramatically increasing in incidence as a result of changes in human behavior and increased body mass index. Yet, the pathological consequences of these disorders that involve the both the neuronal and vascular systems are intimately linked through the pathways that mediate oxidative stress. Here we highlight some of the relevant oxidative pathways that determine insulin resistance through reactive oxygen species, mitochondrial dysfunction, uncoupling proteins, and endoplasmic reticulum stress. These pathways are ultimately linked to protein kinase B (Akt) and the insulin signaling pathways that determine the initial onset of glucose intolerance and the subsequent course to apoptotic cell injury. Through the elucidation of these targets, improvement in current strategies as well as the development of future clinical applications can move forward for both the prevention and treatment of Type 1 and Type 2 DM.
Keywords: Apoptosis, Akt, diabetes, endoplasmic reticulum stress, erythropoietin, exercise, mitochondria, oxidative stress, uncoupling proteins
THE GLOBAL SCOPE OF DIABETES MELLITUS
Diabetes mellitus (DM) is a condition that leads to elevated levels of serum glucose and is believed to affect at least 16 million Americans and approximately 170 million individuals worldwide (Quinn, 2001). When data is extrapolated on the prevalence of diabetes for the year 2030, approximately 366 million individuals may be afflicted by the disease. These projections are considered to be underestimated, since they assume levels of obesity will remain constant (Wild et al., 2004). Type 2 DM is recognized as the etiology of over 80 percent of all diabetics and is dramatically increasing in incidence as a result of changes in human behavior and increased body mass index (Laakso, 2001). However, it is Type 1 insulin-dependent diabetes mellitus (IDDM) which accounts for only 5-10 percent of all diabetics that represents a highly significant health concern, since this disorder begins early in life and leads to long-term complications throughout the body involving cardiovascular, renal, and nervous system disease (Daneman, 2006). Interestingly, disease of the nervous system can become the most debilitating and affect sensitive cognitive regions of the brain, such as the hippocampus that modulates memory function, resulting in significant functional impairment and dementia (Awad et al., 2004, Gerozissis, 2003). Furthermore, both focal and generalized neuropathies, especially in conjunction with vascular disease, can result in severe disability (Perkins and Bril, 2002). The incidence of undiagnosed diabetes and impaired glucose tolerance in the young raises further concerns. Individuals with impaired glucose tolerance are at 2.5 times greater risk for the development of diabetic complications than individuals with normal glucose tolerance (Harris and Eastman, 2000). As a result, healthcare costs for diabetic complications are a significant driver for government resource consumption with costs of $214.8 million for outpatient expenditures and $1.45 billion for inpatient expenditures (Maciejewski and Maynard, 2004). If one examines cognitive impairments resulting from diabetes in the general population that can mimic Alzheimer's disease (Chong et al., 2005d), annual costs equal $100 billion (Maiese and Chong, 2004, McCormick et al., 2001, Mendiondo et al., 2001).
Type 1 DM is associated with the presence of alleles of the Human leukocyte antigen (HLA) class II genes within the major histocompatibility complex (MHC). The disorder is an autoimmune disease resulting from inflammatory infiltration of the islets of Langerhans and the selective destruction of β-cells in the pancreas that leads to insulin loss (Bonner-Weir, 2000). Monogenic inheritance does not appear to lead to Type 1 DM with work illustrating that multiple loci with possible epistatic interactions among other loci may be responsible for genetic transmission (Bottino and Trucco, 2005, Davies et al., 1994). Yet, a variety of environmental factors also may play a significant role with Type 1 DM. For example, some studies have suggested that Type 1 DM in monozygotic twins can occur with a cumulative risk from birth to 35 years of age of 70% (Kyvik et al., 1995, Melanitou 2005). In addition, other work suggests a concordance between monozygotic twins to be approximately 50% (Redondo et al., 2001) illustrating that environmental factors form a platform for the predisposition of Type 1 DM.
In Type 1 DM, activation of T-cell clones that are capable of recognizing and destroying β-cells lead eventually to severe insulin deficiency. These T-cell clones are able to escape from control of the thymus during circumstances that yield high affinity for major histocompatibility complex (MHC) molecules with T-cell receptors but incorrect low affinity for self-peptides. Once released into the bloodstream, these T-cell clones can become activated to destroy self-antigens. Upon initial diagnosis, approximately ninety percent of individuals with Type 1 DM have elevated titers of autoantibodies (Type 1A DM). The remaining ten percent of Type 1 DM individuals do not have serum autoantobodies and are described as having maturity-onset diabetes of the young (MODY) that can be a result of β-cell dysfunction with autosomal-dominant inheritance (Type 1B DM) (Permutt et al., 2005). Other variables reported in patients with Type 1 DM include the presence of insulin resistance that is usually characteristic of Type 2 DM and can lead to neurological and vascular disease (Kernan et al., 2002, Orchard et al., 2003). It is important to note that there is an converse overlap with Type 1 and Type 2 DM, since almost ten percent of Type 1 DM patients may have elevated serum autoantibodies (Pietropaolo et al., 2000).
Loss of autoimmunity in Type 1 DM can be precipitated by a number of exogenous events, such as the exposure to infectious agents (Luppi et al., 1995). In most cases but not all, the insulin gene (INS) and the human MHC or HLA complex are believed to contain the loci with IDDM1 and IDDM2 to account for the susceptibility to Type 1 DM with defective antigen presentation (Awata et al., 1997, Baisch et al., 1990). Interestingly, a HLA class II molecule has been linked to Type 1 DM inheritance. Specifically, HLA-DQ that lacks a charged aspartic acid (Asp-57) in the β-chain is believed to lead to the ineffective presentation of autoantigen peptides during thymus selection of T-cells (Todd et al., 1987). Animal models that involve the nonobese diabetic (NOD) mice further support these findings, since these mice spontaneously develop diabetes with the human predisposing HLA-DQ corresponding molecule of H2 I-Ag. Yet, NOD mice without H2 I-Ag do not develop diabetes (Lund et al., 1990).
In contrast to Type 1 DM, Type 2 DM, also called noninsulin-dependent diabetes mellitus (NIDDM), is characterized by insulin resistance with significant metabolic dysfunction that include obesity, impaired insulin function and secretion, and increased endogenous glucose output. It should be noted that although insulin resistance forms the basis for the development of Type 2 DM, elevated serum glucose levels are also a result of the concurrent impairment in insulin secretion. This abnormal insulin secretion may be a result of defective β-cell function, chronic exposure to free fatty acids and hyperglycemia, and the loss of inhibitory feedback through plasma glucagon levels (Del Prato and Marchetti, 2004). Type 2 DM is the most prevalent form of diabetes and generally occurs more often in individuals over 40 years of age. The disorder is characterized by a progressive deterioration of glucose tolerance with early β-cell compensation for insulin resistance (achieved by β-cell hyperplasia) and subsequently followed by progressive decrease in β-cells mass. Another type of diabetes, gestational diabetes mellitus (GDM), is defined as a state of glucose intolerance during some cases of pregnancy, but usually subsides after delivery.
