

Abstract
Leptin is an adiopokine that plays a pivotal role in the progression of liver fibrogenesis and carcinogenesis. Recently, leptin was shown to be mitogenic in human liver cancer cell lines HepG2 and Huh7. Whether leptin can act as a mitogen in normal hepatocytes is unclear. Methionine adenosyltransferase (MAT) is an essential enzyme that catalyzes the formation of S-adenosylmethionine (SAMe), the principal methyl donor and precursor of polyamines. Two genes (MAT1A and MAT2A) encode for the catalytic subunit of MAT, whereas a third gene (MAT2β) encodes for a regulatory subunit that modulates the activity of MAT2A-encoded isoenzyme. The aims of this study were to examine whether leptin’s mitogenic activity involves MAT2A and MAT2β and whether this can be modulated. We found that leptin is mitogenic in HepG2 cells but not in primary human or mouse hepatocytes. Leptin induced the expression of MAT2A and MAT2β in HepG2 cells and normal human and mouse hepatocytes, but although it increased SAMe level in HepG2 cells, it had no effect on SAMe level in normal hepatocytes. Leptin-mediated induction of MAT genes and growth in HepG2 cells required activation of extracellular signal-regulated kinase and phosphatidylinositol-3-kinase signaling pathways. Treatment with SAMe or its metabolite methylthioadenosine (MTA) lowered expression of MAT2A and MAT2β and blocked leptin-induced signaling, including an increase in MAT gene expression and growth. Increased expression of MAT2A and MAT2β is required for leptin to be mitogenic, although by entirely different mechanisms.
Conclusion
Leptin induces MAT2A and MAT2β expression in HepG2 cells and normal hepatocytes but is mitogenic only in HepG2 cells. Pharmacological doses of SAMe or MTA lower expression of both MAT2A and MAT2β and interfere with leptin signaling.
Leptin, the product of the obese (Ob) gene, is a 16-kDa circulating hormone secreted by white adipocytes that acts as an important signaling molecule in energy regulation and food intake.1 Leptin may also play an important role in the process of initiation and progression of human cancers.2,3 In the liver, emerging evidence has suggested a role of leptin in hepatic inflammation1 and fibrogenesis.4 Serum leptin levels are higher in alcoholic5 and liver cirrhosis patients.6 Leptin induces the proliferation of primary hepatic stellate cells4 and enhances α2(I) collagen messenger RNA (mRNA) production.7 Recently, leptin was shown to play a pivotal role in the progression of fibrogenesis and carcinogenesis in a nonalcoholic steatohepatitis animal model.8 Furthermore, leptin can act as a mitogen in the human hepatoblastoma cell line HepG2 and the human hepatocarcinoma–derived cell line Huh7 and promote their invasion and migration.9 However, whether leptin can act as a mitogen in normal hepatocytes is unclear.
Methionine adenosyltransferase (MAT) is an essential cellular enzyme that catalyzes the formation of S-adenosylmethionine (SAMe), the principal biological methyl donor in mammalian cells and donor of propylamine moieties required for polyamine biosynthesis.10 In the biosynthesis of polyamines from SAMe, methylthioadenosine (MTA) is generated as a byproduct that is a known inhibitor of methylation.11 MTA can be converted back to SAMe via the methionine salvage pathway.10 Three distinct forms of MAT (MATI, MATII, and MATIII) exist in mammalian tissues that are the products of two different genes (MAT1A and MAT2A).12 The gene MAT1A encodes the α1 catalytic subunit, which organizes into dimers (MATIII) or tetramers (MATI).12 The gene MAT2A encodes for the α2 catalytic subunit in the MATII isoform. A third gene, MAT2β, encodes for a β regulatory subunit that regulates the activity of MATII by lowering inhibition constant (Ki) for SAMe and Michaeli’s constant (Km) for methionine.13 MAT1A is expressed mostly in adult liver,12,14 whereas MAT2A is widely distributed.15 MAT2A is also expressed by fetal liver but is replaced by MAT1A during development.14 Although MAT isoenzymes catalyze the same reaction, they are differentially regulated by their product, SAMe. SAMe maintains MAT1A expression in hepatocytes but inhibits MAT2A expression.10 In addition, pharmacological doses of SAMe and its metabolite MTA are proapoptotic in liver cancer cells but antiapoptotic in normal hepatocytes.16
In adult liver, increased expression of MAT2A is associated with increased growth, dedifferentiation, and malignant degeneration.10,17,18 It has been shown that MAT2A is induced by hepatocyte growth factor (HGF), and the up-regulation of MAT2A is required for HGF’s mitogenic response.19 However, the molecular mechanism was not explored. Increased expression of MAT2β also provides a growth advantage in hepatoma cells, and although it is not expressed in normal liver, its expression is increased in liver cirrhosis and hepatocellular carcinoma.20
Because leptin signaling was shown to induce the growth and invasive potential of liver cancer cells9 and MAT2A and MAT2β genes are associated with liver cell proliferation, we hypothesized that leptin’s mitogenic effect in liver cancer cells may require induction of MAT2A and MAT2β genes. Our results show that leptin is a mitogen in liver cancer cell line HepG2 but not in primary cultures of human or mouse hepatocytes. The mitogenic effect in HepG2 cells requires induction of both MAT2A and MAT2β, and pharmacologic doses of SAMe and MTA can down-regulate the expression of both genes and block leptin-mediated signaling. Interestingly, although knockdown of MAT2A may have compromised cell growth by limiting polyamines available for growth, knockdown of MAT2β interrupted leptin signaling. This is a novel action of MAT2β not previously described and greatly broadens the role of this gene in biology.
Materials and Methods
Materials
Leptin, MTA, PD98059 [extracellular signal-regulated kinase (ERK) inhibitor], and LY294002 [phosphatidylinositol-3-kinase (PI3-K) inhibitor] were obtained from Sigma (St. Louis, MO). Lyophilized leptin was dissolved according to the manufacturer’s instructions at 1 mg/mL. Endotoxin contamination in leptin was less than 0.1 ng/μg of protein according to Sigma. SAMe in the form of disulfate p-toluenesulfonate dried powder was generously provided by Gnosis SRL (Cairate, Italy). All other reagents were analytical-grade and were obtained from commercial sources.
Cell Lines and Primary Cultures
HepG2 cells and primary hepatocytes isolated from 6-month-old male C57/B6 mice (plated on collagen-coated dishes) were obtained from the Cell Culture Core of the University of Southern California Research Center for Liver Diseases. HepG2 cells were cultured according to instructions from the American Type Culture Collection (Rockville, MD). Primary human hepatocytes were obtained in a suspension culture from CellzDirect (Pittsboro, NC). The cells were centrifuged at 2000 rpm for 5 minutes at 4°C in a Beckman GS-15R centrifuge and plated on collagen-coated dishes with minimal essential medium supplemented with 10% fetal bovine serum.
Cell Treatment Conditions
For all experiments, HepG2 cells or primary human and mouse hepatocytes were serum-starved for 24 hours and treated with 100 ng/mL leptin for 14 or 24 hours unless otherwise mentioned. SAMe and MTA were used at concentrations of 1 mM for 14 hours in HepG2 cells and for 24 hours in primary human hepatocytes. When combined with leptin, SAMe and MTA were added to the medium 30 minutes before the addition of leptin. At 1 mM SAMe or MTA and with the duration of treatment up to 16 hours in the current study, no toxicity was evident (not shown). For inhibitor assays, cells were pretreated with PD98059 at 6 μM and LY294002 at 600 nM for 1 hour.
Northern Blot Analysis
RNA (10 μg) from HepG2 cells and human hepatocytes were subjected to northern blot analysis with specific MAT2A and MAT2β complementary DNA probes as we described.21 For housekeeping control, membranes were rehybridized with a 32P-labeled β-actin complementary DNA probe as described.21 Autoradiography and densitometry were used to quantitate relative RNA as described.21 Results of northern blot analysis were normalized to β-actin.
