

Abstract
Immunotherapy for cancer is often limited by weak immunogenicity of tumor antigens. However, immune systems are usually strong and effective against foreign invading antigens. To test whether the destructive effect of adaptive immunity against foreign antigens can be redirected to tumors for cancer therapy, we immunized mice with adenovector expressing LacZ (Ad/CMV-LacZ). Subcutaneous syngeneic tumors were then established in the immunized animals or in naïve animals. The immune response against adenovirus or LacZ was redirected to tumors by intratumoral injection of Ad/CMV-LacZ. We found that immunization and treatment with the adenovector dramatically reduced the tumor growth rate compared with intratumoral administration of adenovector in naïve mice. Complete tumor regression was observed in about 50% of the immunized animals but not in the naïve animals. Similar effects were observed when oncolytic vaccinia virus was used to immunize and treat tumors. Lymphocyte infiltration in tumors was dramatically increased in the immunized group when compared with other groups. Moreover, immunity against parental tumor cells was induced in the animals cured with immunization and treatment with Ad/CMV-LacZ, as evidenced by the lack of tumor growth when the mice were challenged with parental tumor cells. Taken together, these results suggest that redirecting adaptive immunity against foreign antigens is a potential approach for anticancer therapy and that pre-existing immunity could enhance virotherapy against cancers.
Keywords: Gene therapy, cancer vaccine, virotherapy, immunotherapy
Introduction
The presence of tumor antigens derived from mutant proteins, dysregulated or overexpressed proteins, and viral oncogenic proteins are the cornerstones of tumor immunology and together are the driving force of cancer immunotherapy. Because multiple gene mutations and dysregulated gene expression are involved in the development of malignant phenotypes, the identification of tumor antigens and the use of the immune system for cancer therapy have been intensively investigated.1,2 Both passive and active immunotherapies targeted to tumor antigens have been explored for cancer treatment. Whereas passive immunotherapy uses antibodies,3 cytotoxic T lymphocytes specific to tumor antigens,4 or both, active immunotherapy uses tumor antigens for vaccination, co-stimulatory molecules for enhancement of antigen signals, cytokines for activation and proliferation of immunoactive cells, dendritic cells for effective processing and presentation of tumor antigens, and alloantigens, xenoantigens, and various adjuvants for enhancement of the immune response.5-9 All these therapeutic strategies rely on effective immunity directly against tumor antigens. However, because of the heterogenecity of tumor cells among patients and within a tumor mass, some of these strategies need to be individualized for clinical application or the tumor cells may easily acquire resistance. Moreover, because most tumor antigens are derived from or are similar to self-antigens, the immune response to these antigens is often suppressed or too weak to be effective.8,9
Immune systems, even those of cancer patients, can recognize and mount a response to foreign materials, such as viruses and bacteria, sufficiently enough to destruct and eliminate them. The destructive effect of adaptive immunity against foreign invaders would be beneficial for cancer therapy if this effect could be redirected to tumors. Because the destruction of foreign antigen-expressing tumor cells is carried out by active immune cells, tumor antigens present in tumor cells may be effectively processed and presented back to immune systems, thereby inducing tumor antigen-specific immunity and eliminating other tumor cells through immune response-mediated bystander effects.
To test whether adaptive immunity against foreign antigens can be used for cancer therapy, we immunized immunocompetent mice with foreign antigens by virus-mediated gene transfer. Syngeneic tumors were then established in both naïve and immunized animals, which were subsequently treated with viral vector encoding the foreign genes. The results showed that the antitumor effect of virus-mediated foreign gene transfer is more effective in immunized animals than in naïve animals. Moreover, the cured immunized animals rejected parental tumor cells, which suggested that antitumor immunity can be induced by this approach.
Materials and Methods
Cell lines and cell culture
Murine fibrosarcoma cell line UV2237, melanoma cell line K1735, breast cancer cell line LM2, and colorectal adenocarcinoma cell line CT26 were maintained in our laboratory 10 or obtained from American Type Culture Collection (Manassas, VA). Murine lung carcinoma M109 cell line was obtained from Frederick Cancer Research and Development Center, National Cancer Institute (Frederick, MD). The cells were routinely cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum, 100 units/mL penicillin, and 100 mg/mL streptomycin. All cells were maintained in the presence of 5% CO2 at 37°C.
Viruses
Adenovirus expressing the LacZ gene (Ad/CMV-LacZ) or the green fluorescent protein (GFP) gene (Ad/CVM-GFP) has been described previously.11,12 Oncolytic vaccinia virus (vSP) has also been described previously.13 The expansion, purification, titration, and quality analyses of both vectors were performed at the Vector Core Facility of The University of Texas M. D. Anderson Cancer Center as previously described.13,14 For adenovirus, the titer used in this study was determined by the absorbancy of the dissociated virus at A260nm (1 A260nm unit = 1 × 1012 viral particles [VP]/mL), and the infectious units (IU) determined by the median tissue culture infective dose (TCID50) assay 14 were used as additive information. The VP:IU ratio was usually between 30:1 and 100:1. For vSP, the titer used in this study was the IU determined by the TCID50 assay.
