

Abstract
Purpose
Radioresistance may be caused by cancer stem cells (CSCs). Because CSCs require telomerase to proliferate, a telomerase-specific oncolytic adenoviral vector carrying apoptotic TRAIL and E1A gene (Ad/TRAIL-E1) may preferentially target CSCs.
Experimental Design
We established two pairs of parental and radioresistant (R) esophageal carcinoma cell lines (Seg-1, Seg-1R and TE-2, TE-2R) by fractionated irradiation (FIR). Stem cell markers were measured by Western blotting and flow cytometry. Serial sorting was used to enrich stem-like side population (SP) cells. Telomerase activity, transgene expression, antitumor activity, apoptosis induction, and viral replication were determined in vitro and/or in vivo.
Results
Expression of the stem cell markers β-catenin, Oct3/4, and β1-integrin in Seg-1R cells was 29.4%, 27.5%, and 97.3%, respectively, compared with 4.8%, 14.9%, and 45.3% in Seg-1 cells (P < 0.05). SP levels in Seg-1R and TE-2R cells were 14.6% and 2.7%, respectively, compared with 3.4% and 0.3% in Seg-1 and TE-2 cells. Serial sorting of Seg-1R SP cells demonstrated enrichment of the SP cells. Telomerase activity in Seg-1R, Seg-1R SP and TE-2R cells were significantly higher than Seg-1, Seg-1R non-SP and TE-2 cells respectively (P < 0.05). Seg-1R and TE-2R cells were more sensitive to Ad/TRAIL-E1 than were parental cells. Increased Coxsackie-adenovirus receptor (CAR) and elevated transgene expressions were found in the radioresistant cells. Ad/TRAIL-E1 resulted in significant tumor growth suppression and longer survival in Seg-1R-bearing mice (P < 0.05) with no significant toxicity.
Conclusion
Radioresistant cells established by FIR display CSC-like cell properties. Ad/TRAIL-E1 preferentially targets radioresistant CSC-like cells.
Keywords: Radioresistance, cancer stem-like cells, telomerase-specific oncolytic adenovirus, esophageal, Ad/TRAIL-E1
Ionizing radiation plays a crucial role in the combined modality management for advanced esophageal carcinoma. However, only 28% of patients achieve a pathological complete response after combined chemoradiotherapy (1). Radiation dose escalation increases toxicity without improving clinical outcome (2). The underlying mechanisms of radioresistance remaines elusive. Esophageal carcinoma frequently recurs or progresses after radiation as small foci, suggesting that only a fraction of the tumor cells is responsible for regrowth. Because cancer cells are clonal in origin, they may undergo processes similar to the self-renewal and differentiation of normal stem cells (3). Multi-potent cancer stem cells (CSCs) may explain the histologic heterogeneity found in tumors (4-6). In addition, cancer progression and metastasis may involve the escape of CSCs from innate somatic niche regulators. Identification of a crucial cellular subpopulation of esophageal carcinoma cells with potent tumorigenicity supports the CSC hypothesis in solid tumors (7).
β-Catenin is an essential component of both intercellular junctions and the canonical Wnt signaling pathway, which has been implicated in stem cell survival (8). Overexpression of β-catenin leads to self-renewal of stem cells in vitro and in vivo (9) and has been linked with stem cell survival and tumorigenesis (10-12). OCT4 may also play a role in the maintenance of viability of the mammalian germline, functioning as a ‘stem cell survival’ factor (13,14), and in the induction of pluripotency in somatic cells (15). Several investigators have reported that the β1-integrin might be a stem cell marker in certain tissues, including epidermis (16-18), testis (19), and colonic crypts (20). Recently, some human primary cancers and cell lines have been shown to possess “side population” (SP) cells that have been described as CSCs (21-25). In addition, SP cells appear to be enriched in stem cells, which play a pivotal role in normal development and cancer biology. Thus, they could provide a useful tool and a readily accessible source for stem cell studies in both the normal and cancerous settings.
Radiation oncologists have been advocating the existence of stem cells in normal tissues and cancers for decades (26). We therefore reasoned that radioresistant esophageal carcinoma cells established by continuous FIR might harbor esophageal CSCs. Telomerase is required for stem cells including esophageal cancer to proliferate (27-29). The human telomerase reverse transcriptase promoter (hTERT) can be useful for targeted therapy of CSCs. Our recent study showed that the adenoviral vector Ad/TRAIL-F/RGD under control of the hTERT promoter can sensitize lung cancer and esophageal cancer cells to radiation (30,31). Recently, we constructed a TRAIL-expressing oncolytic adenoviral vector (Ad/TRAIL-E1), in which the expressions of both the TRAIL and E1A genes are under the control of a synthetic promoter consisting of sequences from the hTERT promoter and a minimal cytomegalovirus early promoter. We hypothesized that Ad/TRAIL-E1 may preferentially target and eliminate esophageal CSCs. This is the first report of establishing radioresistant CSC-like cells with high telomerase activity and specific targeting by an hTERT promoter–controlled replicating oncolytic adenoviral vector carrying apoptotic TRAIL and E1A genes.
Materials and Methods
Cell Lines and Cell Culture
Cells of the human esophageal adenocarcinoma cancer cell line Seg-1 were obtained from David Beer (University of Michigan, Ann Arbor, MI). The human esophageal squamous cancer cell line TE-2 was obtained from the Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer (Tohoku University, Sendai, Japan) (32). The two cell lines were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 1% glutamine, and 1% antibiotics and cultured at 37°C in a humidified incubator containing 5% CO2. Normal human fibroblast cells (NHFB) (Clonetics, San Diego, CA) were cultured in medium recommended by the manufacturer.
Establishment of Radioresistant Cell Lines
The cells were first grown to approximately 50% confluence in 10-cm dishes. Cells were treated with 2 Gy of γ-radiation in a Cs-137 unit (model E-0103; U. S. Nuclear Corp., Burbank, CA) at room temperature and then returned to the incubator. When they reached approximately 80% confluence, the cells were trypsinized and subcultured into new dishes. When they reached approximately 50% confluence, the cells were again irradiated (second fraction). The fractionated irradiations were continued until the total dose reached 60 Gy. The parental cells were subjected to identical trypsinization, replating, and culture conditions but were not irradiated. For all assays on irradiated cells, there was at least a 2-week interval between the last 2-Gy FIR and the experiment.