Quantitative Polymerase Chain Reaction (PCR) Analysis
Total RNA was subjected to reverse transcription (RT) with Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA). Two microliters of RT product was subjected to quantitative real-time PCR analysis. The primers and TaqMan probes for human and mouse MAT2A and MAT2β and universal PCR master mix were purchased from ABI (Foster City, CA). Hypoxanthine phosphoribosyl-transferase 1 (HPRT1) was used as a housekeeping gene as described.22 The thermal profile consisted of 1 cycle at 95°C for 15 minutes followed by 40 cycles at 95°C for 15 seconds and at 60°C for 1 minute. The expression of MAT2A and MAT2β was checked by normalization of the cycle threshold (Ct) of these genes to that of the control housekeeping gene (HPRT1).23 The delta Ct obtained was used to find the relative expression of MAT2A or MAT2β genes according to the following formula: relative expression =2−ΔΔCt, where ΔΔCt = ΔCt of MAT genes in treated cells −ΔCt of MAT genes in control cells
RNA Interference (RNAi) Analysis
RNAi experiments in HepG2 cells were performed by the reverse transfection method with Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s protocol. Small interfering RNA (siRNA) oligonucleotides for MAT2A, MAT2β, and scrambled siRNA were synthesized by the University of Southern California Norris Comprehensive Cancer Center Microchemical Core Laboratory and annealed to form duplexes. The following siRNA sequences were used: si-MAT2A, 5′-GUGAGAGAGAGCUAUUAGATT-3′(sense) and 5′-UCUAAUAGCUCUCUCUCACTC-3′ (antisense); si-MAT2β, 5′-GAAUGCUGGAUCCAUCAAUTT-3′ (sense) and 5′-AUUGAUGGAUCCAGCAUUCTC-3′(antisense); and si-control with a scrambled sequence (negative control siRNA having no perfect matches to known human genes), 5′-UUCUCCGAACGUGUCACAUdTdT-3′ (sense) and 5′-AUGUGACACGUUCGGAGAAdTdT-3′ (antisense). Transfection was allowed to proceed for 48 hours, and cells were processed for different assays. The siRNA transfection efficiency of Lipofectamine RNAiMax in HepG2 cells was determined by the BLOCK-iT Alexa Fluor red fluorescent oligo protocol (Invitrogen).
Transient Transfection Assays
The human MAT2A promoter construct −571/+60-LUC in pGL-3 enhancer vector has been described previously.24,25 For transient transfection assays, 2 × 105 HepG2 cells were plated in 12-well dishes and transfected with 0.5 μg of MAT2A promoter construct and control pGL3-enhancer vector for 2 hours with the Superfect transfection reagent (Qiagen, Valencia, CA). To control for transfection efficiency, cells were cotransfected with 20 ng of Renilla phRL-TK vector from Promega. After the treatment, cells were lysed with 1× passive lysis buffer (Promega), and the Firefly and Renilla luciferase activity was measured with the Dual-Luciferase Reporter Assay system (Promega).
Cell Proliferation Assay
To assay for cell proliferation, HepG2 cells and human and mouse hepatocytes were plated at a density of 104 per well on a 96-well plate (approximately 30% confluent) and serum-starved for 24 hours. Bromodeoxyuridine (BrDU) was added to each well at a dilution of 1:2000. The BrDU incorporation (a measure of DNA synthesis and growth) under different treatment conditions was measured with the BrDU cell proliferation assay kit (CalBiochem, San Diego, CA).
Western Blot Analysis
For western blot analysis of ERK1/2 or AK strain transforming (AKT) phosphorylation, HepG2 cells were serum-starved for 48 hours. Cells were then pretreated with SAMe, MTA, or vehicle for 30 minutes, and this was followed by treatment with leptin or fetal bovine serum for 30 minutes. For a time course study of signal transducers and activators of transcription 3 (STAT3) phosphorylation, cells were serum-starved for 16 hours and then treated with leptin for the indicated times. For siRNA experiments, HepG2 cells were transfected with siRNA in a serum-containing medium for 18 hours and then serum-starved for 36 hours. Leptin was added during the last 30 minutes of knockdown for ERK1/2 or AKT phosphorylation and during the last 1 hour for STAT3 phosphorylation. For western blot analysis of the leptin receptors, HepG2 cells or mouse hepatocytes were cultured for 24 hours in a serum-containing medium. Western blotting was performed following standard protocols (Amersham BioSciences, Piscataway, NJ) with primary antibodies for phosphorylated ERK1/2, total ERK2, and leptin receptor long and short forms (Ob-R H-300; Santa Cruz Biotechnologies, Santa Cruz, CA) or phosphorylated AKT-Ser473, total AKT, phosphorylated STAT3-Tyr705, and total STAT3 (Cell Signaling Technologies, Danvers, MA).
Determination of SAMe and MTA Levels
Cellular SAMe and MTA levels were measured by high-performance liquid chromatography as described.26
Statistical Analysis
Data are given as mean ± standard error of the mean. Statistical analysis was performed with analysis of variance followed by Fisher’s test for multiple comparisons. For changes in mRNA levels, ratios of MAT genes to β-actin or HPRT1 densitometric values were compared. For changes in protein levels, ratios of activated ERK, AKT, STAT3 to total ERK, AKT, and STAT3 densitometric values, respectively, were compared. Comparison was done by analysis of variance and significance was defined by P < 0.05.
Results
Leptin and MAT Gene Expression
Leptin treatment increased the steady-state mRNA levels of both MAT2A and MAT2β in HepG2 cells in a time-dependent and dose-dependent fashion (Fig. 1A). Similar results were obtained with real-time PCR (Fig. 1B). Leptin also elicited a time-dependent increase in MAT2A and MAT2β mRNA levels in primary human hepatocytes (Fig. 1C). The results were confirmed by real-time PCR (140%-150% of control; Fig. 1D). Leptin had no effect on the MAT1A mRNA level in HepG2 cells (data not shown).
Fig. 1.
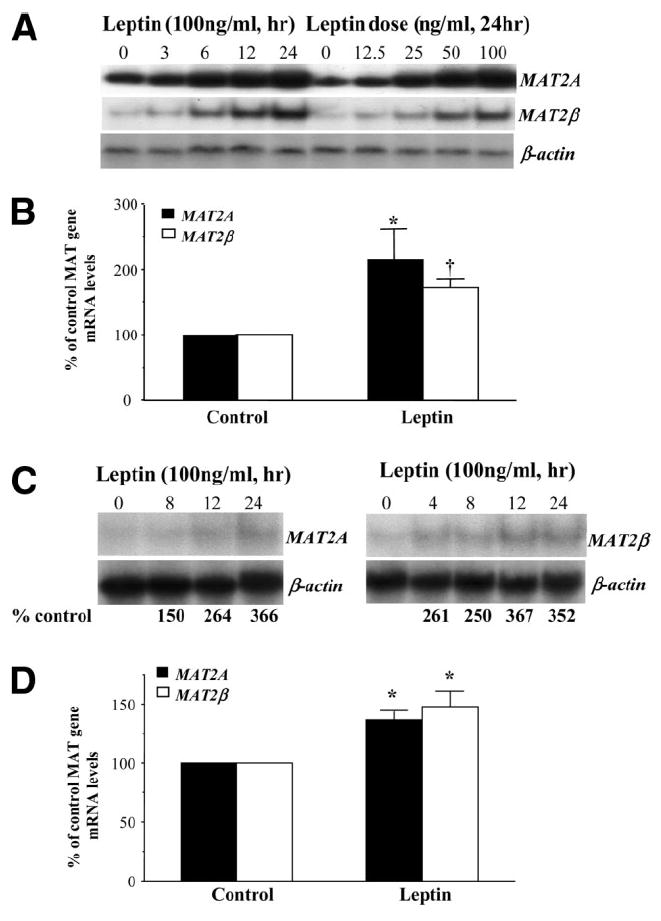
Leptin induces the expression of MAT2A and MAT2β in HepG2 cells and primary human hepatocytes. (A) HepG2 cells were treated with 100 ng/mL leptin for different times or with different doses of leptin for 24 hours. The expression of MAT2A and MAT2β was examined by northern blotting with a 32P-labeled probe specific for these genes as described in the Materials and Methods section. (B) HepG2 cells were treated with leptin for 14 hours, and MAT2A-specific or MAT2β-specific RNA was estimated by quantitative RT-PCR as described in the Materials and Methods section. The mean ± SE of 4 different experiments is shown; *P < 0.05, †P < 0.01 versus control. (C) Primary human hepatocytes were treated with leptin in a time-dependent fashion, and the expression of MAT2A and MAT2β genes was assessed by northern blotting as above. Numbers below the blots refer to a densitometric measure expressed as a percentage of 0 time control. (D) Primary human hepatocytes were treated with leptin for 24 hours, and the expression of MAT2A and MAT2β genes was assessed by real-time PCR. The mean ± SE of 3 different experiments is shown; *P < 0.01 versus control.