Animal Experiments
Six-week-old female C3H mice or Balb/C mice were purchased from Charles River Laboratories (Wilmington, MA) and housed at M. D. Anderson Cancer Center under conventional conditions. Animal experiments were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals (National Institute of Health Publication number 85-23) and the institutional guidelines of M. D. Anderson. For virus-mediated foreign antigen immunization, C3H mice were injected intradermally with 0.1 mL of PBS alone (mock immunization) or with 1 × 1010 VP of Ad/CMV-LacZ or Ad/CVM-GFP. This procedure was repeated every 7 days for 3 weeks. One week after the last immunization, 1 × 106 cells of UV2237, a syngeneic fibrosarcoma cell line derived by repeated exposure to ultraviolet irradiation,15 were subcutaneously injected into the right hind leg of each mouse to establish subcutaneous tumors. When the tumors grew to approximately 3-5 mm in diameter, immunized and naïve mice were randomly divided into subgroups for treatment. The control group was treated with PBS, and the other groups were treated with viruses that were used in the immunizations. Tumors were injected with 0.1 mL of PBS alone or with Ad/CMV-LacZ or Ad/CVM-GFP (5 × 1010 VP/injection) every 3 days for a total of three or four injections. The tumor growth rate was assessed by measuring tumor volume every 2 or 3 days with calipers. The tumor volume was calculated using the formula a(b2) ÷ 2, for which a and b represent the longest and shortest diameters, respectively. The mice were euthanized when their tumors reached 1.5 cm in diameter or the tumor site became ulcerated. This in vivo experiment was performed three times.
To test whether the effects of virus-mediated foreign antigen immunization can occur in other tumor models, C3H mice were injected intradermally with 0.1 mL of PBS alone or with 1 × 1010 VP of Ad/CMV-LacZ every 7 days for 3 weeks. One week after the last immunization, 1 × 106 K1735 murine melanoma tumor cells were subcutaneously injected into the right hind leg of each mouse to establish subcutaneous tumors. When the tumors grew to approximately 3-5 mm in diameter, immunized and naïve mice were randomly divided into subgroups for treatment. The control group was treated with PBS, and the other groups were treated with viruses that were used in the immunizations. Tumors were injected with 0.1 mL of PBS alone or with Ad/CMV-LacZ (5 × 1010 VP/injection) every 3 days for a total of three or four injections, and the tumor growth rate was determined. This in vivo experiment was performed three times.
To test whether the immune response against parental tumor cells was elicited in mice with complete regression of UV2237 tumors, we challenged C3H mice that were not exposed to adenovirus with 1 × 106 parental UV2237 cells. For control animals, we surgically removed the primary tumors from naïve animals or from Ad/CMV-LacZ-immunized animals that did not respond to Ad/CMV-LacZ treatment and then injected the tumor cells on the opposite side of the original treatment site. The growth rate of tumors at this second challenge site was determined. This in vivo experiment was performed three times. To determine whether the immune response against tumor cells was specific for parental tumor cells, a similar experiment was performed in which a similar group of mice was challenged with UV2237 cells or with K1725 cells.
Because human adenovirus does not replicate efficiently in murine tumor cells, we also tested the toxicity of vaccinia virus on several murine cancer cells. Cancer cells were cultured in 96-well plates and then infected with different doses of vSP.13 Multiplicity of infection (MOI) represented the dose, which meant ratio of infectious virus particles to cells. A dose-effect analysis was performed 72 hours after treatment with vSP to determine which cell line was the most sensitive to the oncolytic effects of vSP. This analysis was performed for three separate experiments. The most sensitive cell line was used in the subsequent in vivo evaluation of vSP in which Balb/C mice were injected intradermally with 0.1 mL of PBS alone or with 1 × 106 IU vSP. This procedure was repeated every 7 days for 3 weeks. Four weeks after the first immunization with vSP, blood samples were collected from immunized or naïve mice and tested for neutralization antibodies against vSP. We then inoculated each mouse in the right hind leg with 1 × 106 cancer cells to establish subcutaneous tumors. When the tumors grew to approximately 3-5 mm in diameter, immunized and naïve mice were randomly divided into subgroups for treatment. The control group was treated with PBS, and the other groups were treated with viruses that were used in the immunizations. Tumors were injected with 0.1 mL of PBS alone or with vSP (1 × 107 IU/injection) every 3 days for a total of three injections. The tumor growth rate was assessed.
Cell viability assay
The viability of the cell lines was determined using the sulforhodamine B assay, as previously described.16 Briefly, after fixation of adherent cells with trichloroacetic acid in a 96-well microplate, the protein was stained with sulforhodamine B and the optical density was determined at 570 nm to reflect the number of stained cells, which represented cell viability. The percentage of viable cells was determined relative to the cell viability of the phosphate-buffered saline (PBS) control, which was set at 100%. Each experiment was performed in quadruplicate and repeated at least three times.
Neutralization antibody assay
To assess the immune response to vSP immunization and treatments, we collected blood samples from Balb/C mice before tumor cell inoculation or 5 days after the final injection by cutting the tip of each mouse's tail and measuring the serum neutralization antibody titer. Blood was allowed to clot overnight at 4°C, and antiserum was collected by centrifuging the clotted blood at 16 000 g for 10 minutes at 4°C and heat inactivated at 56°C for 30 minutes. The neutralization antibody titer for virus was determined as described previously.17 Briefly, serially diluted serum was mixed with vSP for 1h at 37°C and infected human cervical cancer Hela Cell line. Two days later, cells were stained by X-gal and counted the number of blue cell.