Adenoviral Vectors
The hTC promoter was created as described previously (33). Ad/CMV-GFP, Ad/TRAIL-RGD, Ad/GFP-E1, and Ad/TRAIL-E1 have been described previously (34,30,31,35,36). The recombinant oncolytic vector Ad/TRAIL-E1 was constructed by placing hTC upstream of both the TRAIL expression cassette and the E1A gene in a shuttle plasmid designated pAd/hTC-TRAIL-E1. The plasmid was then transfected with a ClaI-digested 35-kb fragment of adenovirus type 5 into 293 cells, and Ad/TRAIL-E1 was identified by PCR analysis. The vector was plaque purified twice, expanded in 293K cells, and purified by two cycles of cesium chloride banding. The sequences in the hTC-TRAIL cassette and the hTC-E1 region were verified by DNA sequencing. Vector amplification, purification, titration, and quality tests were performed as previously reported (37). Unless otherwise specified, Ad/CMV-GFP was used as the vector control, and PBS was used as the mock control. Ad/TRAIL-RGD was used as a control of non-oncolytic adenovirus expressing TRAIL gene and Ad/GFP-E1 was used as a control of oncolytic adenovirus without TRAIL.
Cell Viability and Clonogenic Assay
The viability of the cell lines was determined by using the sulforhodamine B colorimetric assay, as previously described (38). For clonogenic assays, cells derived from parental cells or established radioresistant cell were trypsinised, both passed through a 40-μm sieve, and immediately irradiated at room temperature with the 137Cs laboratory irradiator at a dose rate of 3.07 Gy/minute for the time required to generate a dose curve of 0, 2, 4, 6, and 8 Gy. Corresponding controls were sham irradiated. Colony-forming assays were performed immediately after irradiation by replating cells into triplicate 100-mm culture dishes using same method described previously (31, 32). Each experiment was performed in triplicate and repeated at least twice.
To measure long-term cell killing effects of various Adenoviral vectors in vitro, cells were seeded into 6-well plates at a density of 300 cells per well in normal culture medium overnight and were then treated with Ad/CMV-GFP, Ad/TRAIL-F/RGD or Ad/TRAIL-E1 at 100 MOI. Cells treated with PBS alone were used as the control. The cells were then cultured in an incubator containing 5% CO2 at 37°C for 14 days.
Flow Cytometric Assay
We used the flow cytometric assay to determine TRAIL, β-catenin, Oct3/4, β1 integrin, CAR (Coxsackie-adenovirus receptor) expression, and apoptosis. Flow cytometric evaluation of apoptosis was performed as previously described (31,35). Both floating and attached cells were collected and washed twice with cold PBS. The cells were then fixed with cold 70% ethanol and kept overnight at 4°C. Thirty minutes before the flow cytometric assay, propidium iodide staining (1 mL of propidium iodide, 10 μL of RNase, and 9 mL of PBS, for a final concentration of 50 μg/mL) was performed. The percentages of cells in the sub-G1, G1, S, G2, and M phases were calculated using Cell Quest software (Becton Dickinson, San Jose, CA). Fluorescence-activated cell sorting (FACS) analysis for TRAIL or CAR expression on the cell surface was performed as described previously (34,39). In brief, 1 × 106 cells suspended in 100 μL of PBS were incubated at 4°C for 1 hour with a rabbit polyclonal anti-TRAIL antibody (H257, Santa Cruz Biotechnology) or a mouse monoclonal anti-CAR (RmcB, Upstate) at a concentration of 10 μL/mL. The cells were washed twice with 1% fetal bovine serum in PBS and then incubated in the dark at 4°C for 30 minutes with a FITC-labeled goat anti-rabbit or anti-mouse immunoglobulin antibody (PharMingen, San Diego, CA) at a concentration of 10 μL/mL. After two more washes with 1% fetal bovine serum in PBS, the cells were suspended in 1% formaldehyde in PBS and subjected to fluorescence-activated cell sorting analysis. Normal rabbit or mouse IgG (Santa Cruz Biotechnology) was used as a control for primary antibodies; the levels detected by this control antibody were used as a basal background.
Western Blot Analysis
Expression of E1A, β-catenin, β1 integrin, caspase-3 and caspase-8 was analyzed by western blot using same method described previously (30,31,35). Briefly, cells were cultured overnight in 10-cm dishes in normal culture medium and then treated with Ad/CMV-GFP, Ad/TRAIL-F/RGD, or Ad/TRAIL-E1 at an MOI of 100. Two days after the treatment, cells were washed with cold PBS and lysed in Laemmli's lysis buffer. Equal amounts of protein and lysate were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to enhanced chemiluminescence (ECL) membranes (Hybond; Amersham Corp., Arlington Heights, IL). These membranes were blocked, washed and then incubated with primary antibodies. Rabbit anti-human caspase-3 and anti-E1A were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse antihuman β-catenin and caspase-8 were obtained from BD Pharmingen (San Diego, CA). After a second washing, membranes were incubated with peroxidase-conjugated secondary antibodies and developed with an ECL kit (Amersham Bioscience, Buckinghamshire, United Kingdom). β-Actin was used as a loading control.
Side Population Analysis Using Flow Cytometry
The cells were detached from the dishes with trypsin-EDTA (Invitrogen) and suspended at 2 × 106 cells/mL in Hanks' balanced salt solution supplemented with 2% fetal calf serum and 10 mmol/L HEPES. These cells were then incubated at 37°C for 90 minutes with 10 μg/mL Hoechst 33342 (Sigma Chemical, St Louis, MO), either alone or in the presence of 50 μg/mL verapamil (Sigma Chemical). After incubation, μ1 g/mL propidium iodide (BD Pharmingen, San Diego, CA) was added. Cell analysis and purification were performed using MoFlo carrying a triple-laser (DakoCytomation, Fort Collins, CO). Hoechst 33342 was excited with the UV laser at 350 nm and fluorescence emission was measured with 405/BP30 (Hoechst blue) and 570/BP20 (Hoechst red) optical filters. Propidium iodide labeling was measured through the 630/BP30 filter for the discrimination of dead cells.
Telomerase Activity Measurement
Telomerase activity was assessed using the TeloTAGGG Telomerase PCR ELISA kit from Roche (Roche Applied Science, Penzberg, Germany) according to the manufacturer's instructions. In brief, harvested and transfered 2×105 cells per sample, resuspend the pelleted cells in 200 ul Lysis reagent and incubated on ice for 30 min. After centrifugation at 16,000 × g for 20 min at 4°C, the supernatants of lysates were collected. 3 ul cell extract (corresponding to 3× 103 cell equivalents) were then incubated with biotinylated telomerase substrate oligonucleotide (P1-TS primer) at 25°C for 20 min. Telomerase inactivation at 94°C for 5 min. The extended products were amplified by PCR using P1-TS and P2 primers for 30 cycles. Biotinylated telomeric repeat amplification products were incubated for 2 h at 37°C with a digoxigenin-labeled detection probe complementary to the telomeric repeat sequence and immobilized onto streptavidin-coated microtiter plates. Next, the wells were incubated for 30 min at room temperature with peroxidase-labeled anti-digoxigenin polyclonal antibody. Finally, the amount of telomeric repeat amplification products was determined after the addition of the peroxidase substrate (3,3′,5,5′-tetramethylbenzidine). The absorbance of each batch was measured at a wavelength of 450 nm (with a reference wavelength of approx. 690 nm; Vmax Kinetic microplate reader, Molecular Devices, Sunnyvale, CA) within 30 min after addition of the Stop reagent. While 293 cell line as positive control, 293 cell were heat-treated (85°C for 10 min) as negative control. Additionally, samples and controls were tested in the sample batch throughout the study. Each experiment was performed in quadruplicate during the same day and repeated three times.