THE PRECIPITANTS AND CONSEQUENCES OF INSULIN RESISTANCE
Insulin resistance or defective insulin action occurs when a normal level of insulin produces a subnormal physiologic response. Skeletal muscle and liver are two of the primary insulin-responsive organs responsible for maintaining normal glucose homeostasis. Insulin normally lowers the level of blood glucose through suppression of hepatic glucose production and stimulation of peripheral glucose uptake, but a dysfunction in any step of this process can result in insulin resistance. Exposure to high concentrations of glucose and insulin results in insulin resistance, a characteristic feature of Type 2 DM.
Hyperglycemia also has been associated with oxidative stress and increased levels of reactive oxygen species have been proposed to lead to insulin resistance. Oxidative stress occurs when oxygen free radicals are generated in excess through the reduction of oxygen. Reactive oxygen species (ROS) consist of oxygen free radicals and associated entities that include superoxide free radicals, hydrogen peroxide, singlet oxygen, nitric oxide (NO), and peroxynitrite (Chong et al., 2005c). Most species are produced at low levels during normal physiological conditions and are scavenged by endogenous antioxidant systems that include superoxide dismutase (SOD), glutathione peroxidase, catalase, and small molecule substances such as vitamins C and E. Superoxide radical is the most commonly occurring oxygen free radical that produces hydrogen peroxide by dismutation. Other enzymes capable of producing superoxide are xanthine oxidase, NADPH oxidases and cytochrome P450. Superoxide produces hydrogen peroxide through the Haber-Weiss reaction in the presence of ferrous iron by manganese (Mn)-SOD or copper (Cu)-SOD. In the presence of transition elements, a reaction of hydrogen peroxide with superoxide results in the formation of hydroxyl radical, the most active oxygen free radical. Hydroxyl radical alternatively may be formed through an interaction between superoxide radical and NO (Fubini and Hubbard, 2003). NO interacts with superoxide radical to form peroxynitrite that can further lead to the generation of peroxynitrous acid. Hydroxyl radical is produced from the spontaneous decomposition of peroxynitrous acid. NO itself and peroxynitrite are also recognized as active oxygen free radicals. In addition to directly altering cellular function, NO may work through peroxynitrite that is potentially considered a more potent radical than NO itself (Pfeiffer et al., 2001).
Oxidative stress represents an important pathway for the destruction of cells (Chong et al., 2005d, Li et al., 2006a). The production of ROS can lead to cell injury through cell membrane lipid destruction and cleavage of DNA (Vincent and Maiese, 1999, Wang et al., 2003). ROS result in the peroxidation of cellular membrane lipids (Siu and To, 2002), peroxidation of docosahexaenoic acid, a precursor of neuroprotective docosanoids (Greco and Minghetti, 2004), the cleavage of DNA during the hydroxylation of guanine and methylation of cytosine (Lee et al., 2002), and the oxidation of proteins that yield protein carbonyl derivatives and nitrotyrosine (Adams et al., 2001). In addition to the detrimental effects to cellular integrity, ROS can inhibit complex enzymes in the electron transport chain of the mitochondria resulting in the blockade of mitochondrial respiration (Yamamoto et al., 2002).
The pathogenic effect of hyperglycemia, possibly in concert with free fatty acid release, is mediated to a significant extent via increased production of ROS. In addition to their ability to directly inflict damage on macromolecules, ROS indirectly lead to tissue damage by activating a number of cellular stress-sensitive pathways. Oxidative stress may decrease insulin sensitivity and injure the insulin-producing cells within the pancreas. For example, ROS can penetrate through cell membranes and cause damage to β-cells of pancreas (Chen et al., 2005, Lepore et al., 2004). In addition, free fatty acids which can lead to ROS, have been shown to also contribute to mitochondrial DNA damage and impaired pancreatic β-cell function (Rachek et al., 2006). Oxidative stress also is believed to modify a number of the signaling pathways within a cell that can ultimately lead to insulin resistance. As a result, it is possible that activation of oxidative stress pathways plays a key role in the development of not only the late complications in Type 1 and Type 2 DM, but also insulin resistance.
A number of oxidative stress pathways responsible for insulin resistance can be highlighted. In non-diabetic rats, hyperglycemia was shown to lead to a significant decrease in insulin-stimulated glucose uptake, a significant increase in muscle protein carbonyl content (used as an indicator of oxidative stress), and elevated levels of malondialdehyde and 4-hydroxynonenal as an indictor of lipid peroxidation (Haber et al., 2003). These biological markers of oxidative stress and insulin resistance were normalized during the application of the antioxidant N-acetylcysteine or taurine to suggest that oxidative stress contributes to the pathogenesis of hyperglycemia-induced insulin resistance (Haber et al., 2003). Furthermore, hyperglycemia can lead to increased production of ROS in endothelial cells, liver and pancreatic β-cells (Ceriello et al., 1996, Ihara et al., 1999, Ling et al., 2003, Yano et al., 2004). In a model of nonobese Type 2 DM, higher levels of 8-OHdG and HNE-modified proteins in the pancreatic beta-cells of diabetic rats than in the controls were observed with levels increasing with age and fibrosis of the pancreatic islets (Ihara et al., 1999). Elevated glucose has been shown to increase antioxidant enzyme levels in human endothelial cells, suggesting that elevated glucose levels may produce an oxidative stress in the cells (Ceriello et al., 1996). Even during brief period of hyperglycemia, ROS can be generated to lead to oxidative stress such as in vascular cells (Yano et al., 2004) or in neurons (Fig. 1). Recent clinical correlates support these experimental studies with acute glucose elevation acting as a trigger on oxidative stress than chronic sustained hyperglycemia (Monnier et al., 2006).
Fig. 1. Acute hyperglycemia leads to neuronal injury.
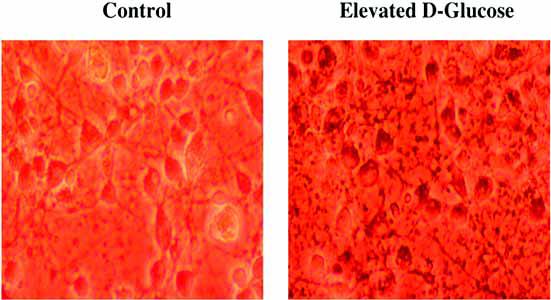
Representative hippocampal neurons obtained from E-19 Sprague-Dawley rat pups were incubated in L-15 growth medium with free serum containing elevated D-glucose of 50 mM for 24 hours at 37°C in a humidified atmosphere of 5% CO2 and 95% room air. Neuronal cell survival was determined by trypan blue exclusion method and reveals significantly increased dye uptake in injured neurons during hyperglycemia (right panel), but not in untreated control neurons (left panel). Note: Acute neuronal injury with elevated glucose can occur at significantly lower glucose concentrations of 20 mM, but a glucose concentration of 50 mM was chosen for dramatic visualization of cell injury.