Effect of SAMe and MTA on Leptin-Mediated MAT Gene Up-Regulation and Growth
We previously showed that SAMe can inhibit MAT2A gene expression in HepG2 cells.25 Figure 2A shows that treatment of HepG2 cells with SAMe or MTA can also lower MAT2β mRNA levels, and both agents were able to block leptin-mediated increases in MAT2A and MAT2β mRNA levels (Fig. 2B). Furthermore, leptin is mitogenic in HepG2 cells, as indicated by increased BrDU incorporation, and both SAMe and MTA blocked this mitogenic effect (Fig. 2C). However, leptin did not exert a mitogenic effect in primary human hepatocytes [control = 0.34 ± 0.02, leptin = 0.35 ± 0.02; results represent the mean ± standard error (SE) from three independent experiments, each performed in quadruplicate]. SAMe or MTA did not exert any effect on basal BrDU incorporation in HepG2 cells when treatment was limited to 14 hours. However, at 24 hours, both agents reduced BrDU incorporation in HepG2 cells (data not shown) because of their differential proapoptotic effects in liver cancer cells.16
Fig. 2.
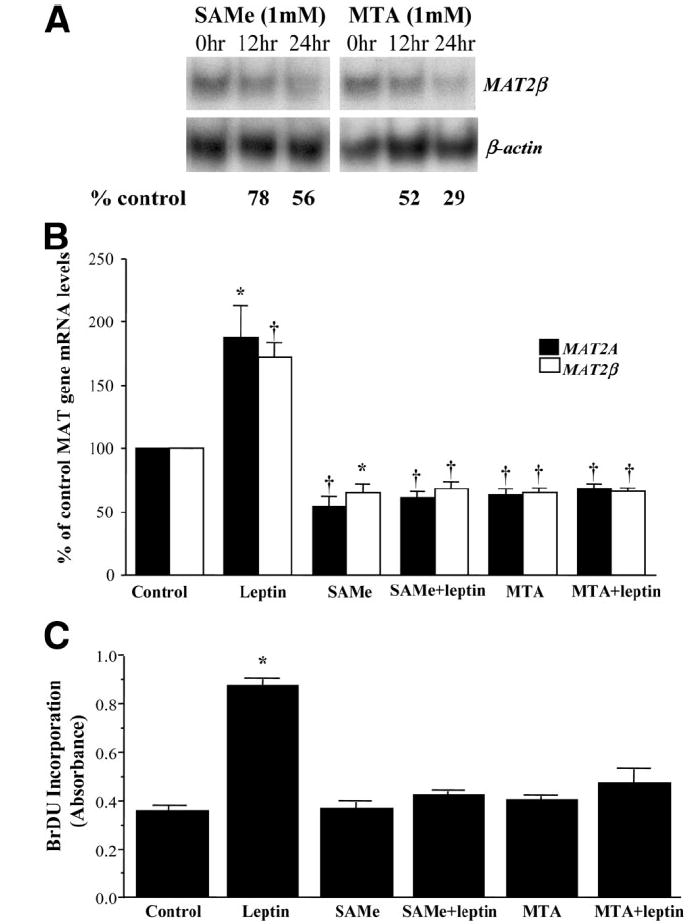
Effect of SAMe and MTA on leptin-mediated MAT gene up-regulation and growth in HepG2 cells. (A) HepG2 cells were treated with 1 mM SAMe or MTA, and the expression of MAT2β gene was assessed by northern blotting as above. Numbers below the blots refer to a densitometric measure expressed as a percentage of 0 time control. (B) HepG2 cells were treated with leptin (100 ng/mL) for 14 hours, SAMe (1 mM), MTA (1 mM), or a combination of leptin with SAMe or MTA as described in the Materials and Methods section. Total RNA from treated cells was subjected to quantitative RT-PCR analysis, and mRNA levels of MAT2A and MAT2β were compared to those of untreated (control) cells. The mean ± SE of 4 to 7 experiments is shown; *P < 0.05, †P < 0.01 versus control. (C) HepG2 cells were treated as in part B along with BrDU, and its incorporation in treated samples was compared to untreated (control) samples as described in the Materials and Methods section. The mean ± SE of 3 to 4 experiments is shown; *P < 0.001 versus control.
We next evaluated the effect of leptin on the transcriptional activity of the MAT2A gene in transient transfection assays (Fig. 3). We found that leptin induced the promoter activity of the MAT2A gene, whereas SAMe and MTA decreased the promoter activity by 66% and 80%, respectively. SAMe and MTA also completely prevented leptin-mediated induction of promoter activity.
Fig. 3.
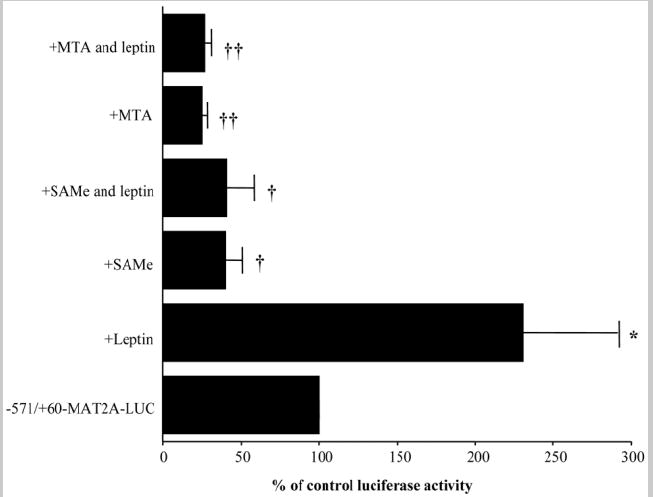
Effect of leptin, SAMe, and MTA on MAT2A promoter activity in HepG2 cells. HepG2 cells were transfected with MAT2A-LUC vector and further treated with leptin, SAMe, MTA, or combinations of leptin with SAMe or MTA as described in the Materials and Methods section. The promoter activity of MAT2A in treated samples was calculated as a percentage of the untreated vector (−571/+60-MAT2A-LUC). The mean ± SE of 5 experiments in triplicate is shown; *P < 0.05, †P < 0.005, ††P < 0.001 versus control −571/+60-MAT2A-LUC.
Leptin Signaling in HepG2 Cells
Leptin induces proliferation and inhibits apoptosis of different cell types via ERK/mitogen-activated protein kinase (MAPK) or PI3-K signaling pathways.3,4,9,27 Hence, we sought to determine whether these survival pathways were also involved in the inductive effects of leptin on MAT genes and growth in HepG2 cells. Figure 4A shows that blockage of ERK/MAPK signaling by PD98059 or PI3-K signaling by LY294002 inhibitor abolished leptin’s inductive effect on both MAT2A and MAT2β mRNA expression. There was a corresponding blockage of leptin-mediated growth of HepG2 cells pretreated with these inhibitors (Fig. 4B).
Fig. 4.
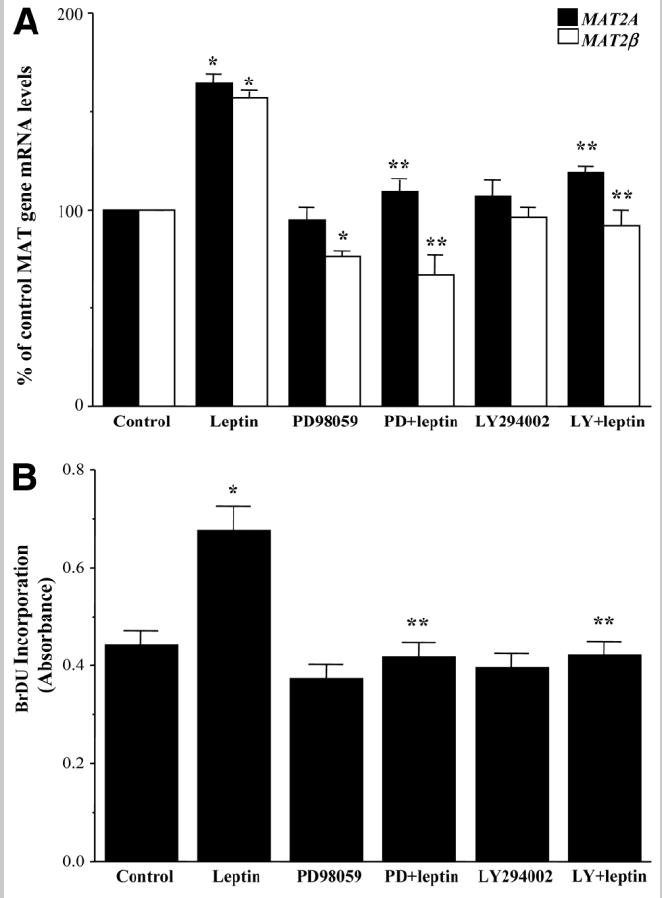
Effect of MAPK or PI3-K inhibition on leptin-mediated MAT gene expression and growth. (A) HepG2 cells were serum-starved for 24 hours and treated with PD98059 (PD) or LY294002 (LY) for 1 hour followed by leptin for 14 hours. Quantitative RT-PCR of MAT gene expression was performed, and the results were compared to those for untreated (control) cells. The mean ± SE of 6 to 8 experiments is shown. *P < 0.001 versus control, **P < 0.005 versus leptin. (B) HepG2 cells were treated as in part A along with BrDU, and its incorporation in treated samples was compared to untreated (control) samples. The mean ± SE of 5 to 7 experiments is shown; *P < 0.005 versus control, **P < 0.001 versus leptin.