Animal Study
All animals were maintained, and animal experiments were performed, under National Institutes of Health and institutional guidelines established for the Animal Core Facility at The University of Texas M. D. Anderson Cancer Center.
Prior to tumor cell inoculation, female nu/nu mice (4–6 weeks old; Charles River Laboratories, Wilmington, MA) were subjected to 3.5 Gy of total body irradiation from a Cs-137 radiation source (model E-0103; U. S. Nuclear Corp.). Seg-1R cells were used to establish subcutaneous tumors in the mice. Briefly, 3 × 106 cells were injected into the right hind leg of each mouse. When the average size of tumors reached 4 to 5 mm in diameter, mice were randomly divided into five groups: PBS, Ad/CMV-GFP, Ad/GFP-E1, Ad/TRAIL-F/RGD, and Ad/TRAIL-E1 (10 mice/group). For each treatment, 100 μL of PBS with or without 5 × 1010 vp adenovirus was injected intratumorally with a 27-gauge needle. The treatments were repeated every 3 days for a total of six treatments. Mice were ear-tagged so that data obtained from individual animals could be traced. Tumor dimensions were measured two times per week using digital calipers. Tumor volume was calculated using the equation V (mm3) = a × b2/2, where a is the largest diameter and b is the smallest diameter. The mice were killed when their tumors reached 15 mm in diameter, in which time, mice was expected to die within 2 weeks. The survival times of mice were estimated based on the date of killing. In another set of experiment, 25 mice bearing Seg-1R were randomly assigned into same five treatment groups (n=5). Each tumor was treated only once, and after 3 days the whole tumor was removed for the adenovirus replication assay and TUNEL assay.
Adenovirus Replication Assay In Vivo
To assess the efficacy of adenovirus replication in vivo, we removed whole tumors from mice 3 days after they were treated only once with various adenoviruses; the tumors were homogenized in 0.5 ml of cold PBS and centrifuged. The viral vector replication in the supernatant of tumor lysate was determined using the tissue culture infectious dose 50 (TCID50) assay in fresh 293 cells as described previously (40).
TUNEL Assay
To detect apoptotic cells in tumors, we used the in situ Cell Death Detection kit, POD (Roche Applied Science, Indianapolis, IN). The cells were stained according to the manufacturer's instructions, counterstained with hematoxylin, and viewed under a light microscope. A brown color indicates apoptotic nuclei as visualized using DAB substrate. Apoptotic cells were counted under a light microscope (×400 magnification) in randomly chosen fields, and the apoptosis index was calculated as a percentage of at least 1,000 scored cells.
Statistical Analysis
The data were expressed as means and 95% confidence intervals (CIs). The statistical significance of the differences in the in vitro was determined by using the Student's t test (two-tailed). A difference was considered statistically significant when the P value was 0.05 or less. Differences in tumor growth in vivo among the treatment groups were assessed by ANOVA with a repeated measurement module. ANOVA was performed to determine statistical significance between treatment groups by using the SAS procedure mixed with SAS version 6.12 software (Statistica, StatSoft, Tulsa, OK). Survival was assessed by using the Kaplan-Meier method.
Results
Identification of CSC-like Cell Characteristics and Expression of Higher Telomerase Activity in Established Radioresistant Esophageal Cancer Cell Lines
To explore the molecular mechanisms involved in radiation-resistance, we established radioresistant esophageal cancer cell lines by applying repeated 2-Gy FIR. The clonogenic survival curves after different total doses for the two pairs of cell lines are shown in Fig. 1A and B. There was a significant increase of radioresistance in Seg-1R compared with Seg-1 cells (P = 0.005) and in TE-2R compared with TE-2 cells (up to 25 times) (P = 0.006). To determine whether radioresistant esophageal cancer cell lines contain candidate CSCs, we measured the stem cell survival factor markers (Fig. 1C). The percentages of cells positive for β-catenin, Oct3/4, and β1-integrin in the Seg-1R cell line were 29.38%, 27.5%, and 97.34%, respectively (Fig. 1C), compared with 4.82%, 14.9%, and 45.26% in the Seg-1 cell line (P < 0.05). Fig. 1D showed increased expression of β-catenin and β1-integrin in the Seg-1R and TE-2R cell lines compared with parental cell lines by Western blot.
Fig.1.

Establishment of radioresistant esophageal cancer cell lines by continuous FIR and identification of radioresistant cells with cancer stem-like cell characteristics. A) Clonogenic survival assay of Seg-1 cells and of Seg-1R cells that were derived from Seg-1 cells by applying continuous 2-Gy fractionated irradiation (total dose of 60 Gy). B) Clonogenic survival assay of TE-2 cells and of TE-2R cells that were derived from TE-2 cells. To determine surviving fractions, counts were normalized using the plating efficiency of the unirradiated corresponding control. Means and 95% confidence intervals are shown for three experiments, each performed in triplicate (n = 9). C) The stem cell survival factor markers β-catenin, Oct3/4, and β1-integrin were measured in Seg-1 cells (white) and Seg-1R cells (black) using flow cytometry and specific FITC-conjugated anti-β-catenin, anti-Oct3/4, and anti-β1 integrin antibodies. Means and 95% confidence intervals of three independent experiments (n = 3) are shown. P values comparing data from Seg-1 and Seg-1R cells were determined using a paired two-sided Student's t test; n.s. = not statistically significant. D) The expression of β-catenin and β1 integrin in Seg-1, Seg-1R, TE-2, and TE-2R cells was analyzed by Western blot. β-Actin was used as a loading control. These experiments were repeated at least three times with similar results.
To further determine whether any of the two pairs of established radioresistant cancer cell lines contained SP cells and whether such SP cells could self-renew, we stained the cells with the fluorescent dye Hoechst 33342, which has been shown to be extruded actively by the SP cells in various tissues by means of verapamil-sensitive ABC transporters; we then analyzed the cells by flow cytometry. The percentages of HoechstLow SP cells were greater in the Seg-1R (14.61%) and TE-2R (2.73%) cell lines than in the Seg-1 (3.47%) and TE-2 (0.30%) lines (Fig. 2A). In each case, the SP population was decreased greatly by treatment with verapamil (Fig. 2A), indicating that the populations were bona fide SPs. Thus, radioresistant cancer cell lines (Seg-1R and TE-2R) possess SP with Hoechst efflux characteristics similar to those defined in hematopoietic stem cells.
Fig.2.