Immunohistochemical assay
Formalin-fixed tumor sections were immersed into preheated antigen-retrieval solution (10 mM Sodium Citrate, pH 6.0) and incubated at 95°C for 20 min. After cooling down to room temperature, the slides were incubated with anti-mouse CD4 (Clone V4, Santa Cruz Biotechnology, Santa Cruz, CA) (1:100 diluted with 5% BSA), or with anti-mouse CD8 (clone 53-6.7, BD Bioscience, San Jose, CA) (1:100 diluted with 5% BSA) at 4°C overnight. The Universal Kit (Vector Laboratory, Burlingame, CA) was used as secondary antibody. Endogenous peroxidase activity was blocked by 20 min incubation in 0.3% hydrogen peroxide. Mouse spleens were used as positive controls and slides with no primary antibody staining were used as negative control. Three sections were evaluated for each tumor.
Statistical analysis
Differences among the treatment groups were assessed by analysis of variance using StatSoft statistical software (Tulsa, OK). Survival analysis was done using the Kaplan-Meier test, and differences between curves were assessed using the log-rank test. P < 0.05 was considered statistically significant.
Results
Regression of fibrosarcoma tumors after immunization and treatment with Ad/CMV-LacZ
To test the feasibility of redirecting adaptive immune response to cancers, we immunized C3H mice with PBS alone or with Ad/CMV-LacZ or Ad/CMV-GFP by intradermal inoculation. Subcutaneous tumors were established by inoculating 1 × 106 cells of the UV2237 fibrosarcoma cell line into the right flank of each mouse. When the tumors reached 3-5 mm in diameter, the animals were intratumorally injected with PBS alone or with Ad/CMV-LacZ or Ad/CMV-GFP, as described in Materials and Methods. We found that animals treated with PBS, regardless of whether they were naïve or immunized, had the same tumor growth rate, suggesting that immunization with adenovector did not affect tumor growth. In contrast, immunization with adenovector dramatically affected the subsequent treatment with adenovector: the tumor growth in animals both immunized and treated with Ad/CMV-LacZ was significantly suppressed compared with that of naïve animals treated with Ad/CMV-LacZ (Figure 1A). Moreover, complete tumor regression (Table 1), which usually occurred at 10-15 days after intratumoral treatment started, and long-term tumor-free survival (Figure 1B) were observed in about 50% of animals that were immunized and treated with Ad/CMV-LacZ. In contrast, no tumor regression was observed in naïve animals treated with Ad/CMV-LacZ (Table 1). No treatment-related toxicity was observed in all groups, including the group immunized and treated with adenovectors. These results demonstrated that acquired immune response to LacZ, GFP, or adenoviral proteins can have an antitumor effect when the same foreign antigen genes are subsequently introduced into tumor cells.
Figure 1.
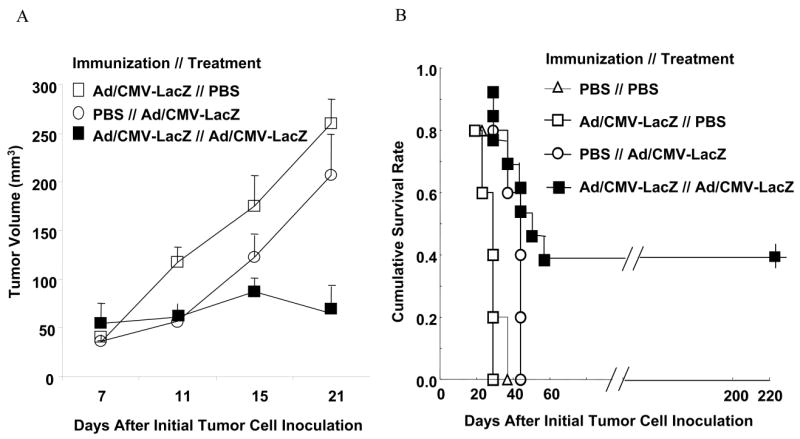
Antitumor activity of Ad/CMV-LacZ treatment in naïve and immunized animals bearing fibrosarcoma. C3H mice were immunized with PBS alone or with Ad/CMV-LacZ. Subcutaneous tumors were then established from UV2237 fibrosarcoma cells and treated by intratumoral injection of PBS alone or with Ad/CMV-LacZ. A) Tumor volume over time. Values are means ± standard errors of 8-10 animals per group. B) Survival curve of 8-15 animals per group. The data are shown from one of three experiments with similar results. The difference between the Ad/CMV-LacZ // Ad/CMV-LacZ group and each of the three other groups was significant (P < 0.05 for each comparison).