Although the precise role of insulin resistance in neuronal injury remains to be established, it has been shown that direct insulin stimulation of neurons may reverse diabetic neuropathy (Brussee et al., 2004). It suggested that defective insulin signaling in peripheral neurons could partly contribute to the development of diabetic neuropathy. Direct intrathecal delivery of doses insufficient to reduce glycemia of insulin or equimolar insulin growth factor-I improved and reversed slowing of motor and sensory conduction velocity in streptozotocin (STZ) -induced diabetic rats. However, intrathecal saline or subcutaneous insulin did not have this effect. Interestingly, the neurotrophic and neuroprotective actions of insulin appear to enhance mitochondrial inner membrane potential and increase ATP levels (Huang et al., 2005). Huang et al. used real-time whole cell fluorescence video microscopy to analyze mitochondrial inner membrane potential (Δψm) in cultured adult sensory neurons. Compared with control, insulin and other neurotrophic factors induced a 2-fold increase in Δψm over a 24 hour period. Insulin was also found to phosphorylate the cAMP response element binding protein (CREB).
Proper cellular function during a number of disorders including diabetes requires the maintenance of mitochondrial membrane potential (Maiese et al., 2005a). Mitochondria are a significant source of superoxide radicals that are associated with oxidative stress (Smeitink et al., 2004). Blockade of the electron transfer chain at the flavin mononucleotide group of complex I (NADPH ubiquinone oxidoreductase) or at the ubiquinone site of complex III (ubiquinone-cytochrome c reductase) results in the active generation of free radicals which can impair mitochondrial electron transport and enhance free radical production (Floyd and Hensley, 2002). Furthermore, mutations in the mitochondrial genome have been associated with the potential development of a host of disorders, such as hypertension, hypercholesterolemia, and hypomagnesemia (Wilson et al., 2004). Loss of mitochondrial membrane potential during a variety of conditions through the opening of the mitochondrial permeability transition pore represents a significant determinant for cell injury and the subsequent induction of the apoptotic cascade (Chong et al., 2003a, Deng et al., 2002, Ieraci and Herrera, 2006, Lin et al., 2000).
As a result, mitochondrial dysfunction plays a role in the development of diabetes and insulin resistance. In patients with Type 2 DM, ADH:O(2) oxidoreductase activity and citrate synthase activity were found to be depressed with skeletal muscle mitochondria being smaller than in control subjects (Kelley et al., 2002). Recently, a decrease in the levels of mitochondrial proteins and mitochondrial DNA in adipocytes was correlated with the development of Type 2 DM (Choo et al., 2006). Furthermore, insulin resistance in the elderly has been associated with elevation in fat accumulation, and reduction in mitochondrial oxidative and phosphorylation activity (Petersen et al., 2003). In addition, an association exists with insulin resistance and the impairment of intramyocellular fatty acid metabolism in young insulin-resistance offspring of parents with Type 2 DM (Petersen et al., 2004).
In addition to the importance of the presence and integrity of mitochondria, uncoupling proteins (UCPs) are carriers expressed in the mitochondrial inner membrane that uncouple oxygen consumption by the respiratory chain from ATP synthesis and can play a significant role during diabetes. These proteins are a family of carrier proteins found in the inner membrane of mitochondria that can control ROS. They catalyze an inducible proton conductance and disperse the proton electrochemical potential gradient across the mitochondrial inner membrane (Douette and Sluse, 2006). This uncoupling of respiration results in the activation of substrate oxidation and dissipation of oxidation energy as heat instead of ATP. In addition, mild uncoupling of respiration may play a significant role in regulating ATP synthesis, and fatty acids and glucose oxidation. Members of UCP family include UCP-1, 2,3,4,5 in mammals (Criscuolo et al., 2006). Members of the UCP family are distinctly distributed among different tissues. UCP-1 and UCP-4 are exclusively expressed in brown adipose tissue and in the brain, respectively. UCP-2 is expressed in most tissues while UCP-5 is present in multiple tissues with an especially high level in the brain and testis. UCP-3 is expressed predominantly in skeletal muscle.
UCP-1 is important for controlling the dissipation of oxidation energy as heat. Overexpression of UCP-1 is potentially beneficial for diabetes by reducing excessive energy in obesity. Indeed, muscle-specific overexpression of UCP for skeletal muscle can increase energy expenditure and enhance insulin action to protect in animal models from high-fat diet induced insulin resistance (Li et al., 2000). In addition, skeletal muscle respiratory uncoupling was also shown to enhance insulin sensitivity in genetic obesity (Bernal-Mizrachi et al., 2002). Similar beneficial effects were also observed in specific overexpression of UCP-1 in white adipose tissues (Kopecky et al., 1995).
Uncoupling protein 2 (UCP-2) and uncoupling protein 3 (UCP-3) are expressed in tissues important for thermogenesis and/or in substrate oxidation, such as adipose tissue and skeletal muscles. UCP-2 is a member of the multigenic UCP family that is expressed in a wide range of tissues and organs. Possible functions of UCP-2 include control of ATP synthesis, regulation of fatty acid metabolism and control of reactive oxygen species production. UCP-2 expression in tissues involved in lipid and energy metabolism and mapping of the gene to a region linked to obesity and hyperinsulinemia has furthered investigations with UCP-2 and diabetes. In human adipose tissue and skeletal muscle, UCP-2 expression is increased during fasting. The carrier was shown to be under the control of fatty acids and thyroid hormones in vivo. There are reports implicating UCP-2 in the pathogenesis of diabetes. Overexpression of UCP-2 in isolated pancreatic islets results in decreased ATP content and blunted glucose-stimulated insulin secretion while UCP-2-deficient mice show an increased ATP level and an enhanced insulin secretion. Lack of UCP-2 dramatically improves insulin secretion and decreases hyperglycemia in leptin-deficient mice (Zhang et al., 2001). Furthermore, overexpression of UCP-2 could enhance resistance of beta-cells hydrogen peroxide toxicity (Li et al., 2001). It is of note that elevated expression of UCP-2 was also reported to exert substantial negative regulation of β-cell insulin secretion and contributes to the impairment of β-cell function (Chan et al., 2001). A role for UCP-3 in carbohydrate metabolism and in Type 2 DM also has been suggested. Mice overexpressing UCP-3 in skeletal muscle showed reduced fasting plasma glucose levels, improved glucose tolerance after an oral glucose load, and reduced fasting plasma insulin levels. It was found that UCP-3 levels were at least twice as low in patients with Type 2 DM compared with controls, suggesting a role for UCP-3 in glucose homeostasis (Schrauwen et al., 2001). In addition, UCP-3 may function to facilitate fatty acid oxidation and minimize ROS production (MacLellan et al., 2005).