Identification of SP cells with stem cell-like characteristics in established radioresistant esophageal cancer cell lines. A) Seg-1, Seg-1R, TE-2, and TE-2R cell lines were labeled with Hoechst 33342 dye and analyzed by flow cytometry before and after treatment with verapamil. Data from one of three independent experiments are shown. B) Seg-1R cell line sorted SP and non-SP cells recovered in culture and photographed with an inverted ×10 phase-contrast microscope. SP cells from the Seg-1R cell line formed tight colonies after 4-7 days in culture, whereas non-SP cells were scattered and did not proliferate. C) Seg-1R sorted SP cells were cultured for 7–10 days, re-sorted by flow cytometry, recovered for an additional 7–10 days, and then re-analyzed by flow cytometry. Each successive sort demonstrated the enrichment of SP cells and the presence of non-SP (NSP) cells. D) Telomerase activity was assessed using the TeloTAGGG Telomerase PCR ELISA kit in Seg-1, Seg-1R, SP/1(SP from sort#1), NSP/1(non-SP from sort#1), SP/2, NSP/2, SP/3, NSP/3 cells and TE-2, TE-2R cells, and NHFB cells. 293 cells as positive control, 293 cells were heat-treated (85°C for 10 min) as negative control. Each experiment was performed in quadruplicate during the same day and repeated three times.
To compare the ability of Seg-1R SP cells and non-SP cells in producing SP cells, we stained Seg-1R cells with Hoechst 33342 and sorted them into SP and non-SP fractions by flow cytometry, and we then cultured equal numbers of SP and non-SP cells separately. As they proliferated, the cells in the two populations had different morphologies. SP cells formed characteristic compact circular colonies with a cobblestone appearance and survived numerous passages (Fig. 2B). Non-SP cells were sparse and failed to proliferate beyond 2 weeks (Fig. 2B). These differences were not a consequence of prolonged Hoechst retention in the non-SP cells because propidium iodide was used to gate out all nonviable cells. When we restained the cells with Hoechst 33342 and re-analyzed them by flow cytometry, we found that the cultures initiated with SP cells contained both SP and non-SP cells. Serial sorting and reanalysis (total passages = 3) of SP cells demonstrated enrichment of the SP cells (from 11.57% to 19.80% to 35.03%) and the presence of non-SP cells (Fig. 2C), suggesting asymmetric division occurred during culture. Thus, SP cells are able to self-renew, be enriched, and produce non-SP cells when recovered and serially sorted in culture.
To demonstrate the difference of telomerase between parental and radioresistant, SP and non-SP cells, we measured the telomerase activity using the TeloTAGGG telomerase PCR-ELISA kit. As shown in Fig. 2D, the telomerase activity of Seg-1R, Seg-1R SP and TE-2R cells was significantly higher than Seg-1, Seg-1R non-SP and TE-2 cells respectively (P < 0.05). We further tested the telomerase activity of the serial sorted cells. Fig. 2D showed that the telomerase activity of SP/1(SP from sort#1), SP/2(SP from sort#2), and SP/3(SP from sort#3) was also significantly higher than NSP/1, NSP/2, and NSP/3 respectively (P < 0.05). However, the telomerase activity of SP/3 was only slightly higher than that of SP/1 and SP/2 with no significant difference (P > 0.05). Certainly, the telomerase activity of these cancer cell lines was very significantly higher than NHFB cells (P < 0.001).
Preferentially Targeted Radioresistant Esophageal CSC-like Cells by Telomerase-specific Oncolytic Adenovirus
To detect the cell-killing effects of Ad/TRAIL-E1, we treated Seg-1, Seg-1R and TE-2, TE-2R, and NHFB cells with Ad/CMV-GFP, Ad/TRAIL-F/RGD, and Ad/TRAIL-E1 at MOIs ranging from 30 to 3000 vp. Treatment with Ad/TRAIL-F/RGD or Ad/TRAIL-E1 resulted in a dose-dependent cell-killing effect at MOIs of 30 to 3,000 vp in all cancer cell lines tested but not in NHFB cells (Fig. 3A). The result also showed that the cell-killing effects of Ad/TRAIL-E1 on esophageal carcinoma cell lines were dramatically better than those of Ad/TRAIL-RGD. Increased cell-killing effects on radioresistant cells by Ad/TRAIL-E1 compared with the parental cell line were observed. These findings support the notion that Ad/TRAIL-E1 selectively targets radioresistant CSC-like cells. In NHFB cells, however, minimal toxicity was observed in treatment with Ad/TRAIL-E1 at MOIs of 1000 to 3000 vp. This suggests that Ad/TERT-TRAIL-E1 is highly oncolytic but minimally toxic to normal cells.
Fig. 3.

High cell-killing and long-term effects of Ad/TRAIL-E1 on radioresistant esophageal cancer cells. A) Cell viability determined by sulforhodamine B colorimetric assay in Seg-1, Seg-1R, TE-2, TE-2R, and NHFB cells on the fourth day after treatment by Ad/CMV-GFP (GFP), Ad/TRAIL-F/RGD (TRAIL), or Ad/TRAIL-E1 (TRAIL-E1) at MOIs ranging from 0 to 3,000 vp. Cells treated with PBS was used as a control, with their viability set at 1. Each experiment was performed in quadruplicate and repeated at least twice. Points, mean of quadruplicate assay results; bars, SD. B) The relative clonogenic survival of the four cancer cell lines after exposure to PBS, Ad/CMV-GFP, Ad/TRAIL-F/RGD, or Ad/TRAIL-E1 at 300 MOI. Each experiment was performed in triplicate and repeated at least twice. Columns, mean results of triplicate assays; bars, SD.
To further determine the long-term effects of various vectors in vitro, we performed a clonogenic formation assay. Ad/TRAIL-E1 inhibited colony formation of esophageal carcinoma cells and radioresistant CSC-like cells significantly more than did Ad/TRAIL-RGD or Ad/CMV-GFP (Fig. 3B). We also found that long-term effects of Ad/TRAIL-E1 on radioresistant CSC-like cells (Seg-1R and TE-2R) were more effective than on parental esophageal carcinoma cells (Seg-1 and TE-2).
To test whether the enhanced cell killing produced by Ad/TRAIL-E1 was associated with apoptosis, we quantified the sub-G1 population of cells after treatment with various vectors in FACS. Seg-1 and Seg-1R, TE-2 and TE-2R, and NHFB cells were treated with our tested viruses at MOIs of 100 and 1000 vp/cell for 48 hours. The result showed that treatment with Ad/TRAIL-E1 or Ad/TRAIL-RGD induced apoptosis in all four cancer cell lines tested, as evidenced by a marked increase of sub-G1 cells compared with cells treated with Ad/CMV-GFP or PBS (Fig. 4A and B), whereas the cells treated with PBS or Ad/CMV-GFP had only background levels of apoptosis. No obvious apoptosis was found in NHFB cells treated with the vectors compared with PBS. In all four cancer cell lines tested, Ad/TRAIL-E1 elicited apoptosis at an MOI of 100 vp/cell as effectively as Ad/TRAIL-RGD induced apoptosis at an MOI of 1000 vp/cell, suggesting that the cell-killing potency of Ad/TRAIL-E1 is at least 10-fold higher than that of Ad/TRAIL-RGD. Notably, both Ad/TRAIL-E1 and Ad/TRAIL-RGD induced higher apoptosis in radioresistant cancer stem-like cells (Seg-1R and TE-2R) than in the parental esophageal carcinoma cells (Seg-1 and TE-2) at the same MOIs.