Table 1. UV2237 tumor regression in C3H mice immunized and treated with adenovirus or PBS.
| Immunization | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ad/CMV-LacZ | Ad/CMV-GFP | PBS (Naïve) | |||||||
| Treatment | No. of mice | No. of regression | % | No. of mice | No. of regression | % | No. of mice | No. of regression | % |
| Ad/CMV-LacZ | 42 | 22 | 52 | 25 | 0 | 0 | |||
| Ad/CMV-GFP | 24 | 6 | 25 | 5 | 2 | 40 | |||
| PBS | 20 | 0 | 0 | 7 | 0 | 0 | |||
Regression of melanoma tumors after immunization and treatment with Ad/CMV-LacZ
To test whether the effects of immunization we observed in fibrosarcoma tumors can occur in other tumor models, we immunized C3H mice with PBS alone or with Ad/CMV-LacZ. The animals were then inoculated subcutaneously with 1 × 106 K1735 murine melanoma tumor cells. When the tumors reached 3-5 mm in diameter, the animals were intratumorally injected with PBS alone or with Ad/CMV-LacZ, as described in Materials and Methods. As we observed for UV2237 tumors, immunization and treatment with Ad/CMV-LacZ dramatically improved the tumor-free survival rate (Figure 2). Thus, redirecting acquired immunity to cancer cells can be used for treatment of different types of tumors.
Figure 2.
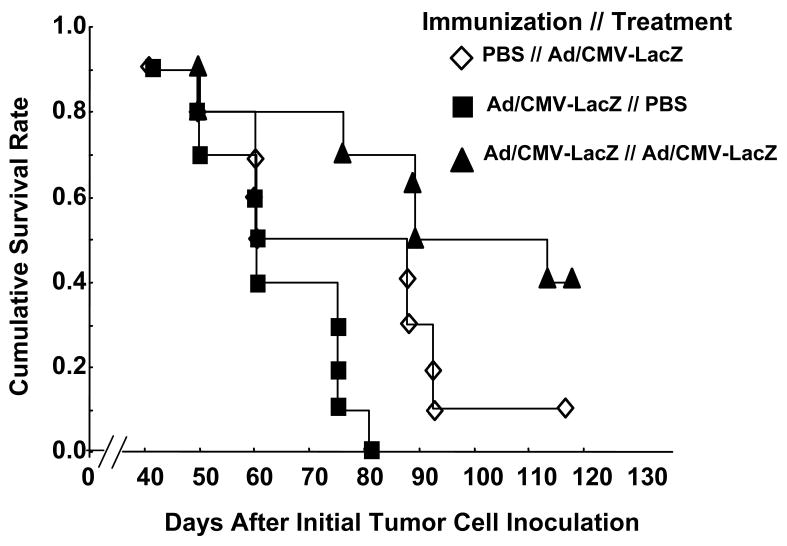
Effects of immunization on melanoma. C3H mice were immunized with PBS alone or with Ad/CMV-LacZ. Subcutaneous tumors were then established from K1735 melanoma cells, and treated by intratumoral injection of PBS alone or with Ad/CMV-LacZ. The data represent the accumulative survival of 10 animals per group. The difference between the Ad/CMV-LacZ // Ad/CMV-LacZ group and each of the other two groups was significant (P < 0.05 for each comparison).
Induction of anticancer immunity
Because intratumoral injection of adenovector seldom leads to transduction of all tumor cells, the complete regression of established tumors we observed in 52% of the C3H mice with UV2237 tumors indicated that this approach might induce bystander effects against untransduced cells. To test whether the immune response against parental tumor cells was elicited in animals immunized with Ad/CMV-LacZ that had complete tumor regression, we challenged these animals with 1 × 106 parental UV2237 cells that were not exposed to Ad/CMV-LacZ or any other adenovector. For control animals, primary tumors from naïve or Ad/CMV-LacZ-immunized mice that did not respond to Ad/CMV-LacZ treatment were surgically removed, and then the tumor cells were injected on the opposite side of the original treatment site. This second challenge was rejected in all the animals that were both immunized and treated with Ad/CMV-LacZ and had complete regression of their original tumors, whereas tumor growth on the second challenged site was noted in all other animals (Table 2). These results suggested that redirecting adaptive immunity against foreign antigens to tumor may indeed induce tumor-specific immunity in some animals.
Table 2. Second challenge rejection by C3H mice injected with parental UV2237 cells.
| Immunization//
Treatment |
Primary UV2237
Tumor Regression |
Challenge Rejections | ||
|---|---|---|---|---|
| No. of mice | No. of rejections | % | ||
| PBS//Ad/CMV-LacZ | No | 3 | 0 | 0 |
| Ad/CMV-LacZ// Ad/CMV-LacZ | No | 7 | 0 | 0 |
| Ad/CMV-LacZ// Ad/CMV-LacZ | Yes | 12 | 12 | 100 |
To test whether the immune response against tumor cells was specific for parental tumor cells, we performed a separate experiment to challenge animals with either UV2237 cells or K1735 cells whose UV2237 tumors had regressed. Animals whose primary tumors were removed by excision were used as the control. The results showed that the animals whose UV2237 tumors had regressed rejected the second challenge of UV2237 cells but not the challenge of K1735 cells (Table 3). In contrast, challenges from both UV2237 cells and K1735 cells formed tumors in control animals. These results demonstrated that the immune response specific for parental tumor cells was elicited by redirecting the acquired immunity against foreign antigens.