Endoplasmic reticulum (ER) stress also is a feature of peripheral insulin resistance. ER stress is characterized by the accumulation of unfolded proteins in the lumen of the ER (Robertson et al., 2006). It is associated with an unfolded protein response (UPR) or by excessive protein traffic that may be triggered by events such as viremia. UPR regulates ER function and functions to coordinate the activity and participation of the processing and degradation pathways for unfolded proteins (Hampton, 2000). Obesity is associated with induction of ER stress predominantly in liver and adipose tissues. Under hyperglycemic conditions, the production of glucosamine by hexosamine pathway may initiate ER stress. This stress can promote a c-Jun N-terminal kinase (JNK)-dependent serine phosphorylation of insulin receptor substrate 1 (IRS-1), which results in suppression of insulin-receptor signaling pathway. In addition, the absence of X-box-binding protein-1 (XBP-1), a transcription factor that modulates the ER stress response, promotes insulin resistance in mice (Ozcan et al., 2004). Furthermore, gene silencing of oxygen-regulated protein 150 (ORP150), a molecular chaperone resident in the ER, has been shown to promote insulin resistance in non-diabetic control mice. In contrast, overexpression of ORP150 significantly decreases insulin resistance and markedly improves glycemic control in the liver of obese diabetic mice. Although not entirely clear, these observations may be associated with phosphorylation state of IRS-1 and protein kinase B (Akt) as well as the expression levels of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase (Nakatani et al., 2005).
TARGETED PATHWAYS OF AKT AND ITS EFFECTORS AGAINST OXIDATIVE STRESS AND DIABETES
The protein Akt, also known as protein kinase B (Chong et al., 2004b, Chong et al., 2005b) is ubiquitously expressed in mammals. Three family members of this serine/threonine kinase are now known to exist that were termed Akt after the molecular cloning of the oncogene v-Akt and two human homologues (Staal, 1987, Staal et al., 1988). They are PKBα or Akt1, PKBβ or Akt2, and PKBγ or Akt3 (Chong et al., 2005b). Akt is part of the AGC (cAMP-dependent kinase/protein kinase G/protein kinase C) superfamily of protein kinases and consists of three functional domains.
Defective glucose transport and insulin resistance are believed to be closely linked to the activity of Akt. Insulin controls glucose transport through insulin receptor substrate-1 (IRS-1) and in part through Akt as well as atypical protein kinase C. Perturbations in this pathway can substantially alter hepatic glucose output and triglyceride release (Farese et al., 2005). Akt signaling also has a central function to insulin sensitivity in diabetes. In response to insulin, Akt leads to glucose uptake in adipocytes through its stimulation of glucose transporter 4 (GLUT-4) translocation and increased synthesis of GLUT-1 (Kohn et al., 1996). The elevated glucose influx is accompanied by increased lipid synthesis and decreased glycogen synthesis. Conversely, impairment of Akt kinase activity was accompanied by impairment in insulin-stimulated glucose transport in muscle and adipocytes both from obese rats and patients with diabetes (Carvalho et al., 2000, Krook et al., 1998). In addition, mutations in Akt2 are linked to the development of severe insulin resistance and diabetes (George et al., 2004). Reduced activity of Akt1 also leads to impaired glucose tolerance and diabetes (Bernal-Mizrachi et al., 2004).
Akt also is considered a central modulator to prevent apoptotic cell injury during late genomic DNA destruction and early apoptotic signaling with membrane phosphatidylserine (PS) exposure (Chong et al., 2005d, Maiese and Chong, 2004). Overexpression of Akt in cells prevents apoptosis during growth factor withdrawal (Datta et al., 1997) and paradigms with oxidative stress (Chong et al., 2004a, Li et al., 2006b). Further work has demonstrated that Akt is necessary for the survival of cells, since expression of a dominant-negative Akt or inhibition of phosphoinositide 3 kinase (PI 3-K), necessary for Akt activation, precipitates cell death during oxidative stress (Kang et al., 2003a, Kang et al., 2003b). Increased Akt activity can foster cell survival during free radical exposure (Chong et al., 2003b, Matsuzaki et al., 1999), matrix detachment (Rytomaa et al., 2000), neuronal axotomy (Namikawa et al., 2000), DNA damage (Chong et al., 2004a, Chong et al., 2002, Henry et al., 2001, Kang et al., 2003a), anti-Fas antibody administration (Suhara et al., 2001), oxidative stress (Chong et al., 2003b, Kang et al., 2003a, Kang et al., 2003b, Yamaguchi and Wang, 2001), hypoxic preconditioning (Wick et al., 2002), ß-amyloid (Aß) exposure (Martin et al., 2001), metabotropic receptor signaling (Chong et al., 2005a, Chong et al., 2006, Maiese et al., 2005a), and cell metabolic pathways (Chong et al., 2005e, Maiese and Chong, 2003). Activation of Akt also can prevent membrane PS exposure on injured cells and block the activation of microglia during oxidative stress (Chong et al., 2005a, Kang et al., 2003a, Kang et al., 2003b).
In a number of experimental models, modulation of Akt activity can critically affect cell survival during hyperglycemia and the outcome of diabetic complications. During chronic hyperglycemic stress, inhibition of Akt leads to increased endothelial cell injury (Okouchi et al., 2006). In STZ diabetic rats, vascular control and integrity (Shah and Singh, 2006) as well as mitochondrial function (Di Noia et al., 2006) were improved with an agent that leads to activation of Akt. Furthermore, ER stress inducers can lead to dephosphorylation and inactivation of Akt with subsequent cell death (Hyoda et al., 2006). On the converse side, overexpression of Akt, such as in endothelial cells, can protect cells from injury during elevated glucose concentrations (Varma et al., 2005).
Given the therapeutic potential of targeting Akt to treat both Type 1 and Type 2 DM and the complications of these disorders, recent work brings to light a number of options. Physical exercise is one of the important lifestyle interventions and generally recommended for patient with diabetes. Regular exercise has shown beneficial effects on glycemic control, weight loss and insulin resistance. An exercise results in pronounced improvement in glycemic control usually over relatively short of time (Maiorana et al., 2002). Furthermore, in diet-induced obesity rats, a single session of exercise improved insulin sensitivity by reversing the increased activity of protein tyrosine phosphatase-1B (PTP1B) and enhanced serine phosphorylation of IRS-1 insulin signaling (Ropelle et al., 2006). Other mechanisms that may account for the benefits of physical activity during diabetes involve Akt. In the immediate periods following acute exercise, Akt phosphorylation and activity has been shown to be increased (Howlett et al., 2006).
Combined with the benefits of exercise, erythropoietin (EPO) may another consideration for diabetic therapeutic strategies. EPO is a trophic factor that is approved by the Food and Drug Administration for the treatment of anemia, but a body of recent work has revealed that EPO is not required only for erythropoiesis and that EPO and its receptor exist in other organs and tissues outside of the liver and the kidney, such as the brain and heart. As a result, EPO has been identified as a possible candidate for disorders that involve both cardiac and nervous system diseases (Maiese et al., 2004, Maiese et al., 2005c). Protection by EPO in a number of cellular systems can block apoptotic injury from a number of sources, such as reduced or absent oxygen tension, excitotoxicity, and free radical exposure. Since apoptotic injury involves both genomic DNA destruction and membrane PS externalization that involve stroke, inflammation, Alzheimer's disease, and myocardial infarction (Han and Suk, 2005, Li et al., 2004, Li et al., 2006b), EPO can be considered unusual in its ability to prevent both the exposure of membrane PS residues and inhibit the committed stages of genomic DNA destruction in several experimental models to potentially offer protection against microglial phagocytosis and thrombotic injury (Cai and Semenza, 2004, Chong et al., 2003b, Chong et al., 2002, Chong et al., 2003c, Grimm et al., 2002, Parsa et al., 2003).