Fig. 4.

Apoptosis induction and transgene expression of Ad/TRAIL-E1 in radioresistant esophageal cancer cells. A) and B) The sub-G1 population of Seg-1, Seg-1R, TE-2, TE-2R, and NHFB cells after treatment with various vectors at MOIs of 100 and 1000 vp/cell for 48 hours in a fluorescence-activated cell sorting analysis. C) Western blot analysis of expression of E1A and cleavage of caspase-8 and caspase-3 in Seg-1 and Seg-1R cells infected with 100 MOI of Ad/CMV-GFP, Ad/TRAIL-RGD, or Ad/TRAIL-E1 for 48 hours. D) The expression of TRAIL was measured in Seg-1 and Seg-1R cells using flow cytometry and specific FITC-conjugated anti- TRAIL antibody. Columns, mean results of triplicate assays; bars, SD.
To verify that tumor killing is associated with expression of the apoptotic genes TRAIL and E1A, Seg-1 and Seg-1R cells infected with 100 MOI of Ad/CMV-GFP, Ad/TRAIL-RGD, or Ad/TRAIL-E1 were incubated for 48 hours, harvested, and then analyzed by western blotting. Ad/CMV-GFP and Ad/TRAIL-RGD do not contain E1A and are not capable of replicating on their own, whereas Ad/TRAIL-E1 treated cells highly expressed E1A (Fig. 4C). The expression of the TRAIL protein on the cell surface was determined by FACS. Increased TRAIL expression was observed in Seg-1 and Seg-1R cells treated with 100 MOI of Ad/TRAIL-RGD and Ad/TRAIL-E1 vectors but not in cells treated with PBS or Ad/CMV-LacZ (Fig. 4D). Moreover, both Seg-1 and Seg-1R cells treated with Ad/TRAIL-E1 had higher expression levels of TRAIL than did cells treated with Ad/TRAIL-RGD, and expression levels of both E1A and TRAIL were higher in Seg-1R cells compared with Seg-1 cells. To confirm the induction of apoptosis by Ad/TRAIL-E1, we tested caspase-3 and caspase-8 by Western blot analysis as well. After the cells were treated with various vectors at 100 MOI for 48 hours, cleaved caspase-3 and cleaved caspase-8 were observed in cells treated with Ad/TRAIL-RGD or Ad/TRAIL-E1 (Fig. 4C). However, in comparison with cells treated with Ad/TRAIL-RGD, both Seg-1 and Seg-1R cells treated with Ad/TRAIL-E1 had notably increased cleavage of caspase-3 and caspase-8. Moreover, Seg-1R cells treated with either Ad/TRAIL-E1 or Ad/TRAIL-RGD were detected more expression of cleaved caspase-3 and cleaved caspase-8 than that of Seg-1 cells.
To explore the other mechanism that radioresistant cancer cells were more sensitive to adenovirus than parental cells, we analyzed the expression of CAR, αVβ3, αVβ5-integrins, and DR4 in these cell lines using FACS. As shown in Fig. 5A and 5B, the expression of CAR, but not expressions of αVβ3, αVβ5-integrins, or DR4, was significantly higher in the two radioresistant cancer cell lines than in the parental cells (P < 0.05). Thus, we first elucidated the reason that adenovirus efficacy varied among different cell lines is related to that radioresistant cancer cells is associated with higher expression of CAR. We further compared the CAR levels in the SP with the non-SP population of cells. Interestingly, the expression of CAR in the SP fraction was significantly higher than that of the non-SP population of cells (Fig. 5C, P < 0.05).
Fig. 5.

Higher expression of CAR in radioresistant esophageal cancer cells. A) The expressions of CAR, αVβ3, αVβ5 integrins, and DR4 were measured in Seg-1 cells (white) and Seg-1R cells (black) using flow cytometry and specific FITC-conjugated anti-CAR, anti-αVβ3, anti-αVβ5, and anti-DR4 antibodies. B) The expressions of CAR, αVβ3, αVβ5 integrins, and DR4 were measured in TE-2 cells (white) and TE-2R cells (black). C) The expression of CAR in the SP fraction (Seg-1RSP) and the non-SP (Seg-1RNSP) population of Seg-1R cells. Means and 95% confidence intervals of three independent experiments (n = 3) are shown. P values comparing data from Seg-1 and Seg-1R cells were determined using a paired two-sided Student's t test; n.s. = not statistically significant.
Ad/TRAIL-E1 Suppressed Tumor Growth and Prolonged Survival in Mice Bearing Radioresistant Esophageal CSC-like Cells
To further evaluate the antitumor activity of Ad/TRAIL-E1 in vivo, we established human radioresistant esophageal adenocarcinoma xenografts in 6-week-old female nude mice by inoculating them with Seg-1R cells. As shown in Fig. 6A and B, Ad/TRAIL-RGD led to significant tumor suppression and prolonged survival time in tumor-bearing mice compared with mock treatment or control vector (Ad/CMV-GFP) treatment (P = 0.0008 for tumor volume and P = 0.0002 for survival). Ad/GFP-E1 also led to significant tumor suppression and prolonged survival time in tumor-bearing mice compared with mock treatment or control vector (Ad/CMV-GFP) treatment (P = 0.0025 for tumor volume and P = 0.0334 for survival). Remarkably, treatment with Ad/TRAIL-E1 suppressed tumor growth and enhanced survival in tumor-bearing mice compared with mock treatment or control vector (Ad/CMV-GFP) treatment (P = 0.00004 for tumor volume and P = 0.00002 for survival). Furthermore, treatment with Ad/TRAIL-E1 significantly retarded tumor growth and prolonged survival time compared with treatment with Ad/TRAIL-RGD (P = 0.00196 for tumor volume and P = 0.02620 for survival) and Ad/GFP-E1 (P = 0.00103 for tumor volume and P = 0.0008 for survival). However, treatment with Ad/TRAIL-RGD was no significantly different compared with Ad/GFP-E1 (P = 0.33 for tumor volume and P = 0.057 for survival). The mean survival times for mice treated with PBS or Ad/CMV-GFP, Ad/TRAIL-RGD, Ad/GFP-E1, Ad/TRAIL-E1 were 55, 64, 86, 74 and 125 days, respectively. Treatment with Ad/TRAIL-E1 led to tumor-free survival in 40% mice for 180 days, the longest time point observed in this study. We also evaluated possible treatment-related toxicity by measuring serum liver enzymes and blood cell counts (RBC, WBC, and platelets) 5 days after the final treatment injection. All of the results were within the reference ranges, and no substantial differences were found between the groups (data not shown). In addition, no weight loss was noted in the mice.