Table 3. Challenge rejection by C3H mice injected with parental UV2237 cells or injected with K1735 cells.
| Immunization//Treatment | Primary UV2237 Tumor Regression | UV2237 Challenge Rejection | K1735 Challenge Rejection | ||||
|---|---|---|---|---|---|---|---|
| No. of mice | No. of rejection | % | No. of mice | No. of rejection | % | ||
| PBS//PBS | no | 4 | 0 | 0 | 4 | 0 | 0 |
| PBS//Ad/CMV-LacZ | no | 4 | 0 | 0 | 4 | 0 | 0 |
| Ad/CMV-LacZ//Ad/CMV-LacZ | yes | 4 | 4 | 100 | 4 | 0 | 0 |
Enhancement of the oncolytic effect of vSP by immunization
Because oncolytic virotherapy, which uses viral replication and subsequent cell killing for cancer treatment, ultimately results in the expression of viral antigen in cancer cells, we hypothesized that redirecting acquired immunity against an oncolytic virus enhances the therapeutic effects of oncolytic virotherapy. Because human adenovirus does not replicate efficiently in murine tumor cells, we tested the toxicity of vSP on five murine cancer cell lines that were infected with different doses of vSP.13 A dose-effect analysis performed 72 hours after treatment showed that the colorectal adenocarcinoma tumor cell line CT26 was the most sensitive to the oncolytic effects of vSP (Figure 3A). Therefore, we used CT26 cells in the subsequent in vivo evaluation of vSP.
Figure 3.
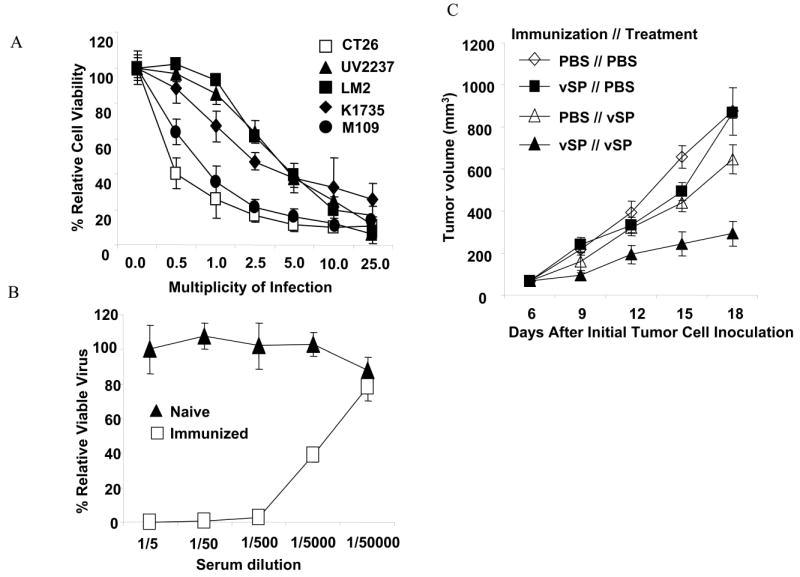
Effect of immunization on vSP therapy. A) Cytotoxicity of vSP in five murine cancer cell lines. Cells were treated with different doses of vSP for 72 hours, and cell viability was determined using the sulforhodamine B assay. Cells treated with PBS were used as the control, and their viability was set at 100%. Each data point represents the means ± standard deviations of three independent experiments. B) Serum neutralization antibody against vSP. Serum was obtained 4 weeks after the initial immunization with vSP or PBS. The neutralization antibody titer was determined as described in Materials and Methods. Values represent the means ± standard deviations of 3 animals per group. C) Tumor growth over time. CT26 murine colorectal adenocarcinoma cells were inoculated subcutaneously in BALB/c mice immunized with PBS or vSP and then treated with PBS or vSP, respectively. Tumor volume was monitored over time. Values are means ± standard errors of 5-7 animals per group. The difference in overall tumor growth rate growth was significant between the vSP // vSP group and each of the other three groups (P < 0.05 for each comparison).
Balb/C mice were immunized with vSP as described in Materials and Methods. Four weeks after the first immunization, blood samples were collected from immunized or naïve mice and tested for neutralization antibodies against vSP. Anti-vSP neutralizing antibody was detected in all immunized mice (average titer 1:7800) but not in naïve mice (average titer < 1:5), suggesting that the anti-vSP immune response was successfully induced (Figure 3B). We then inoculated immunized and naïve mice with CT26 cells to establish subcutaneous tumors. When the tumors grew to 3-5 mm in diameter, the animals were treated with PBS alone or with vSP. Our results showed that in naïve mice, treatment with vSP suppressed tumor growth compared with treatment with PBS but that the vSP-mediated tumor-suppressing effect was more dramatic in immunized mice than in naïve mice (Figure 3C). The difference was significant (P < 0.05). Tumor regression was observed in 2 of the 7 animals that were both immunized and treated with vSP but in none of the 17 mice of the other immunization and treatment groups. No treatment-related toxicity were observed in all groups, including the group immunized and treated with vSP. These results demonstrated that an existing immune response against an oncolytic virus enhances the oncolytic virotherapy of cancers.
Enhanced lymphocyte infiltration in tumors of immunized animals
Enhanced vSP antitumor activity in immunized group indicated roles of immune response in vSP virotherapy. To test whether there is difference in immune response inside tumors, we harvested tumors at two days after the last treatment as described in Materials and Methods. Immnunohistochemical staining with anti-mouse CD4 and anti-mouse CD8 antibodies showed a dramatic increase of CD4+ lymphocytes and, to a less degree, of CD8+ lymphocytes in tumors of animals immunized and treated with vSP when compared with all other groups (Figure 4). Moreover, increased tumor cell nuclear condensation and fragmentation, hallmarks of apoptosis/necrosis, were observed in vSP-immunized and treated group when compared with all other groups. This result demonstrate that preimmunization with a viral vector followed by intratumoral delivery of the same viral vector will enhance infiltration of immune active lymphocytes in tumor sides and enhance tumor cell killing.