Involvement of MAT Signaling in Leptin’s Mitogenic Activity
To see whether induction of MAT2A and MAT2β genes is required for leptin’s mitogenic action, we used an siRNA-based knockdown approach that decreased MAT2A or MAT2β expression in HepG2 cells by 60% or 80%, respectively. The siRNA transfection efficiency of Lipofectamine RNAiMax for these two genes was found to be 93.5%. Figure 5A shows that leptin-mediated induction of MAT2A and MAT2β was prevented by knockdown of these genes in HepG2 cells in comparison with untreated (control) and scrambled siRNA–treated cells. Correspondingly, leptin-mediated growth of HepG2 cells was completely prevented when leptin’s inductive effect on MAT2A or MAT2β gene expression was blocked (Fig. 5B).
Fig. 5.
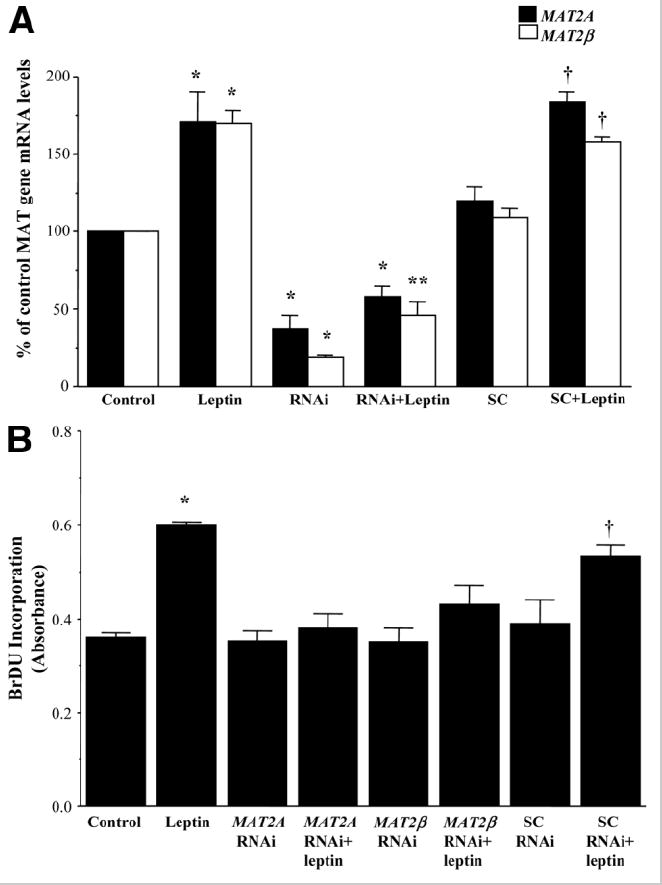
Effect of MAT2A and MAT2β knockdown on leptin’s proliferative potential in HepG2 cells. Knockdown of MAT genes in HepG2 cells was performed as described in the Materials and Methods section, and cells were treated with leptin. (A) Quantitative RT-PCR analysis showing the knockdown of these genes in HepG2 cells and the effect of leptin compared to untreated control cells and scrambled-RNAi control (SC) cells. The mean ± SE of 4 to 5 experiments is shown; *P < 0.005 versus respective control, **P < 0.05 versus RNAi and control, †P < 0.005 versus SC. (B) BrDU incorporation of siRNA-treated cells with or without leptin was compared to untreated and scrambled RNAi-treated cells. The mean ± SE of 4 experiments is shown; *P < 0.005 versus control, †P < 0.05 versus scrambled.
SAMe and MTA Inhibit Leptin Signaling in HepG2 Cells
To see if exogenous SAMe and MTA can interfere with leptin signaling, we examined their influence in leptin-activated signal transduction pathways ERK and PI3-K. Figure 6 shows that leptin activated the ERK/MAPK and PI3-K signal transduction pathways in HepG2 cells as measured by the level of phosphorylation of their downstream components, ERK1/2 and AKT, respectively. SAMe and MTA by themselves had no influence on the basal activity of these signal transduction pathways but blocked leptin-mediated activation of ERK1/2, and MTA also blocked leptin-mediated activation of PI3-K. To see if this inhibitory effect of SAMe or MTA was specific to leptin, we checked the effect of these molecules on serum-mediated ERK or PI3-K activation HepG2 cells. Figure 7 shows that serum-mediated phosphorylation of ERK or AKT was not affected by SAMe treatment but was completely prevented by MTA.
Fig. 6.
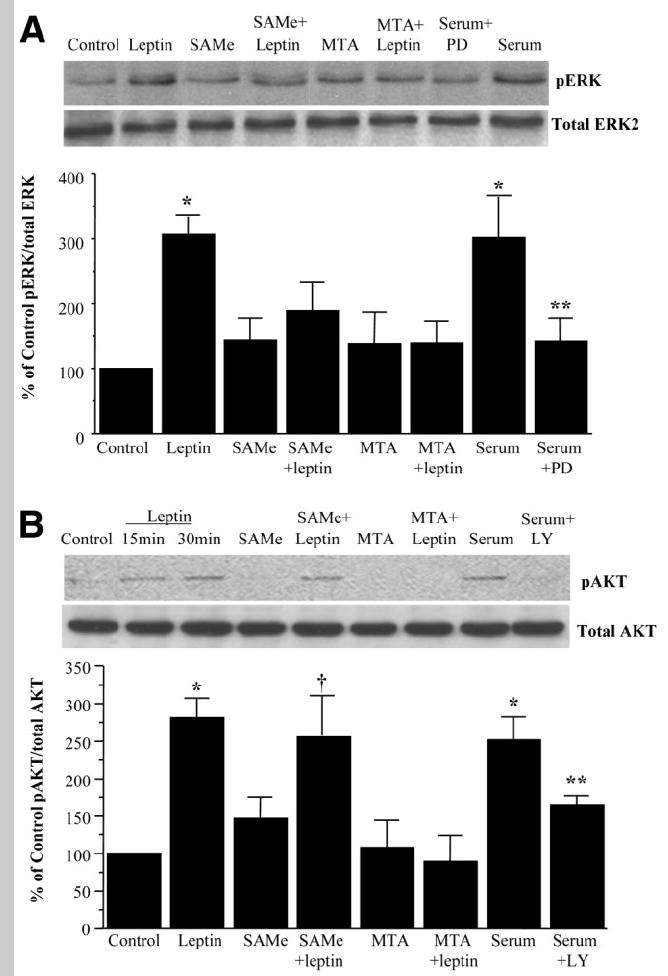
Effect of SAMe or MTA on leptin signaling in HepG2 cells. HepG2 cells were treated with leptin, SAMe, or MTA, and phosphorylation of (A) ERK1/2 and (B) AKT was checked by western blotting as described in the Materials and Methods section. Subsequently, the same protein extracts were analyzed for total ERK2 and total AKT to verify protein levels. Representative photographs from 4 to 6 independent experiments are shown, and results of densitometric analysis (mean ± SE) are shown below each blot; *P < 0.05 versus control, **P < 0.05 versus serum, † P < 0.05 versus SAMe.
Fig. 7.