Interestingly, EPO leads to the phosphorylation of Akt. Once activated, Akt can confer protection against genomic DNA degradation and membrane PS exposure (Chong et al., 2002, Kang et al., 2003b, Wick et al., 2002). Up-regulation of Akt activity during injury paradigms, such as N-methyl-D-aspartate toxicity (Dzietko et al., 2004), cardiomyocyte ischemia (Parsa et al., 2003), hypoxia (Chong et al., 2002), and oxidative stress (Chong et al., 2003a, Chong et al., 2003b, Chong et al., 2003c), is vital for EPO protection, since prevention of Akt phosphorylation blocks cellular protection and anti-inflammatory mechanisms by EPO (Chong et al., 2003a, Chong et al., 2003b, Chong et al., 2003c). EPO employs the Akt pathway to prevent apoptosis by maintaining mitochondrial membrane potential (Δψm), preventing the cellular release of cytochrome c, and modulating caspase activity (Chong et al., 2003a, Chong et al., 2003b, Chong et al., 2002).
In clinical studies, plasma EPO is often low in diabetic patients with (Mojiminiyi et al., 2006) or without anemia (Symeonidis et al., 2006). In these diabetic patients, levels of EPO appear to be determined by hemoglobin levels and degree of microalbuminuria (Mojiminiyi et al., 2006). In addition, the failure to produce erythropoietin in response to a declining hemoglobin level represents an impaired EPO response in diabetic patients (Thomas et al., 2005). In light of the ability of EPO to modulate Akt activity and provide cellular protection during oxidative stress, EPO may be efficacious in patients with diabetes. Investigations involving subcutaneous EPO in diabetics and non-diabetics with severe, resistant congestive heart failure have shown to decrease fatigue, increase left ventricular ejection fraction, and significantly decrease the number of hospitalization days (Silverberg et al., 2003). Yet, one must be cautious with the introduction of even known therapies for new applications. Some studies suggest that elevated plasma levels of EPO independent of hemoglobin concentration can have a poor prognostic value in individuals with congestive heart failure (van der Meer et al., 2004). The use of EPO in patients with uncontrolled hypertension is contraindicated, since both acute and long-term administration of EPO can precipitate hypertensive emergencies. Maintenance treatment with EPO in cancer patients receiving chemotherapy has been associated with nonfatal myocardial infarction, vascular thrombosis, pyrexia, vomiting, shortness of breath, paresthesias, and upper respiratory tract infection (Henry et al., 2004, Maiese et al., 2005b).
DIRECTIONS FOR THE FUTURE
Knowledge gained in understanding the complex cellular and systemic processes of diabetes provide essential insight into the pathogenesis of diabetes and its complications. Oxidative stress is a principal mechanism in the progression of diabetes and its complications and actively leads to cellular injury in both neuronal and vascular cells that can precede the onset of many diabetic complications. Elucidating the mechanisms that determine insulin resistance as well as glucose signaling and cell survival via Akt may begin to lay new strategies for both the treatment and prevention of Type 1 and Type 2 DM.
ACKNOWLEDGEMENTS
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, Nelson Foundation Award, NIH NIEHS (P30 ES06639), and NIH NINDS/NIA (NS053946).
REFERENCES
- Adams S, Green P, Claxton R, Simcox S, Williams MV, Walsh K, Leeuwenburgh C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front Biosci. 2001;6:A17–24. doi: 10.2741/adams. [DOI] [PubMed] [Google Scholar]
- Awad N, Gagnon M, Messier C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J Clin Exp Neuropsychol. 2004;26:1044–80. doi: 10.1080/13803390490514875. [DOI] [PubMed] [Google Scholar]
- Awata T, Kurihara S, Kikuchi C, Takei S, Inoue I, Ishii C, Takahashi K, Negishi K, Yoshida Y, Hagura R, Kanazawa Y, Katayama S. Evidence for association between the class I subset of the insulin gene minisatellite (IDDM2 locus) and IDDM in the Japanese population. Diabetes. 1997;46:1637–42. doi: 10.2337/diacare.46.10.1637. [DOI] [PubMed] [Google Scholar]
- Baisch JM, Weeks T, Giles R, Hoover M, Stastny P, Capra JD. Analysis of HLA-DQ genotypes and susceptibility in insulin-dependent diabetes mellitus. N Engl J Med. 1990;322:1836–41. doi: 10.1056/NEJM199006283222602. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi C, Weng S, Li B, Nolte LA, Feng C, Coleman T, Holloszy JO, Semenkovich CF. Respiratory uncoupling lowers blood pressure through a leptin-dependent mechanism in genetically obese mice. Arterioscler Thromb Vasc Biol. 2002;22:961–8. doi: 10.1161/01.atv.0000019404.65403.71. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Fatrai S, Johnson JD, Ohsugi M, Otani K, Han Z, Polonsky KS, Permutt MA. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114:928–36. doi: 10.1172/JCI20016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab. 2000;11:375–8. doi: 10.1016/s1043-2760(00)00305-2. [DOI] [PubMed] [Google Scholar]
- Bottino R, Trucco M. Multifaceted therapeutic approaches for a multigenic disease. Diabetes. 2005;54(Suppl 2):S79–86. doi: 10.2337/diabetes.54.suppl_2.s79. [DOI] [PubMed] [Google Scholar]
- Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes. 2004;53:1824–30. doi: 10.2337/diabetes.53.7.1824. [DOI] [PubMed] [Google Scholar]
- Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109:2050–3. doi: 10.1161/01.CIR.0000127954.98131.23. [DOI] [PubMed] [Google Scholar]
- Carvalho E, Rondinone C, Smith U. Insulin resistance in fat cells from obese Zucker rats--evidence for an impaired activation and translocation of protein kinase B and glucose transporter 4. Mol Cell Biochem. 2000;206:7–16. doi: 10.1023/a:1007009723616. [DOI] [PubMed] [Google Scholar]
- Ceriello A, dello Russo P, Amstad P, Cerutti P. High glucose induces antioxidant enzymes in human endothelial cells in culture. Evidence linking hyperglycemia and oxidative stress. Diabetes. 1996;45:471–7. doi: 10.2337/diab.45.4.471. [DOI] [PubMed] [Google Scholar]
- Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, Tsushima RG, Pennefather PS, Salapatek AM, Wheeler MB. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. 2001;50:1302–10. doi: 10.2337/diabetes.50.6.1302. [DOI] [PubMed] [Google Scholar]