Fig. 6.

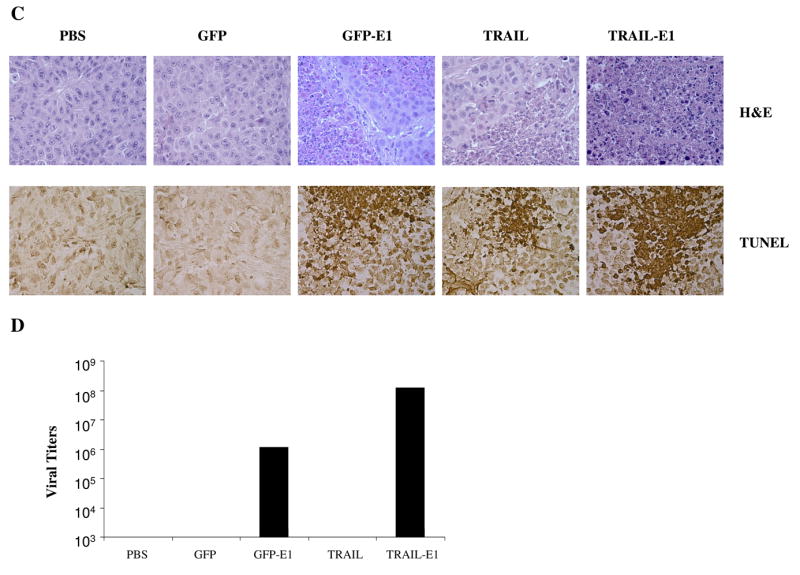
Ad/TRAIL-E1 suppressed tumor growth and prolonged survival in mice bearing radioresistant esophageal adenocarcinoma cells. Seg-1R cells were grown as xenograft tumors in nude mice, and mice bearing tumors that reached 4 to 5 mm in diameter were intratumorally injected with PBS, Ad/CMV-GFP, Ad/TRAIL-F/RGD, Ad/GFP-E1 or Ad/TRAIL-E1 for a total of six injections, with vectors of 5 × 1010 vp each time. Arrows indicate times when treatments were given. A) Tumor volume was monitored over time after the inoculation of tumor cells. Points, means; bars, SE. ANOVA was performed to determine statistical significant differences between treatment groups. B) Kaplan-Meier survival curves for the treatment groups. C) Representative fields showing hematoxylin and eosin–stained sections (upper) and tumor cell apoptosis by TUNEL assay (lower) in Seg-1R tumors from mice. Mice received the various treatments only once, and tumors were removed 3 days after the treatment. Brown color indicates apoptotic nuclei as visualized using DAB substrate. Apoptotic cells were counted under a light microscope in randomly chosen fields. D) The viral vector replication in supernatants of tumor lysates was determined using the tissue culture infectious dose 50 (TCID50) assay in fresh 293 cells.
Adenovirus Replication and Apoptosis Induction by Ad/TRAIL-E1 In Vivo
As shown in Fig. 6C, H&E staining revealed necrosis within tumors from mice treated with Ad/TRAIL-RGD and Ad/GFP-E1. Furthermore, Ad/TRAIL-E1 induced more tumor necrosis. To assess apoptosis induction in vivo, we performed TUNEL staining on tumor sections. As shown in Fig. 6C, Ad/TRAIL-E1 resulted in a significantly higher apoptotic index (43.7%) compared with Ad/TRAIL-F/RGD (19.6%) or Ad/GFP-E1 (17.5%), after subtracting the background level of 3.5% (P = 0.00269). To assess the efficacy of adenovirus replication in vivo, we determined the viral vector replication in supernatants of tumor lysate using the tissue culture infectious dose 50 (TCID50) assay in fresh 293 cells. We found 107 to 108 infectious viral units in the lysates of tumor from mice treated with Ad/TRAIL-E1 and 106 to 107 infectious viral units in the lysates of tumor from mice treated with Ad/GFP-E1 (Fig. 6D). No detectable infectious units (i.e., <103, the lowest dilution the assay can measure) were found in the lysates of tumors from mice treated with Ad/CMV-GFP or Ad/TRAIL-RGD, demonstrating that the oncolytic vectors but not the E1-defective vectors were replicating in those tumor cells.
Discussion
The existence of CSCs has profound implications for cancer biology and therapy (41). It has been proposed that CSCs may be particularly resistant to chemotherapy and radiation therapy, although good evidence supporting this notion has been lacking (42). Recently one published report suggested that glioblastoma stem cells are radioresistant and may therefore contribute to treatment failures (43); another paper demonstrated that breast cancer–initiating cells are relatively radioresistant compared with the remainder of breast cancer cells (44). We examined whether radioresistant cell lines could reflect biological characteristics of stem-like cells. We found that the putative stem cell markers β-catenin, Oct3/4, and β1-integrin expression were significantly increased in radioresistance cells. In addition, we noticed that several so-called “stemness genes” were upregulated and apoptosis genes were downregulated in Seg-1R cells compared with Seg-1 cells in microarray analysis (data not shown). Moreover, serial sorting and reanalysis of SP cells from the Seg-1R cell line demonstrated enrichment of the SP and the presence of non-SP cells, suggesting that asymmetric division occurred during culture. These results indicate that a tumor hierarchy exists in which SP cells can generate both SP and non-SP cells. SP cells appear to be able to self-renewal and proliferate but not in non-SP cells. This is in accordance with previous observations that the SP fraction can divide asymmetrically and display a capacity for self-renewal (23,45,46). Finally, higher telomerase activity was detected in radioresistant CSCs, particularly SP cells compared with parental cells, indicating CSCs may present a reservoir with unlimited proliferation potential for generating cancer cells. Our results demonstrate that radioresistant stem-like solid cancer cell lines can be established in radioresistant esophageal cancer cells by continuous FIR. It would be interesting to see whether the accumulation of CSCs after FIR can also be documented in esophageal cancer patients treated with standard (50.4-Gy) radiotherapy and chemotherapy and whether the percentage of CSCs within cancer tissues predicts radiosensitivity.
The radioresistance may be caused by radiation-induced gene mutation, altered gene expression, or enrichment of CSCs since CSCs may be resistant to radiotherapy. FIR mimics the clinical situation of development of radioresistance after standard chemoradiotherapy. Elimination of radioresistance is crucial to achieving a pathological complete response and a cure. There are a variety of markers and techniques to isolate and eliminate these rare CSCs (21,45-48). In primary breast cancer, it has been demonstrated that the CSCs are telomerase positive (48). The telomerase inhibitors being developed might therefore target CSCs as well as the more mature cancer cells (27). In the current study, we found that oncolytic adenoviral vector Ad/TRAIL-E1 appears to preferentially targets esophageal cancer cells, particularly CSCs.