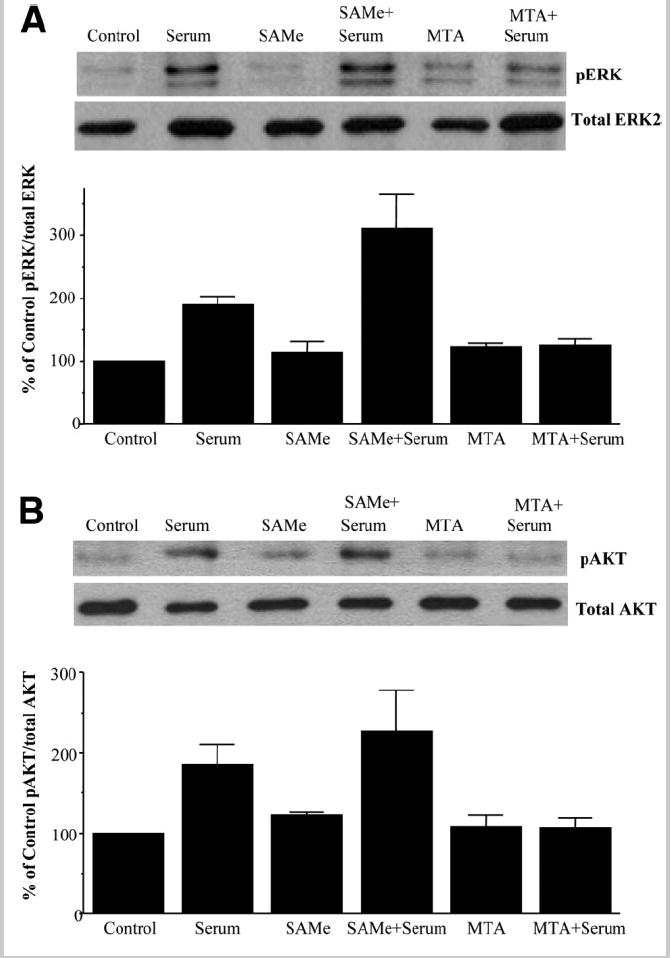
Effect of SAMe or MTA on serum-mediated ERK or AKT phosphorylation. HepG2 cells were treated with serum, SAMe, or MTA, and phosphorylation of (A) ERK1/2 and (B) AKT was checked by western blotting as described in the Materials and Methods section. Subsequently, the same protein extracts were analyzed for total ERK2 and total AKT to verify protein levels. Representative photographs from 2 independent experiments are shown, and results of densitometric analysis are shown below each blot as the mean ± SE.
Leptin Signaling in HepG2 Cells Is Dependent on Expression of the MAT2β Gene
To delineate the components of the leptin signaling pathway that are dependent on MAT gene expression, we knocked down MAT2A and MAT2β genes in HepG2 cells to study whether this affects ERK or AKT phosphorylation. Figure 8 shows that leptin-mediated ERK or AKT phosphorylation was completely blocked by MAT2β siRNA in comparison with untreated and scrambled treated controls. However, knockdown of MAT2A did not affect leptin signaling leading to ERK or AKT phosphorylation. Leptin-mediated activation of its receptor leads to STAT3 phosphorylation, which lies upstream of ERK and PI3-K.4 In Fig. 9A, we confirmed that leptin induces STAT3 phosphorylation in serum-starved HepG2 cells (16-hour starvation) in a time-dependent manner (Fig. 9A). Because there was still a substantial level of basal STAT3 phosphorylation (Fig. 9A, lane 0 time), we increased serum starvation to 36 hours to reduce baseline signal. Figure 9B shows that MAT2β knockdown completely prevented leptin-mediated induction of STAT3 phosphorylation.
Fig. 8.
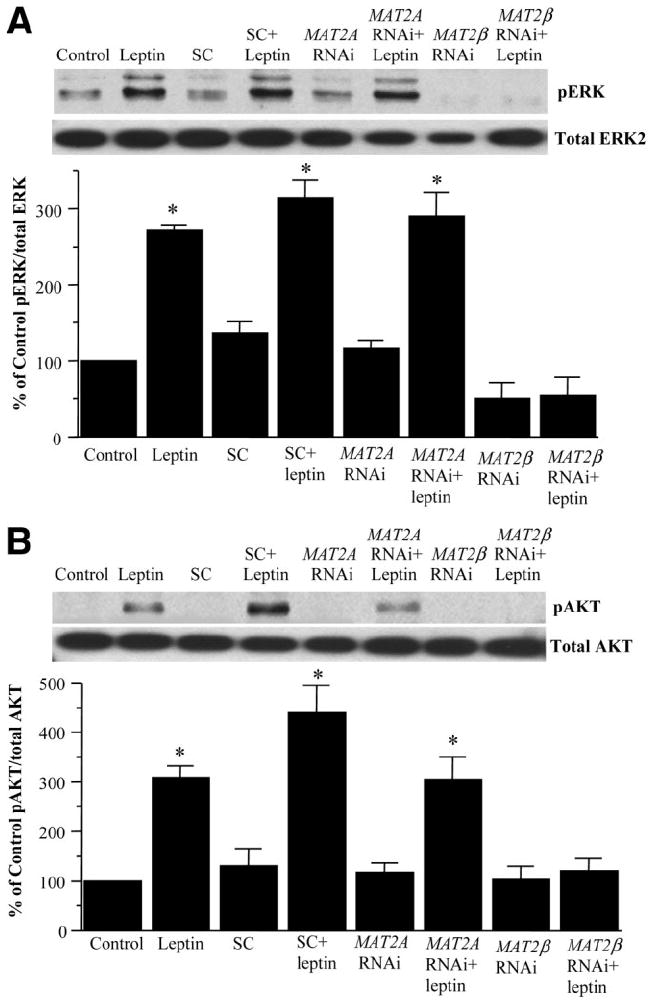
Effect of MAT gene knockdown on leptin signaling in HepG2 cells. (A) HepG2 cells were treated with leptin, scrambled RNAi (SC), MAT2A RNAi, or MAT2β RNAi alone or together, and phosphorylation of (A) ERK1/2 and (B) AKT was checked by western blotting as described in the Materials and Methods section. Subsequently, the same protein extracts were analyzed for total ERK2 and total AKT to verify protein levels. Representative photographs from 3 to 4 independent experiments are shown, and results of densitometric analysis (mean ± SE) are shown below each blot; *P < 0.05 versus respective controls.
Fig. 9.
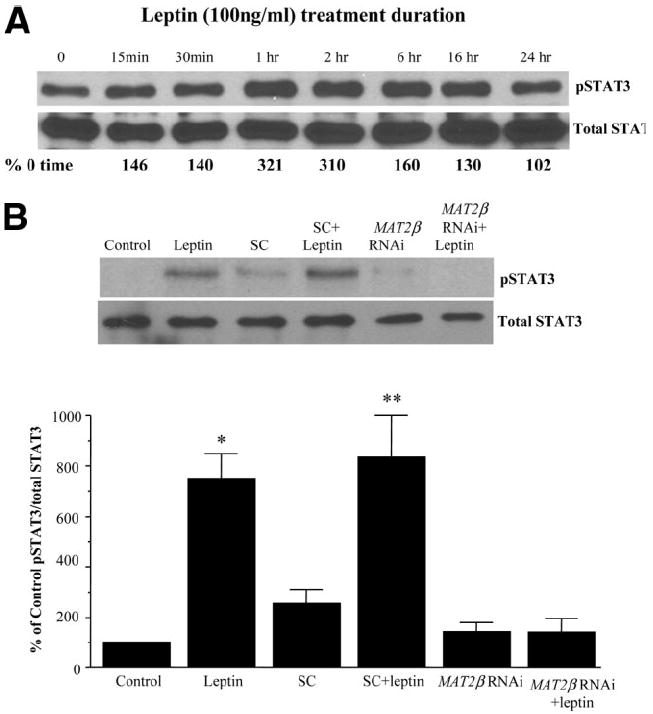
Leptin-mediated STAT3 signaling in HepG2 cells. (A) HepG2 cells were treated with leptin for the indicated times, and the phosphorylation of STAT3 was checked by western blotting as described in the Materials and Methods section. Subsequently, the same protein extracts were analyzed for total STAT3. Representative photographs from two independent experiments are shown, and numbers below the blot refer to average densitometric values expressed as a percentage of 0 time control. (B) Effect of MAT2β knockdown on leptin-mediated STAT3 activation. HepG2 cells were treated with scrambled RNAi (SC) and MAT2β RNAi alone or together with leptin, and phosphorylation of STAT3 was checked by western blotting as described in the Materials and Methods section. Representative photographs from 4 independent experiments are shown, and results of densitometric analysis (mean ± SE) are shown below; *P < 0.01 versus control, **P < 0.05 versus SC.