- Chen H, Li X, Epstein PN. MnSOD and catalase transgenes demonstrate that protection of islets from oxidative stress does not alter cytokine toxicity. Diabetes. 2005;54:1437–46. doi: 10.2337/diabetes.54.5.1437. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang J, Li F, Maiese K. mGluRI targets microglial activation and selectively prevents neuronal cell engulfment through Akt and caspase dependent pathways. Curr Neurovasc Res. 2005a;2:197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004a;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J Cereb Blood Flow Metab. 2003a;23:320–30. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003b;138:1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106:2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid Redox Signal. 2004b;6:277–87. doi: 10.1089/152308604322899341. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005b;20:299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Group I metabotropic receptor neuroprotection requires Akt and its substrates that govern FOXO3a, Bim, and beta-catenin during oxidative stress. Curr Neurovasc Res. 2006;3:107–17. doi: 10.2174/156720206776875830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005c;75:207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer's disease. Brain Res Brain Res Rev. 2005d;49:1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res. 2003c;71:659–69. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Li F, Maiese K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through Akt, Bad, PARP, and mitochondrial associated “anti-apoptotic” pathways. Curr Neurovasc Res. 2005e;2:271–85. doi: 10.2174/156720205774322584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, Yoon YS, Yoon G, Choi KM, Ko YG. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia. 2006;49:784–91. doi: 10.1007/s00125-006-0170-2. [DOI] [PubMed] [Google Scholar]
- Criscuolo F, Mozo J, Hurtaud C, Nubel T, Bouillaud F. UCP2, UCP3, avUCP, what do they do when proton transport is not stimulated? Possible relevance to pyruvate and glutamine metabolism. Biochim Biophys Acta. 2006;1757:1284–91. doi: 10.1016/j.bbabio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Daneman D. Type 1 diabetes. Lancet. 2006;367:847–58. doi: 10.1016/S0140-6736(06)68341-4. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davies JL, Kawaguchi Y, Bennett ST, Copeman JB, Cordell HJ, Pritchard LE, Reed PW, Gough SC, Jenkins SC, Palmer SM, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature. 1994;371:130–6. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- Del Prato S, Marchetti P. Beta- and alpha-cell dysfunction in type 2 diabetes. Horm Metab Res. 2004;36:775–81. doi: 10.1055/s-2004-826163. [DOI] [PubMed] [Google Scholar]
- Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia MA, Van Driesche S, Palmieri F, Yang LM, Quan S, Goodman AI, Abraham NG. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome C oxidase activity in experimental diabetes. J Biol Chem. 2006;281:15687–93. doi: 10.1074/jbc.M510595200. [DOI] [PubMed] [Google Scholar]
- Douette P, Sluse FE. Mitochondrial uncoupling proteins: new insights from functional and proteomic studies. Free Radic Biol Med. 2006;40:1097–107. doi: 10.1016/j.freeradbiomed.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Dzietko M, Felderhoff-Mueser U, Sifringer M, Krutz B, Bittigau P, Thor F, Heumann R, Buhrer C, Ikonomidou C, Hansen HH. Erythropoietin protects the developing brain against N-methyl-D-aspartate receptor antagonist neurotoxicity. Neurobiol Dis. 2004;15:177–87. doi: 10.1016/j.nbd.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Standaert ML. Insulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): actions and defects in obesity and type II diabetes. Exp Biol Med (Maywood) 2005;230:593–605. doi: 10.1177/153537020523000901. [DOI] [PubMed] [Google Scholar]
- Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34:1507–16. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O'Rahilly S, Barroso I. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–8. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerozissis K. Brain insulin: regulation, mechanisms of action and functions. Cell Mol Neurobiol. 2003;23:1–25. doi: 10.1023/A:1022598900246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco A, Minghetti L. Isoprostanes as biomarkers and mediators of oxidative injury in infant and adult central nervous system diseases. Curr Neurovasc Res. 2004;1:341–354. doi: 10.2174/1567202043362036. [DOI] [PubMed] [Google Scholar]
- Grimm C, Wenzel A, Groszer M, Mayser H, Seeliger M, Samardzija M, Bauer C, Gassmann M, Reme CE. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat Med. 2002;8:718–24. doi: 10.1038/nm723. [DOI] [PubMed] [Google Scholar]
- Haber CA, Lam TK, Yu Z, Gupta N, Goh T, Bogdanovic E, Giacca A, Fantus IG. N-acetylcysteine and taurine prevent hyperglycemia-induced insulin resistance in vivo: possible role of oxidative stress. Am J Physiol Endocrinol Metab. 2003;285:E744–53. doi: 10.1152/ajpendo.00355.2002. [DOI] [PubMed] [Google Scholar]
- Hampton RY. ER stress response: getting the UPR hand on misfolded proteins. Curr Biol. 2000;10:R518–21. doi: 10.1016/s0960-9822(00)00583-2. [DOI] [PubMed] [Google Scholar]
- Han HS, Suk K. The function and integrity of the neurovascular unit rests upon the integration of the vascular and inflammatory cell systems. Curr Neurovasc Res. 2005;2:409–23. doi: 10.2174/156720205774962647. [DOI] [PubMed] [Google Scholar]
- Harris MI, Eastman RC. Early detection of undiagnosed diabetes mellitus: a US perspective. Diabetes Metab Res Rev. 2000;16:230–6. doi: 10.1002/1520-7560(2000)9999:9999<::aid-dmrr122>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Henry DH, Bowers P, Romano MT, Provenzano R. Epoetin alfa. Clinical evolution of a pleiotropic cytokine. Arch Intern Med. 2004;164:262–76. doi: 10.1001/archinte.164.3.262. [DOI] [PubMed] [Google Scholar]
- Henry MK, Lynch JT, Eapen AK, Quelle FW. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood. 2001;98:834–41. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- Howlett KF, Sakamoto K, Yu H, Goodyear LJ, Hargreaves M. Insulin-stimulated insulin receptor substrate-2-associated phosphatidylinositol 3-kinase activity is enhanced in human skeletal muscle after exercise. Metabolism. 2006;55:1046–52. doi: 10.1016/j.metabol.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Verkhratsky A, Fernyhough P. Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol Cell Neurosci. 2005;28:42–54. doi: 10.1016/j.mcn.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochem Biophys Res Commun. 2006;340:286–90. doi: 10.1016/j.bbrc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Ieraci A, Herrera DG. Nicotinamide Protects against Ethanol-Induced Apoptotic Neurodegeneration in the Developing Mouse Brain. PLoS Med. 2006;3:e101. doi: 10.1371/journal.pmed.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara Y, Toyokuni S, Uchida K, Odaka H, Tanaka T, Ikeda H, Hiai H, Seino Y, Yamada Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes. 1999;48:927–32. doi: 10.2337/diabetes.48.4.927. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003a;74:37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003b;64:557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–50. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kernan WN, Inzucchi SE, Viscoli CM, Brass LM, Bravata DM, Horwitz RI. Insulin resistance and risk for stroke. Neurology. 2002;59:809–15. doi: 10.1212/wnl.59.6.809. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–8. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- Kopecky J, Clarke G, Enerback S, Spiegelman B, Kozak LP. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J Clin Invest. 1995;96:2914–23. doi: 10.1172/JCI118363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook A, Roth RA, Jiang XJ, Zierath JR, Wallberg-Henriksson H. Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes. 1998;47:1281–6. doi: 10.2337/diab.47.8.1281. [DOI] [PubMed] [Google Scholar]