Factors determining the susceptibility of the targeting radioresistant cancer cells to the adenovirus are not entirely known. In this study, we observed that radioresistant cancer cells were more sensitive to adenovirus than were parental cells. Overexpression of telomerase activity in esophageal radioresistant CSCs-like cells may explain partially the increased sensitivity of esophageal radioresistant cancer cell lines to Ad/TRAIL-E1. Further study revealed increased CAR expression and elevated transgene expression in the radioresistant cancer cell lines. Increased CAR expression on the cell surface of radioresistant cancer cells may contribute to the efficiency of adenovirus-mediated gene transfer. Generally, one major weakness of adenovirus treatment is that adenoviral infection efficiency depends on the expression of CAR, which is not highly expressed on the cell surface of many types of cancer cells (49,50). However, our study showed that the CAR was highly expressed on the cell surface of radioresistant CSC-like cells. In addition, we found that the telomerase activity of SP/1(SP from sort#1), SP/2(SP from sort#2), and SP/3(SP from sort#3) was significantly higher than NSP/1, NSP/2, and NSP/3 respectively (Fig.2D). We also found that the expression of CAR in the SP fraction was significantly higher than in the non-SP population of cells (Fig.5C). It appears that both increased telomerase activity and expression of CAR play role in the efficacy of Ad/TRAIL-E1. Certainly, the relative contribution of these two processes and detail mechanism in the tumoricidal effect of Ad/TRAIL-E1 remains to be further elucidated.
In terms of toxicity, our previous report showed that hTERT promoter activity was low in mesenchymal stem cells and Ad/TRAIL-F/RGD was associated with very low toxicity to these mesenchymal stem cells in vitro even at high doses (36). In the current study, we evaluated potential treatment-related toxicity of adenovirus vectors in nude mice model using local injection. We found no substantial differences among the treatment groups indicating low toxicity of Ad/TRAIL-E1. However, the potential toxicity of Ad/TRAIL-E1 using intravenous injection could be higher and further study is needed.
The Coxsackie Adenovirus Receptor (CAR) has primarily been studied in its role as the initial cell surface attachment receptor for Coxsackie and group C adenoviruses. However, CAR is a multi-faced molecule expressed by many cell types and fulfilling many but yet poorly defined cellular functions (51). It behaves typically as an adhesion molecule promoting cell aggregation and may play role in embryogenesis and stem cell lineage (51). Overexpression of CAR in radiation resistant cancer stem-like cell may be caused by preferential killing of cancer cells with low CAR expression by radiation or radiation induced CAR expression. The role of tumorigenesis of CAR remains controversial. Recent data indicated that silencing surface CAR expression abrogated xenograft tumorigenesis in vivo and colony formation in vitro (52). These data indicated that CAR expression may be needed for the efficient formation of tumors by a subset of cancer cells. It would be interesting to further explore the relationship between CAR-mediated signaling pathways and cancer stem cell properties.
Taken together, our results indicated that radioresistant cells could be established by repeated FIR and that they display CSC-like properties. Ad/TRAIL-E1 specifically targeted esophageal radioresistant CSC-like cells and prolonged survival. Identification of the esophageal CSCs would provide a critical step in advancing the development of future anti-CSCs targeted therapies.
Acknowledgments
We thank Dr. Raymond E Meyn for his advice and review of the article, Drs. James D. Cox and Jack A. Roth for their support of this project and the Department of Scientific Publications for their assistance in the preparation of this article.
Grant support: Radiology Society of North America Research Scholar Award (J.Y. C.), Career Development Award from The University of Texas Lung Cancer Specialized Programs of Research Excellence grant from National Cancer Institute (J.Y. C.), National Cancer Institute grants RO1 CA 092487 and RO1 CA 098582 (B. F.), National Institutes of Health Core Grant CA-16672, and Lockton Grant-Matching Funds (B. F.).
References
- 1.Cooper JS, Guo MD, Herskovic A, et al. Chemoradiotherapy of locally advanced esophageal cancer: long-term follow-up of a prospective randomized trial (RTOG 85-01). Radiation Therapy Oncology Group. JAMA. 1999;281:1623–7. doi: 10.1001/jama.281.17.1623. [DOI] [PubMed] [Google Scholar]
- 2.Minsky BD, Pajak TF, Ginsberg RJ, et al. INT 0123 (Radiation Therapy Oncology Group 94-05) phase III trial of combined-modality therapy for esophageal cancer: high-dose versus standard-dose radiation therapy. J Clin Oncol. 2002;20:1167–74. doi: 10.1200/JCO.2002.20.5.1167. [DOI] [PubMed] [Google Scholar]
- 3.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Poste G, Greig R. On the genesis and regulation of cellular heterogeneity in malignant tumors. Invasion Metastasis. 1982;2:137–76. [PubMed] [Google Scholar]
- 5.Poste G, Tzeng J, Doll J, et al. Evolution of tumor cell heterogeneity during progressive growth of individual lung metastases. Proc Natl Acad Sci U S A. 1982;79:6574–8. doi: 10.1073/pnas.79.21.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tu SM, Lin SH, Logothetis CJ. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002;3:508–13. doi: 10.1016/s1470-2045(02)00820-3. [DOI] [PubMed] [Google Scholar]
- 7.Diehn M, Clarke MF. Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst. 2006;98:1755–7. doi: 10.1093/jnci/djj505. [DOI] [PubMed] [Google Scholar]
- 8.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–51. [PubMed] [Google Scholar]
- 9.Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 10.Theodosiou NA, Tabin CJ. Wnt signaling during development of the gastrointestinal tract. Dev Biol. 2003;259:258–71. doi: 10.1016/s0012-1606(03)00185-4. [DOI] [PubMed] [Google Scholar]
- 11.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Welm B, Podsypanina K, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci U S A. 2003;100:15853–8. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–91. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 14.Gidekel S, Pizov G, Bergman Y, Pikarsky E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell. 2003;4:361–70. doi: 10.1016/s1535-6108(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 16.Jones PH, Watt FM. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73:713–24. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- 17.Jones PH, Harper S, Watt FM. Stem cell patterning and fate in human epidermis. Cell. 1995;80:83–93. doi: 10.1016/0092-8674(95)90453-0. [DOI] [PubMed] [Google Scholar]