Effect of Leptin, SAMe, MTA, MAT2A, and MAT2β RNAi Treatments on SAMe and MTA Levels
Increased MAT2A expression could lead to increased SAMe biosynthesis, but increased MAT2β might prevent SAMe accumulation because the β subunit lowers the Ki for SAMe.13 Because leptin induces the expression of MAT2A and MAT2β, we next determined whether it would affect the levels of SAMe and MTA and whether a lack of these genes would affect it. Table 1 shows that leptin increased SAMe level by 43% in HepG2 cells. Knockdown of MAT2A decreased SAMe levels by 71%, and leptin was unable to raise SAMe levels in cells treated with MAT2A RNAi. On the other hand, knockdown of MAT2β increased SAMe levels by 86%, and leptin had no further effect on SAMe levels in cells treated with MAT2β RNAi. Exogenous SAMe and MTA treatments raised SAMe levels by 450% and 214%, respectively. Leptin, MAT2A, and MAT2β knockdowns had no influence on MTA levels, but exogenous SAMe and MTA treatments increased MTA levels by 70 to 80%, respectively. In primary human hepatocytes, leptin treatment did not influence steady-state SAMe levels after 24 hours (results not shown).
Table 1.
SAMe and MTA Levels in HepG2 Cells After Various Treatments
| Treatment | SAMe | MTA |
|---|---|---|
| Control | 0.35 ± 0.03 | 0.10 ± 0.018 |
| Leptin | 0.50 ± 0.03* | 0.07 ± 0.002 |
| Scrambled RNAi | 0.32 ± 0.02 | |
| Scrambled RNAi + leptin | 0.43 ± 0.03** | |
| MAT2A RNAi | 0.10 ± 0.03† | 0.08 ± 0.004 |
| MAT2A RNAi + leptin | 0.13 ± 0.03† | 0.09 ± 0.007 |
| MAT2β RNAi | 0.65 ± 0.05† | 0.09 ± 0.004 |
| MAT2β RNAi + leptin | 0.61 ± 0.05† | 0.09 ± 0.006 |
| SAMe | 1.93 ± 0.27† | 0.17 ± 0.010† |
| MTA | 1.10 ± 0.10† | 0.18 ± 0.024† |
The unit for all metabolites is nmol/mg of protein. The results represent the mean ± the standard error of the mean from 6 experiments. HepG2 cells were treated with leptin (100 ng/mL) for 14 hours and scrambled RNAi, MAT2A RNAi, or MAT2β RNAi for 48 hours alone or with leptin during the last 14 hours. SAMe and MTA (both 1 mM) treatment was for 14 hours also.
P < 0.05 versus control.
P < 0.01 versus scrambled RNAi.
P < 0.005 versus control.
Leptin Signaling in Primary Mouse Hepatocytes
Lack of a mitogenic response in human hepatocytes to leptin could be due to factors including time delay in receiving human hepatocytes (typically 36-48 hours), rapid dedifferentiation after isolation, age and gender differences, and other unknown variables. To control for these variables and to examine whether leptin can activate its downstream signals in primary hepatocytes, we studied leptin signaling in primary cultures of mouse hepatocytes within 24 hours of isolation. Both mouse hepatocytes and HepG2 cells expressed the long and short forms of the leptin receptors (Fig. 10A), their sizes being consistent with previous reports.28,29 Furthermore, leptin-mediated signaling via STAT3 activation was evident in mouse hepatocytes as in HepG2 cells, although it was more robust and sustained in HepG2 cells (Figs. 9A and 10B). Leptin induced both MAT2A and MAT2β genes in mouse hepatocytes, with magnitudes very similar to those in human hepatocytes (Fig. 1D). Like human hepatocytes, leptin is not mitogenic in mouse hepatocytes.
Fig. 10.
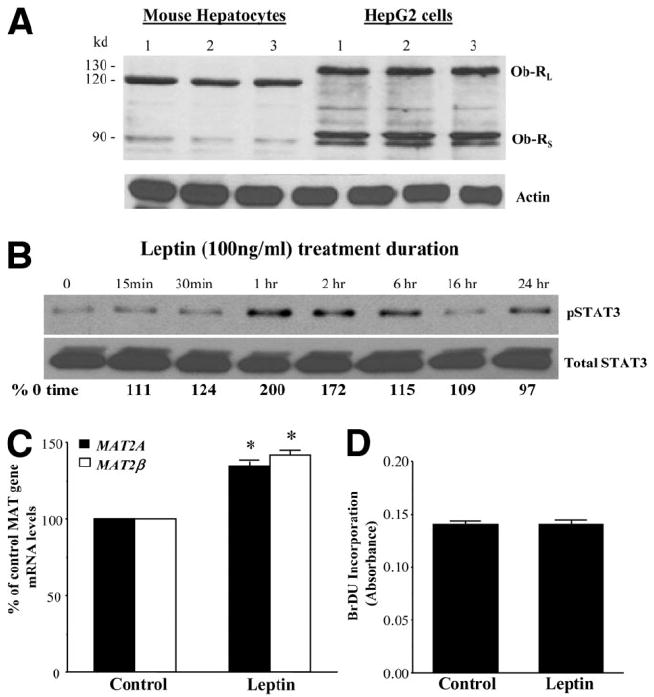
Leptin signaling in primary mouse hepatocytes. (A) Expression of leptin receptors (Ob-RL, long receptor, and Ob-RS, short receptor) in mouse hepatocytes and HepG2 cells. HepG2 cells and mouse hepatocytes were cultured, and western blotting for the long and short forms of leptin receptors was performed as described in the Materials and Methods section. Mouse hepatocytes from three livers and three preparations of HepG2 cells are shown. (B) Leptin-mediated STAT3 signaling in mouse hepatocytes. Primary mouse hepatocytes were treated with leptin for the indicated times, and the phosphorylation of STAT3 was examined as described in the Materials and Methods section. A representative blot from two independent experiments is shown, with the numbers below referring to average densitometric values. (C) Leptin induces MAT2A and MAT2β genes in mouse hepatocytes. Primary mouse hepatocytes were treated with leptin for 24 hours, and the expression of MAT2A and MAT2β genes was assessed by real-time PCR. The mean ± SE of 3 different experiments is shown; *P < 0.001 versus control. (D) Leptin is not mitogenic in mouse hepatocytes. Mouse hepatocytes were treated as in part C along with BrDU, and its incorporation in leptin-treated samples was compared to untreated (control) samples. The mean ± SE from 2 different experiments performed in triplicate is shown.
Discussion
The mitogenic potential of leptin in cancer growth is well documented.2,3,9,27 The expression of MAT genes in the liver is a determinant of liver cell growth and differentiation.10 Although MAT1A is a marker of differentiation, MAT2A and MAT2β are markers of growth and dedifferentiation.10,17-20 Because leptin is a known mitogen for liver cancer cells and may facilitate progression to liver cancer in vivo,8,9 we wondered if leptin’s mitogenic potential might involve MAT genes. We also examined whether exogenous SAMe and MTA might interfere with leptin’s effect.
Leptin increased the mRNA levels of MAT2A and MAT2β in HepG2 cells and primary human and mouse hepatocytes. However, leptin induced growth only in HepG2 cells. Leptin-mediated up-regulation of MAT genes and growth was completely abrogated by inhibition of cell survival pathways ERK/MAPK and PI3-K. Leptin is known to activate these two pathways in many cell types.3,4,9 Here we find that leptin also induces these pathways in HepG2 cells and that activation of these signal transduction pathways is responsible for increased MAT2A and MAT2β gene expression and growth. The fact that inhibition of either one of these pathways can inhibit the functional outcome, growth, and increased expression of MAT genes is consistent with previous reports showing cross-talk between the two signal transduction pathways.30 Thus, inhibition in ERK/MAPK can down-regulate PI3-K and vice versa.31
We next examined whether SAMe or MTA can interfere with leptin signaling. Interestingly, we found that both agents lowered MAT2A and MAT2β mRNA levels and blocked leptin-mediated increases in MAT gene expression and growth in HepG2 cells. How can this be explained? We previously showed that SAMe inhibited growth of HepG2 cells18 and blocked HGF-mediated growth of primary rat hepatocytes.32 The molecular mechanism for growth inhibition in HepG2 cells was not explored.18 In primary cultures of rat hepatocytes, SAMe’s inhibitory effect on HGF’s mitogenic effect was not via inhibiting ERK activation33 but rather through blocking HGF-mediated activation of adenosine monophosphate kinase, which led to nuclear-to-cytoplasmic translocation of human RNA binding (HuR), an mRNA binding protein that stabilized cyclins involved in cell cycle progression.32 However, HGF and SAMe had no effect on adenosine monophosphate kinase in HepG2 cells.32 These observations suggest that although exogenous SAMe can inhibit or block mitogen-induced growth in both liver cancer cells and primary rat hepatocytes, they occur by different mechanisms. Our current study shows that these agents can interfere with leptin signaling in HepG2 cells by blocking ERK and PI3-K activation. Although SAMe blocked leptin signaling in HepG2 cells but had no effect on serum-mediated activation of ERK or PI3-K, MTA is a more general inhibitor of these survival pathways. Activation of these pathways is responsible for leptin’s profibrogenic effect,4 and SAMe has been shown to block fibrosis in an animal model of fibrogenesis.34 It will be interesting to see if SAMe and MTA can block leptin-mediated activation of these pathways in hepatic stellate cells.