Figure 4.
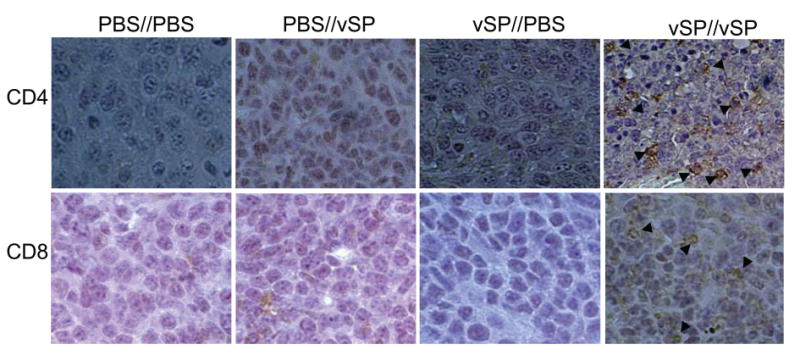
Representative pictures of lymphocyte infiltration in tumor. Immunization//Treatment were indicated on top of each panel. Arrows indicate positive stained cells. Note, tumor cell nuclear condensation and fragmentation in vSP//vSP group.
Discussion
The use of foreign antigens in anticancer therapy has been investigated for more than a century. The successful clinical application with foreign antigens is exemplified by the treatment of superficial bladder cancer with bacillus Calmette-Guerin (BCG).18 Local, intravesical instillation of BCG leads to induction of various chemokines and inflammatory cytokines, up-regulation of adhesion molecule expression, and infiltration of lymphocytes, macrophages, and neutrophils, all of which result in elimination of cancer cells in a small group of bladder cancer patients.18-20 Moreover, BCG can be engineered to express cytokines to improve its efficacy.21 Genetically attenuated bacteria and viruses have also been explored for cancer treatment directly or as vehicles of cancer immunotherapy or vaccination.22-24 Indeed, regression of hematopoietic malignancies have been observed after viral infection or vaccination,25,26 suggesting that a foreign invader may trigger systemic anticancer effects. Nevertheless, the treatment regimen for most virus- or bacteria-based immunotherapies is usually local or locoregional application without pre-immunization. It is not certain whether induction of the immune response to those foreign antigens before local application affects treatment outcomes. Because cancer regions may be immune privileged,27-30 direct application of foreign antigen to the cancer site without pre-immunization may reduce the immune response to these antigens. Moreover, massive infiltration of neutrophils may attenuate the immune response by changing the profiles of cytokine secretion. 31 Furthermore, because most bacterial antigens stay outside of tumor cells, the initial local response is usually against the foreign antigen without direct interaction of immune effector cells and tumor cells: the cancer cells are killed by a bystander effect of local inflammation rather than by a direct attack by immune active cells. Under these circumstances, the processing and presentation of tumor antigens back to the immune system may not be effectively mobilized.
The therapeutic approach of redirecting acquired immunity against foreign antigens to cancer cells may solve such problems. Because the immunization step is not involved in the immune privilege of cancer sites, eliciting a strong immune response against foreign antigens is possible in cancer patients. Once an immune response is established, gene transfer technology can be used to induce ectopic expression of these foreign antigens in cancer cells. Tumor-specific expression of foreign antigens is also possible by controlling transgene expression with tumor-specific promoters, such as telomerase promoter, 11,12,32 or other targeted gene delivery technologies. 33,34 Because this ectopic expression of foreign antigens attracts direct attack by professional immune components, the processing and presentation of possible tumor antigens existing in cancers back to the immune system may be more effective than when cancer cells are killed by the bystander inflammatory response.
It is well documented that gene therapy with a viral vector containing viral genes can result in immune response to transduced cells expressing transgene and/or viral genes. Eliminating transduced cells by host immune systems, either cellular or humoral or both, was reported to be the causal factor of transient transgene expression of adenovirus-mediated gene therapy in immunocompetent animals.35-37 Intratracheal instillation of an E1-deleted adenovirus led to substantial peribronchial and perivascular infiltrates of CD4+ and CD8+ T cells. 38 Cytotoxic T-lymphocytes against major histocompatibility complex-matched target cells infected with an adenovirus not expressing any transgene was induced by administration of an E1-deleted adenovirus, suggesting that adenovirus-specific cellular immune response can be stimulated that leads to destruction of transduced cells.36 However, if transduced cells are malignant, this elimination is beneficial for treatment. This may explain why intratumoral administration of Ad/CMV-LacZ can induce mild antitumor activity in immunocompetent mice. Nevertheless, our results demonstrated that antitumor activity mediated by such a mechanism can be dramatically augmented by preimmunization followed by intratumoral delivery with the same viral vectors. We also found that preimmunization with Ad/CMV-LacZ enhanced antitumor activity of subsequent intratumoral administration of Ad/CMV-GPF. Because both Ad/CMV-LacZ and Ad/CMV-GFP are conventional E1-deleted vectors and are known to express the same adenoviral antigens, enhanced antitumor activity is attributed to preimmunization with viral gene products but not the transgene product. Moreover, our results demonstrated that preimmunization followed by intratumoral delivery of the same foreign antigen genes can also eliminate or reject parental tumor cells that do not express the foreign antigen. This observation is consistent with a previous report by others that LacZ gene expression in a highly tumorigenic murine cancer line abolished tumorigenicity of transduced cells in immunocompetent mice and induced immunity against parental cells. 39 Nevertheless, our results showed that immunity against tumor cells that do not express the foreign antigens is parental tumor antigen-dependent and is not mediated by non-specific cytokine response because the mice only rejected parental tumor cells but not a different tumor cell line. Further more, our results showed that pre-existing immunity against an oncolytic virus could enhance the antitumor activity of that virus. These proof-of-concept results may influence the future design of anticancer gene vectors or the clinical evaluation of oncolytic virotherapy for cancer.