- 18.Li A, Simmons PJ, Kaur P. Identification and isolation of candidate human keratinocyte stem cells based on cell surface phenotype. Proc Natl Acad Sci U S A. 1998;95:3902–7. doi: 10.1073/pnas.95.7.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinohara T, Avarbock MR, Brinster RL. beta1- and alpha6-integrin are surface markers on mouse spermatogonial stem cells. Proc Natl Acad Sci U S A. 1999;96:5504–9. doi: 10.1073/pnas.96.10.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimoto K, Beauchamp RD, Whitehead RH. Identification and isolation of candidate human colonic clonogenic cells based on cell surface integrin expression. Gastroenterology. 2002;123:1941–8. doi: 10.1053/gast.2002.37065. [DOI] [PubMed] [Google Scholar]
- 21.Al Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haraguchi N, Utsunomiya T, Inoue H, et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506–13. doi: 10.1634/stemcells.2005-0282. [DOI] [PubMed] [Google Scholar]
- 23.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101:781–6. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wulf GG, Wang RY, Kuehnle I, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood. 2001;98:1166–73. doi: 10.1182/blood.v98.4.1166. [DOI] [PubMed] [Google Scholar]
- 25.Seigel GM, Campbell LM, Narayan M, Gonzalez-Fernandez F. Cancer stem cell characteristics in retinoblastoma. Mol Vis. 2005;11:729–37. [PubMed] [Google Scholar]
- 26.Lange CS, Gilbert CW. Studies on the cellular basis of radiation lethality. 3. The measurement of stem-cell repopulation probability. Int J Radiat Biol Relat Stud Phys Chem Med. 1968;14:373–88. doi: 10.1080/09553006814551221. [DOI] [PubMed] [Google Scholar]
- 27.Ju Z, Rudolph KL. Telomeres and telomerase in cancer stem cells. Eur J Cancer. 2006;42:1197–203. doi: 10.1016/j.ejca.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 28.Chang JY. Telomerase: a potential molecular marker and therapeutic target for cancer. J Surg Oncol. 2004;87:1–3. doi: 10.1002/jso.20076. [DOI] [PubMed] [Google Scholar]
- 29.Yu HP, Xu SQ, Lu WH, et al. Telomerase activity and expression of telomerase genes in squamous dysplasia and squamous cell carcinoma of the esophagus. J Surg Oncol. 2004;86:99–104. doi: 10.1002/jso.20050. [DOI] [PubMed] [Google Scholar]
- 30.Chang JY, Zhang X, Komaki R, Cheung R, Fang B. Tumor-specific apoptotic gene targeting overcomes radiation resistance in esophageal adenocarcinoma. Int J Radiat Oncol Biol Phys. 2006;64:1482–94. doi: 10.1016/j.ijrobp.2005.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Cheung RM, Komaki R, Fang B, Chang JY. Radiotherapy sensitization by tumor-specific TRAIL gene targeting improves survival of mice bearing human non-small cell lung cancer. Clin Cancer Res. 2005;11:6657–68. doi: 10.1158/1078-0432.CCR-04-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukuda K, Sakakura C, Miyagawa K, et al. Differential gene expression profiles of radioresistant oesophageal cancer cell lines established by continuous fractionated irradiation. Br J Cancer. 2004;91:1543–50. doi: 10.1038/sj.bjc.6602187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis JJ, Wang L, Dong F, et al. Oncolysis and suppression of tumor growth by a GFP-expressing oncolytic adenovirus controlled by an hTERT and CMV hybrid promoter. Cancer Gene Ther. 2006;13:720–3. doi: 10.1038/sj.cgt.7700944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong F, Wang L, Davis JJ, et al. Eliminating established tumor in nu/nu nude mice by a tumor necrosis factor-alpha-related apoptosis-inducing ligand-armed oncolytic adenovirus. Clin Cancer Res. 2006;12:5224–30. doi: 10.1158/1078-0432.CCR-06-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin T, Gu J, Zhang L, et al. Targeted expression of green fluorescent protein/tumor necrosis factor-related apoptosis-inducing ligand fusion protein from human telomerase reverse transcriptase promoter elicits antitumor activity without toxic effects on primary human hepatocytes. Cancer Res. 2002;62:3620–5. [PubMed] [Google Scholar]
- 36.Jacob D, Davis J, Zhu H, et al. Suppressing orthotopic pancreatic tumor growth with a fiber-modified adenovector expressing the TRAIL gene from the human telomerase reverse transcriptase promoter. Clin Cancer Res. 2004;10:3535–41. doi: 10.1158/1078-0432.CCR-03-0512. [DOI] [PubMed] [Google Scholar]
- 37.Gu J, Kagawa S, Takakura M, et al. Tumor-specific transgene expression from the human telomerase reverse transcriptase promoter enables targeting of the therapeutic effects of the Bax gene to cancers. Cancer Res. 2000;60:5359–64. [PubMed] [Google Scholar]
- 38.Pauwels B, Korst AE, de Pooter CM, et al. Comparison of the sulforhodamine B assay and the clonogenic assay for in vitro chemoradiation studies. Cancer Chemother Pharmacol. 2003;51:221–6. doi: 10.1007/s00280-002-0557-9. [DOI] [PubMed] [Google Scholar]
- 39.Pearson AS, Koch PE, Atkinson N, et al. Factors limiting adenovirus-mediated gene transfer into human lung and pancreatic cancer cell lines. Clin Cancer Res. 1999;5:4208–13. [PubMed] [Google Scholar]
- 40.Fang B, Ji L, Bouvet M, Roth JA. Evaluation of GAL4/TATA in vivo. Induction of transgene expression by adenovirally mediated gene codelivery. J Biol Chem. 1998;273:4972–5. doi: 10.1074/jbc.273.9.4972. [DOI] [PubMed] [Google Scholar]
- 41.Clarke MF, Dick JE, Dirks PB, et al. Cancer Stem Cells--Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 42.Diehn M, Clarke MF. Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst. 2006;98:1755–7. doi: 10.1093/jnci/djj505. [DOI] [PubMed] [Google Scholar]
- 43.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 44.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–85. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 45.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 46.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–9. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hadnagy A, Gaboury L, Beaulieu R, Balicki D. SP analysis may be used to identify cancer stem cell populations. Exp Cell Res. 2006;312:3701–10. doi: 10.1016/j.yexcr.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 48.Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 49.Ito H, Aoki H, Kuhnel F, et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst. 2006;98:625–36. doi: 10.1093/jnci/djj161. [DOI] [PubMed] [Google Scholar]
- 50.Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–3. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 51.Hauwel M, Furon E, Gasque P. Molecular and cellular insights into the coxsackie-adenovirus receptor: Role in cellular interactions in the stem cell niche. Brain Research Reviews. 2005;48:265–272. doi: 10.1016/j.brainresrev.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 52.Qin M, Escuadro B, Dohadwala M, Sharma S, Batra RK. A novel role for the coxsackie adenovirus receptor in mediating tumor formation by lung cancer cells. Cancer Res. 2004;64:6377–80. doi: 10.1158/0008-5472.CAN-04-1490. [DOI] [PubMed] [Google Scholar]