Leptin treatment increased cellular SAMe levels in HepG2 cells. This occurred despite a similar increase in both MAT2A and MAT2β gene expression. This suggests that there is more α2 subunit than β subunit in HepG2 cells so that SAMe level can rise. The increase in SAMe level may facilitate growth by feeding into the polyamine biosynthesis pathway.10 Knockdown of MAT2A lowered SAMe level, and leptin was unable to exert its mitogenic effect, consistent with the idea that increased SAMe is required for growth. However, knockdown of MAT2β actually raised SAMe level, and yet it also blocked leptin-induced growth. This is consistent with the β subunit acting to lower Ki of MATII13 for SAMe so that when its expression falls, steady-state SAMe level should rise. It also suggests that MAT2β’s regulation of leptin’s mitogenic effect is independent of SAMe. Indeed, MAT2β expression is necessary for leptin to activate STAT3 and its downstream signaling pathways ERK and PI3-K. This is an aspect of MAT2β never reported as its encoded protein has been thought only to act as a regulatory subunit for MATII. The molecular mechanism(s) for this effect will require further study and is beyond the scope of this work.
If a leptin-mediated increase in intracellular SAMe level is required for increased growth in HepG2 cells, why would exogenous SAMe treatment block growth? The most plausible reason for this seemingly contradictory finding lies in the fact that at high pharmacologic doses of SAMe, the effect may be mediated at least in part by MTA. SAMe is unstable and converts to MTA spontaneously and in the polyamine pathway. Although SAMe is a methyl donor and precursor of polyamines, MTA inhibits methylation and polyamine synthesis.11 The fact that SAMe treatment raised intracellular MTA levels supports this notion. Thus, although a physiological increase in SAMe (without a change in MTA) stimulates growth in liver cancer cells, a pharmacologic increase may inhibit growth because of increased MTA level. Alternatively, it is possible that supraphysiologic SAMe levels achieved with pharmacologic doses may be deleterious. It should be noted that the situation is quite different in normal hepatocytes, in which a fall in SAMe level has been shown to occur prior to the cell cycle being entered.17 This fall is believed to be necessary in order to release the inhibitory tone that it exerts on mitogens.10 Thus, a fall in SAMe level allows normal hepatocytes to grow, whereas an increase appears necessary for liver cancer cells to grow. The finding that leptin treatment had no effect on SAMe levels in primary human hepatocytes is consistent with its lack of a mitogenic effect. Even though leptin induced the expression of MAT2A and MAT2β in both primary human and mouse hepatocytes, MAT1A continues to be expressed at a higher level, and this may have prevented a change in SAMe. Furthermore, leptin receptor expression and leptin signaling through STAT3 were evident in primary mouse hepatocytes, and normal human hepatocytes also express leptin receptors.29 Therefore, lack of mitogenic response cannot be attributed to the absence of signaling in normal liver cells.
In summary, we have demonstrated that leptin induces MAT2A and MAT2β gene expression in both HepG2 cells and human and mouse hepatocytes but acts as a mitogen only in HepG2 cells. In HepG2 cells, leptin’s inductive effect on MAT genes and growth requires activation of ERK/MAPK and PI3-K signal transduction pathways. Furthermore, the mitogenic effect also requires increased MAT2A and MAT2β expression. Although MAT2A induction is necessary to provide SAMe for growth, MAT2β interacts with leptin signaling so that knockdown of this gene prevents leptin-mediated STAT3 activation as well as ERK and AKT activation. Although a physiologic increase in SAMe facilitates growth in HepG2 cells, exogenous treatment with SAMe at pharmacological doses actually inhibits leptin signaling and growth. Figure 11 summarizes these findings.
Fig. 11.
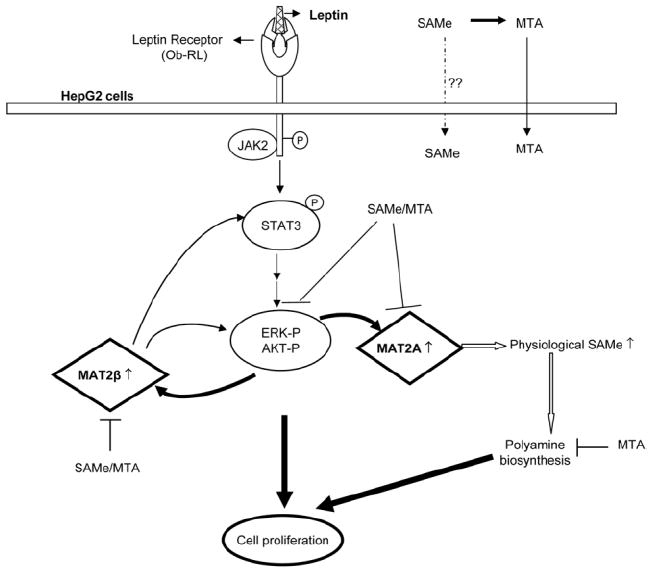
Schematic diagram for the role of MAT genes and SAMe in leptin-induced mitogenic response in HepG2 cells. Upon leptin interaction with its receptor, Ob-RL, activation of Janus kinase 2 (JAK2) occurs via trans-phosphorylation of the receptor as well as subsequent phosphorylation of the STAT proteins. This leads to a series of downstream events including activation of ERK/MAPK and PI3-K/AKT survival pathways in HepG2 cells. Leptin signaling induces MAT2A and MAT2β genes via activation of these survival pathways, which further leads to enhanced cell proliferation. The MAT2A-encoded protein induces leptin’s mitogenic response by raising intracellular SAMe levels, leading to polyamine biosynthesis and growth. The MAT2β gene interacts with leptin signaling components and modulates cell growth. Pharmacological doses of SAMe and MTA act as inhibitors of leptin signaling and growth in HepG2 cells.
Acknowledgments
Supported by National Institutes of Health grants DK51719 (to S.C.L.) and AA12677, AA13847, and AT1576 (to S.C.L. and J.M.M.), Plan Nacional de I+D SAF 2005-00855, HEPADIP-EULSHM-CT-205 (to J.M.M.), the Training Program in Alcoholic Liver and Pancreatic Diseases through a postdoctoral fellowship (T32 AA07578 to K.R.), and Centro de Investigación Cooperativa en Biosciencías through a postdoctoral fellowship (to A.I.A.).
HepG2 cells were provided by the Cell Culture Core of the University of Southern California Research Center for Liver Diseases (DK48522).
Abbreviations
- AKT
AK strain transforming
- BrDU
bromodeoxyuridine
- Ct
cycle threshold
- ERK
extracellular signal-regulated kinase
- HGF
hepatocyte growth factor
- HPRT1
hypoxanthine phosphoribosyl-transferase 1
- JAK2
Janus kinase 2
- MAPK
mitogen-activated protein kinase
- MAT
methionine adenosyltransferase
- mRNA
messenger RNA
- MTA
methylthioadenosine
- Ob
obese
- PCR
polymerase chain reaction
- PI3-K
phosphatidylinositol-3-kinase
- RNAi
RNA interference
- RT
reverse transcription
- SAMe
S-adenosylmethionine
- SE
standard error
- siRNA
small interfering RNA
- STAT
signal transducers and activators of transcription
Footnotes
Published online in Wiley InterScience (www.interscience.wiley.com).