Acknowledgments
We thank Elizabeth Hess for editorial review, Debbie Smith for assistance in preparing the manuscript, and Li Wang for the preparation and quality tests of adenovectors. This work has been supported by National Cancer Institute grants CA 092487 and CA 098582 (both to B. Fang), National Institutes of Health core grant CA-16672, Lockton grant-matching funds, Homer Flower Gene Therapy Research Fund and Charles Rogers Gene Therapy Fund.
References
- 1.Stevanovic S. Identification of tumour-associated T-cell epitopes for vaccine development. Nature Rev, Cancer. 2002(2):514–520. doi: 10.1038/nrc841. [DOI] [PubMed] [Google Scholar]
- 2.van der BP, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den EB, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 3.Leyland-Jones B. Trastuzumab: hopes and realities. Lancet Oncol. 2002;3:137–144. doi: 10.1016/s1470-2045(02)00676-9. [DOI] [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson M, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nabel GJ. Genetic, cellular and immune approaches to disease therapy: past and future. Nature Med. 2004;10:135–141. doi: 10.1038/nm990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui Y, Kelleher E, Straley E, Fuchs E, Gorski K, Levitsky H, Borrello I, Civin CI, Schoenberger SP, Cheng L, Pardoll DM, Whartenby KA. Immunotherapy of established tumors using bone marrow transplantation with antigen gene--modified hematopoietic stem cells. Nature Med. 2003;9:952–958. doi: 10.1038/nm882. [DOI] [PubMed] [Google Scholar]
- 7.Fabre JW. The allogeneic response and tumor immunity. Nature Med. 2001;7:649–652. doi: 10.1038/89008. [DOI] [PubMed] [Google Scholar]
- 8.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–437. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 9.Ko EC, Wang X, Ferrone S. Immunotherapy of malignant diseases. Challenges and strategies. Int Arch Allergy & Immun. 2003;132:294–309. doi: 10.1159/000074897. [DOI] [PubMed] [Google Scholar]
- 10.Bouvet M, Fang B, Ekmekcioglu S, Ji L, Bucana CD, Hamada K, Grimm EA, Roth JA. Suppression of the immune response to an adenovirus vector and enhancement of intratumoral transgene expression by low-dose etoposide. Gene Ther. 1998;5:189–95. doi: 10.1038/sj.gt.3300564. [DOI] [PubMed] [Google Scholar]
- 11.Gu J, Kagawa S, Takakura M, Kyo S, Inoue M, Roth JA, Fang B. Tumor-specific transgene expression from the human telomerase reverse transcriptase promoter enables targeting of the therapeutic effects of the Bax gene to cancers. Cancer Res. 2000;60:5359–5364. [PubMed] [Google Scholar]
- 12.Lin T, Gu J, Zhang L, Huang X, Stephens LC, Curley SA, Fang B. Targeted expression of green fluorescent protein/Tumor necrosis factor-related apoptosis-inducing ligand fusion protein from human telomerase reverse transcriptase promoter elicits antitumor activity without toxic effect on primary human hepatocytes. Cancer Res. 2002;62:3620–3625. [PubMed] [Google Scholar]
- 13.Guo ZS, Naik A, O'Malley ME, Popovic P, Demarco R, Hu Y, Yin X, Yang S, Zeh HJ, Moss B, Lotze MT, Bartlett DL. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- 14.Fang B, Ji L, Bouvet M, Roth JA. Evaluation of GAL4/TATA in vivo. Induction of transgene expression by adenovirally mediated gene codelivery. J Biol Chem. 1998;273:4972–5. doi: 10.1074/jbc.273.9.4972. [DOI] [PubMed] [Google Scholar]
- 15.Kripke ML. Latency, histology, and antigenicity of tumors induced by ultraviolet light in three inbred mouse strains. Cancer Res. 1977;37:1395–1400. [PubMed] [Google Scholar]
- 16.Dong F, Wang L, Davis JJ, Hu W, Zhang L, Guo W, Teraishi F, Ji L, Fang B. Eliminating established tumor in nu/nu nude mice by a tumor necrosis factor-alpha-related apoptosis-inducing ligand-armed oncolytic adenovirus. Clin Cancer Res. 2006;12:5224–5230. doi: 10.1158/1078-0432.CCR-06-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang B, Eisensmith RC, Li XH, Finegold MJ, Shedlovsky A, Dove W, Woo SL. Gene therapy for phenylketonuria: phenotypic correction in a genetically deficient mouse model by adenovirus-mediated hepatic gene transfer. Gene Ther. 1994;1:247–54. [PubMed] [Google Scholar]
- 18.Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol. 1976;116:180–183. doi: 10.1016/s0022-5347(17)58737-6. [DOI] [PubMed] [Google Scholar]