- Kyvik KO, Green A, Beck-Nielsen H. Concordance rates of insulin dependent diabetes mellitus: a population based study of young Danish twins. BMJ. 1995;311:913–7. doi: 10.1136/bmj.311.7010.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso M. Cardiovascular disease in type 2 diabetes: challenge for treatment and prevention. J Intern Med. 2001;249:225–35. doi: 10.1046/j.1365-2796.2001.00789.x. [DOI] [PubMed] [Google Scholar]
- Lee DH, O'Connor TR, Pfeifer GP. Oxidative DNA damage induced by copper and hydrogen peroxide promotes CG-->TT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res. 2002;30:3566–73. doi: 10.1093/nar/gkf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore DA, Shinkel TA, Fisicaro N, Mysore TB, Johnson LE, d'Apice AJ, Cowan PJ. Enhanced expression of glutathione peroxidase protects islet beta cells from hypoxia-reoxygenation. Xenotransplantation. 2004;11:53–9. doi: 10.1111/j.1399-3089.2004.00082.x. [DOI] [PubMed] [Google Scholar]
- Li B, Nolte LA, Ju JS, Han DH, Coleman T, Holloszy JO, Semenkovich CF. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat Med. 2000;6:1115–20. doi: 10.1038/80450. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Cell life versus cell longevity: the mysteries surrounding the NAD(+) precursor nicotinamide. Curr Med Chem. 2006a;13:883–95. doi: 10.2174/092986706776361058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Erythropoietin on a tightrope: balancing neuronal and vascular protection between intrinsic and extrinsic pathways. Neurosignals. 2004;13:265–89. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr Neurovasc Res. 2006b;3:187–201. doi: 10.2174/156720206778018758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V. Uncoupling protein-2 participates in cellular defense against oxidative stress in clonal beta-cells. Biochem Biophys Res Commun. 2001;282:273–7. doi: 10.1006/bbrc.2001.4577. [DOI] [PubMed] [Google Scholar]
- Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J Cereb Blood Flow Metab. 2000;20:1380–91. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- Ling PR, Mueller C, Smith RJ, Bistrian BR. Hyperglycemia induced by glucose infusion causes hepatic oxidative stress and systemic inflammation, but not STAT3 or MAP kinase activation in liver in rats. Metabolism. 2003;52:868–74. doi: 10.1016/s0026-0495(03)00057-x. [DOI] [PubMed] [Google Scholar]
- Lund T, O'Reilly L, Hutchings P, Kanagawa O, Simpson E, Gravely R, Chandler P, Dyson J, Picard JK, Edwards A, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature. 1990;345:727–9. doi: 10.1038/345727a0. [DOI] [PubMed] [Google Scholar]
- Luppi P, Rossiello MR, Faas S, Trucco M. Genetic background and environment contribute synergistically to the onset of autoimmune diseases. J Mol Med. 1995;73:381–93. doi: 10.1007/BF00240137. [DOI] [PubMed] [Google Scholar]
- Maciejewski ML, Maynard C. Diabetes-related utilization and costs for inpatient and outpatient services in the Veterans Administration. Diabetes Care. 2004;27(Suppl 2):B69–73. doi: 10.2337/diacare.27.suppl_2.b69. [DOI] [PubMed] [Google Scholar]
- MacLellan JD, Gerrits MF, Gowing A, Smith PJ, Wheeler MB, Harper ME. Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes. 2005;54:2343–50. doi: 10.2337/diabetes.54.8.2343. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor Neurol Neurosci. 2004;22:87–104. [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol Sci. 2003;24:228–32. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Li F. Driving cellular plasticity and survival through the signal transduction pathways of metabotropic glutamate receptors. Curr Neurovasc Res. 2005a;2:425–46. doi: 10.2174/156720205774962692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin and cancer. JAMA. 2005b;293:1858–1859. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004;25:577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005c;293:90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorana A, O'Driscoll G, Goodman C, Taylor R, Green D. Combined aerobic and resistance exercise improves glycemic control and fitness in type 2 diabetes. Diabetes Res Clin Pract. 2002;56:115–23. doi: 10.1016/s0168-8227(01)00368-0. [DOI] [PubMed] [Google Scholar]
- Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78:1000–8. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem. 1999;73:2037–46. [PubMed] [Google Scholar]
- McCormick WC, Hardy J, Kukull WA, Bowen JD, Teri L, Zitzer S, Larson EB. Healthcare utilization and costs in managed care patients with Alzheimer's disease during the last few years of life. J Am Geriatr Soc. 2001;49:1156–60. doi: 10.1046/j.1532-5415.2001.49231.x. [DOI] [PubMed] [Google Scholar]
- Melanitou E. The autoimmune contrivance: genetics in the mouse model. Clin Immunol. 2005;117:195–206. doi: 10.1016/j.clim.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mendiondo MS, Kryscio RJ, Schmitt FA. Models of progression in AD: predicting disability and costs. Neurology. 2001;57:943–4. doi: 10.1212/wnl.57.6.943. [DOI] [PubMed] [Google Scholar]
- Mojiminiyi OA, Abdella NA, Zaki MY, El Gebely SA, Mohamedi HM, Aldhahi WA. Prevalence and associations of low plasma erythropoietin in patients with Type 2 diabetes mellitus. Diabet Med. 2006;23:839–44. doi: 10.1111/j.1464-5491.2006.01893.x. [DOI] [PubMed] [Google Scholar]
- Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295:1681–7. doi: 10.1001/jama.295.14.1681. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–51. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20:2875–86. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okouchi M, Okayama N, Alexander JS, Aw TY. NRF2-dependent glutamate-L-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia- induced brain endothelial cell apoptosis. Curr Neurovasc Res. 2006;3:249–61. doi: 10.2174/156720206778792876. [DOI] [PubMed] [Google Scholar]