Potential conflict of interest: Nothing to report.
References
- 1.Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, et al. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology. 2001;34:288–297. doi: 10.1053/jhep.2001.26518. [DOI] [PubMed] [Google Scholar]
- 2.Saglam K, Aydur E, Yilmaz M, Göktas S. Leptin influences cellular differentiation and progression in prostate cancer. J Urol. 2003;169:1308–1311. doi: 10.1097/01.ju.0000055903.18400.25. [DOI] [PubMed] [Google Scholar]
- 3.Sharma D, Saxena NK, Vertino PM, Anania FA. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal transduction pathways. Endocr Relat Cancer. 2006;13:629–640. doi: 10.1677/erc.1.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saxena NK, Titus MA, Ding X, Floyd J, Srinivasan S, Sitaram SV, et al. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004;18:1612–1614. doi: 10.1096/fj.04-1847fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henriksen JH, Holst JJ, Møller S, Brinch K, Bendtsen F. Increased circulating leptin in alcoholic cirrhosis: relation to release and disposal. Hepatology. 2003;29:1818–1824. doi: 10.1002/hep.510290601. [DOI] [PubMed] [Google Scholar]
- 6.Comlekci A, Akpinar H, Yesil S, Okan I, Ellidokuz E, Okan A, et al. Serum leptin levels in patients with liver cirrhosis and chronic viral hepatitis. Scand J Gastroenterol. 2003;7:779–786. doi: 10.1080/00365520310003877. [DOI] [PubMed] [Google Scholar]
- 7.Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, et al. Leptin-mediated neovascularization is a prerequisite for progression of non-alcoholic steatohepatitis in rats. Hepatology. 2006;44:983–991. doi: 10.1002/hep.21338. [DOI] [PubMed] [Google Scholar]
- 9.Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, et al. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–2507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mato JM, Corrales FJ, Lu SC, Avila MA. S-adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 11.Clarke SG. Inhibition of mammalian protein methyltransferases by 5′-methylthioadenosine (MTA): a mechanism of action of dietary SAMe? Enzymes. 2006;24:467–493. doi: 10.1016/S1874-6047(06)80018-1. [DOI] [PubMed] [Google Scholar]
- 12.Kotb M, Mudd SH, Mato JM, Geller AM, Kredich NM, Chou JY, et al. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997;13:51–62. doi: 10.1016/s0168-9525(97)01013-5. [DOI] [PubMed] [Google Scholar]
- 13.Halim AB, LeGros L, Geller AM, Kotb M. Expression and functional interaction of the catalytic and regulatory subunits of human methionine adenosyltransferase in mammalian cells. J Biol Chem. 1999;274:29720–29725. doi: 10.1074/jbc.274.42.29720. [DOI] [PubMed] [Google Scholar]
- 14.Gil B, Casado M, Pajares MA, Boscá L, Mato JM, Martĺn-Sanz P, et al. Differential expression pattern of SAM synthetase isoenzymes during rat liver development. Hepatology. 1996;24:876–881. doi: 10.1002/hep.510240420. [DOI] [PubMed] [Google Scholar]
- 15.Horikawa S, Tsukada K. Molecular cloning and developmental expression of a human kidney S-adenosylmethionine synthetase. FEBS Lett. 1992;312:37–41. doi: 10.1016/0014-5793(92)81405-b. [DOI] [PubMed] [Google Scholar]
- 16.Yang HP, Sadda MR, Li M, Zeng Y, Chen LX, Bae WJ, et al. S-Adenosylmethionine and its metabolite induce apoptosis in HepG2 cells: role of protein phosphatase 1 and Bcl-xS. Hepatology. 2004;40:221–231. doi: 10.1002/hep.20274. [DOI] [PubMed] [Google Scholar]
- 17.Huang ZZ, Mao Z, Cai J, Lu SC. Changes in methionine adenosyltransferase during liver regeneration in the rat. Am J Physiol. 1998;275:G14–G21. doi: 10.1152/ajpgi.1998.275.1.G14. [DOI] [PubMed] [Google Scholar]
- 18.Cai J, Mao Z, Hwang JJ, Lu SC. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res. 1998;58:1444–1450. [PubMed] [Google Scholar]
- 19.Pañeda C, Gorospe I, Herrera B, Nakamura T, Fabregat I, Varela-Nieto I. Liver cell proliferation requires methionine adenosyltransferase 2A mRNA upregulation. Hepatology. 2002;35:1381–1391. doi: 10.1053/jhep.2002.32538. [DOI] [PubMed] [Google Scholar]
- 20.Martínez-Chantar ML, García-Trevijano ER, Latasa MU, Martín-Duce A, Fortes P, Caballería J, et al. Methionine adenosyltransferase II β subunit gene expression provides a proliferative advantage in human hepatoma. Gastroenterology. 2003;124:940–948. doi: 10.1053/gast.2003.50151. [DOI] [PubMed] [Google Scholar]
- 21.Yang HP, Magilnick N, Noureddin M, Mato JM, Lu SC. Effect of hepatocyte growth factor on methionine adenosyltransferase genes and growth is cell density-dependent in HepG2 cells. J Cell Physiol. 2007;210:766–773. doi: 10.1002/jcp.20891. [DOI] [PubMed] [Google Scholar]
- 22.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:34.1–34.11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- 24.Yang HP, Sadda MR, Yu V, Zeng Y, Lee TD, Ou XP, et al. Induction of human methionine adenosyltransferase 2A expression by tumor necrosis factor alpha: role of NF-κB and AP-1. J Biol Chem. 2003;278:50887–50896. doi: 10.1074/jbc.M307600200. [DOI] [PubMed] [Google Scholar]
- 25.Yang HP, Huang ZZ, Zeng ZH, Chen CJ, Selby RR, Lu SC. Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am J Physiol. 2001;280:G184–G190. doi: 10.1152/ajpgi.2001.280.2.G184. [DOI] [PubMed] [Google Scholar]
- 26.Farrar C, Clarke S. Altered levels of S-adenosylmethionine and S-adenosylhomocysteine in the brains of L-isoaspartyl (D-aspartyl) O-methyltransferase-deficient mice. J Biol Chem. 2002;277:27856–27863. doi: 10.1074/jbc.M203911200. [DOI] [PubMed] [Google Scholar]
- 27.Hoda MR, Keely SJ, Bertelsen LS, Junger WG, Dharmasena D, Barrett KE. Leptin acts as a mitogenic factor for colonic cancer cells. Br J Surg. 2007;94:346–354. doi: 10.1002/bjs.5530. [DOI] [PubMed] [Google Scholar]
- 28.Hoggard N, Hunter L, Duncan JS, Williams LM, Trayhurn P, Mercer JG. Leptin and leptin receptor mRNA and protein expression in the murine fetus and placenta. Proc Natl Acad Sci U S A. 1997;94:11073–11078. doi: 10.1073/pnas.94.20.11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briscoe CP, Hanif S, Arch JRS, Tadayyon M. Leptin receptor long-form signaling in a human liver cell line. Cytokine. 2001;14:225–229. doi: 10.1006/cyto.2001.0871. [DOI] [PubMed] [Google Scholar]
- 30.Marino M, Acconcia F, Trentalance A. Biphasic estradiol-induced AKT phosphorylation is modulated by PTEN via MAP kinase in HepG2 cells. Mol Biol Cell. 2003;14:2583–2591. doi: 10.1091/mbc.E02-09-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wennstrom S, Downward J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol. 1999;19:4279–4288. doi: 10.1128/mcb.19.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martínez-Chantar ML, Vázquez-Chantada M, Garnacho M, Latasa MU, Varela-Rey M, Dotor J, et al. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology. 2006;131:223–232. doi: 10.1053/j.gastro.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Trevijano ER, Martínez-Chantar ML, Latasa MU, Mato JM, Avila MA. NO sensitizes rat hepatocytes to hepatocyte growth factor-induced proliferation through the modulation of S-adenosylmethionine levels. Gastroenterology. 2002;122:1355–1363. doi: 10.1053/gast.2002.33020. [DOI] [PubMed] [Google Scholar]
- 34.Corrales F, Giménez A, Alvarez L, Caballería J, Pajares MA, Andreu H, et al. S-adenosylmethionine treatment prevents carbon tetrachloride-induced S-adenosylmethionine synthetase inactivation and attenuates liver injury. Hepatology. 1992;16:1022–1027. doi: 10.1002/hep.1840160427. [DOI] [PubMed] [Google Scholar]