- 19.Alexandroff AB, Jackson AM, O'Donnell MA, James K. BCG immunotherapy of bladder cancer: 20 years on. Lancet. 1999;353:1689–1694. doi: 10.1016/S0140-6736(98)07422-4. [DOI] [PubMed] [Google Scholar]
- 20.Bohle A, Brandau S. Immune mechanisms in bacillus Calmette-Guerin immunotherapy for superficial bladder cancer. J Urol. 2003;170:964–969. doi: 10.1097/01.ju.0000073852.24341.4a. [DOI] [PubMed] [Google Scholar]
- 21.Chabalgoity JA, Dougan G, Mastroeni P, Aspinall RJ. Live bacteria as the basis for immunotherapies against cancer. Expert Rev Vaccines. 2002;1:495–505. doi: 10.1586/14760584.1.4.495. [DOI] [PubMed] [Google Scholar]
- 22.Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A, McCormick F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–6. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 23.Huang XF, Ren W, Rollins L, Pittman P, Shah M, Shen L, Gu Q, Strube R, Hu F, Chen SY. A broadly applicable, personalized heat shock protein-mediated oncolytic tumor vaccine. Cancer Res. 2003;63:7321–7329. [PubMed] [Google Scholar]
- 24.Tjuvajev J, Blasberg R, Luo X, Zheng LM, King I, Bermudes D. Salmonella-based tumor-targeted cancer therapy: tumor amplified protein expression therapy (TAPET) for diagnostic imaging. J Controlled Release. 2001;74:313–315. doi: 10.1016/s0168-3659(01)00340-6. [DOI] [PubMed] [Google Scholar]
- 25.Taqi AM, Abdurrahman MB, Yakubu AM, Fleming AF. Regression of Hodgkin's disease after measles. Lancet. 1981;1:1112. doi: 10.1016/s0140-6736(81)92286-8. [DOI] [PubMed] [Google Scholar]
- 26.Hansen RM, Libnoch JA. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch Intern Med. 1978;138:1137–1138. [PubMed] [Google Scholar]
- 27.Green DR, Ferguson TA. The role of Fas ligand in immune privilege. Nature Rev Mol Cell Biol. 2001;2:917–924. doi: 10.1038/35103104. [DOI] [PubMed] [Google Scholar]
- 28.O'Connell J, Bennett MW, O'Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: cancer as a site of immune privilege. Immunol Today. 1999;20:46–52. doi: 10.1016/s0167-5699(98)01382-6. [DOI] [PubMed] [Google Scholar]
- 29.O'Connell J, O'Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med. 1996;184:1075–1082. doi: 10.1084/jem.184.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, Cerottini J, Tschopp J. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 31.Cassatella MA, Meda L, Gasperini S, D'Andrea A, Ma X, Trinchieri G. Interleukin-12 production by human polymorphonuclear leukocytes. Eur J Immunol. 1995;25:1–5. doi: 10.1002/eji.1830250102. [DOI] [PubMed] [Google Scholar]
- 32.Gu J, Fang B. Telomerase promoter-driven cancer gene therapy. Cancer Biol Ther. 2003;2:S64–S70. [PubMed] [Google Scholar]
- 33.Douglas JT, Rogers BE, Rosenfeld ME, Michael SI, Feng M, Curiel DT. Targeted gene delivery by tropism-modified adenoviral vectors. Nature Biotech. 1996;14:1574–1578. doi: 10.1038/nbt1196-1574. [DOI] [PubMed] [Google Scholar]
- 34.Rots MG, Curiel DT, Gerritsen WR, Haisma HJ. Targeted cancer gene therapy: the flexibility of adenoviral gene therapy vectors. J Controlled Release. 2003;87:159–165. doi: 10.1016/s0168-3659(02)00360-7. [DOI] [PubMed] [Google Scholar]
- 35.Fang B, Eisensmith RC, Wang H, Kay MA, Cross RE, Landen CN, Gordon G, Bellinger DA, Read MS, Hu PC, et al. Gene therapy for hemophilia B: host immunosuppression prolongs the therapeutic effect of adenovirus-mediated factor IX expression. Hum Gene Ther. 1995;6:1039–44. doi: 10.1089/hum.1995.6.8-1039. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Nunes FA, Berencsi K, Furth EE, Gonczol E, Wilson JM. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilchrist SC, Ontell MP, Kochanek S, Clemens PR. Immune response to full-length dystrophin delivered to Dmd muscle by a high-capacity adenoviral vector. Mol Ther. 2002;6:359–368. doi: 10.1006/mthe.2002.0675. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Li Q, Ertl HC, Wilson JM. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;69:2004–2015. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abina MA, Lee MG, Descamps V, Cordier L, Lopez M, Perricaudet M, Haddada H. LacZ gene transfer into tumor cells abrogates tumorigenicity and protects mice against the development of further tumors. Gene Ther. 1996;3:212–216. [PubMed] [Google Scholar]