- Orchard TJ, Olson JC, Erbey JR, Williams K, Forrest KY, Smithline Kinder L, Ellis D, Becker DJ. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care. 2003;26:1374–9. doi: 10.2337/diacare.26.5.1374. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, Thompson RB, Petrofski JA, Annex BH, Stamler JS, Koch WJ. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003;112:999–1007. doi: 10.1172/JCI18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins BA, Bril V. Diagnosis and management of diabetic neuropathy. Curr Diab Rep. 2002;2:495–500. doi: 10.1007/s11892-002-0119-x. [DOI] [PubMed] [Google Scholar]
- Permutt MA, Wasson J, Cox N. Genetic epidemiology of diabetes. J Clin Invest. 2005;115:1431–9. doi: 10.1172/JCI24758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer S, Lass A, Schmidt K, Mayer B. Protein tyrosine nitration in mouse peritoneal macrophages activated in vitro and in vivo: evidence against an essential role of peroxynitrite. FASEB J. 2001;15:2355–64. doi: 10.1096/fj.01-0295com. [DOI] [PubMed] [Google Scholar]
- Pietropaolo M, Barinas-Mitchell E, Pietropaolo SL, Kuller LH, Trucco M. Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes. 2000;49:32–8. doi: 10.2337/diabetes.49.1.32. [DOI] [PubMed] [Google Scholar]
- Quinn L. Type 2 diabetes: epidemiology, pathophysiology, and diagnosis. Nurs Clin North Am. 2001;36:175–92. v. [PubMed] [Google Scholar]
- Rachek LI, Thornley NP, Grishko VI, LeDoux SP, Wilson GL. Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes. 2006;55:1022–8. doi: 10.2337/diabetes.55.04.06.db05-0865. [DOI] [PubMed] [Google Scholar]
- Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, Leslie RD. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia. 2001;44:354–62. doi: 10.1007/s001250051626. [DOI] [PubMed] [Google Scholar]
- Robertson LA, Kim AJ, Werstuck GH. Mechanisms linking diabetes mellitus to the development of atherosclerosis: a role for endoplasmic reticulum stress and glycogen synthase kinase-3. Can J Physiol Pharmacol. 2006;84:39–48. doi: 10.1139/Y05-142. [DOI] [PubMed] [Google Scholar]
- Ropelle ER, Pauli JR, Prada PO, de Souza CT, Picardi PK, Faria MC, Cintra DE, Fernandes MF, Flores MB, Velloso LA, Saad MJ, Carvalheira J. Reversal of diet-induced insulin resistance with a single bout of exercise: The role of PTP1B and IRS-1 serine phosphorylation. J Physiol. 2006;577:997–1007. doi: 10.1113/jphysiol.2006.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytomaa M, Lehmann K, Downward J. Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: inhibition by PKB/Akt but not Raf signalling. Oncogene. 2000;19:4461–8. doi: 10.1038/sj.onc.1203805. [DOI] [PubMed] [Google Scholar]
- Schrauwen P, Hesselink MK, Blaak EE, Borghouts LB, Schaart G, Saris WH, Keizer HA. Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2001;50:2870–3. doi: 10.2337/diabetes.50.12.2870. [DOI] [PubMed] [Google Scholar]
- Shah DI, Singh M. Possible role of Akt to improve vascular endothelial dysfunction in diabetic and hyperhomocysteinemic rats. Mol Cell Biochem. 2006 Jul 14; doi: 10.1007/s11010-006-9273-9. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Silverberg DS, Wexler D, Blum M, Tchebiner JZ, Sheps D, Keren G, Schwartz D, Baruch R, Yachnin T, Shaked M, Schwartz I, Steinbruch S, Iaina A. The effect of correction of anaemia in diabetics and non-diabetics with severe resistant congestive heart failure and chronic renal failure by subcutaneous erythropoietin and intravenous iron. Nephrol Dial Transplant. 2003;18:141–6. doi: 10.1093/ndt/18.1.141. [DOI] [PubMed] [Google Scholar]
- Siu AW, To CH. Nitric oxide and hydroxyl radical-induced retinal lipid peroxidation in vitro. Clin Exp Optom. 2002;85:378–82. [PubMed] [Google Scholar]
- Smeitink JAM, van den Heuvel L, Koopman WJH, Nijtmans LGJ, Ugalde C, Willems P. Cell biological consequences of mitochondrial NADH: Ubiquinone oxidoreductase deficiency. Curr Neurovasc Res. 2004;1:29–40. doi: 10.2174/1567202043480224. [DOI] [PubMed] [Google Scholar]
- Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA. 1987;84:5034–7. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal SP, Huebner K, Croce CM, Parsa NZ, Testa JR. The AKT1 proto-oncogene maps to human chromosome 14, band q32. Genomics. 1988;2:96–8. doi: 10.1016/0888-7543(88)90114-0. [DOI] [PubMed] [Google Scholar]
- Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fasmediated apoptosis via regulation of FLICE- inhibitory protein (FLIP) Circ Res. 2001;89:13–9. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- Symeonidis A, Kouraklis-Symeonidis A, Psiroyiannis A, Leotsinidis M, Kyriazopoulou V, Vassilakos P, Vagenakis A, Zoumbos N. Inappropriately low erythropoietin response for the degree of anemia in patients with noninsulin-dependent diabetes mellitus. Ann Hematol. 2006;85:79–85. doi: 10.1007/s00277-005-1102-9. [DOI] [PubMed] [Google Scholar]
- Thomas MC, Cooper ME, Tsalamandris C, MacIsaac R, Jerums G. Anemia with impaired erythropoietin response in diabetic patients. Arch Intern Med. 2005;165:466–9. doi: 10.1001/archinte.165.4.466. [DOI] [PubMed] [Google Scholar]
- Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- van der Meer P, Voors AA, Lipsic E, Smilde TD, van Gilst WH, van Veldhuisen DJ. Prognostic value of plasma erythropoietin on mortality in patients with chronic heart failure. J Am Coll Cardiol. 2004;44:63–7. doi: 10.1016/j.jacc.2004.03.052. [DOI] [PubMed] [Google Scholar]
- Varma S, Lal BK, Zheng R, Breslin JW, Saito S, Pappas PJ, Hobson RW, 2nd, Duran WN. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:H1744–51. doi: 10.1152/ajpheart.01088.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Nitric oxide induction of neuronal endonuclease activity in programmed cell death. Exp Cell Res. 1999;246:290–300. doi: 10.1006/excr.1998.4282. [DOI] [PubMed] [Google Scholar]
- Wang JY, Shum AY, Ho YJ. Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J Neurosci Res. 2003;72:508–19. doi: 10.1002/jnr.10597. [DOI] [PubMed] [Google Scholar]
- Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–7. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–4. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20:7779–86. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Maruyama W, Kato Y, Yi H, Shamoto-Nagai M, Tanaka M, Sato Y, Naoi M. Selective nitration of mitochondrial complex I by peroxynitrite: involvement in mitochondria dysfunction and cell death of dopaminergic SH-SY5Y cells. J Neural Transm. 2002;109:1–13. doi: 10.1007/s702-002-8232-1. [DOI] [PubMed] [Google Scholar]
- Yano M, Hasegawa G, Ishii M, Yamasaki M, Fukui M, Nakamura N, Yoshikawa T. Short-term exposure of high glucose concentration induces generation of reactive oxygen species in endothelial cells: implication for the oxidative stress associated with postprandial hyperglycemia. Redox Rep. 2004;9:111–6. doi: 10.1179/135100004225004779. [DOI] [PubMed] [Google Scholar]
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–55. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]