

SUMMARY
Yorkie, a transcription factor of the Fat and Hippo signaling pathways, is negatively regulated by the Warts kinase. Here we use Phos-tag gels to characterize Warts-dependent phosphorylation of Yorkie in vivo, and show that Warts promotes phosphorylation of Yorkie at multiple sites. We also show that Warts inhibits Yorkie nuclear localization in vivo, and can promote binding of Yorkie to 14-3-3 proteins in cultured cells. In vivo assessment of the influence of individual upstream regulators of Warts reveals that some mutants (e.g. fat) have only partial effects on Yorkie phosphorylation, and weak effects on Yorkie localization, whereas other genotypes (e.g. ex fat double mutants) have stronger effects on both Yki phosphorylation and localization. We also identify Serine 168 as a critical site through which negative regulation of Yorkie by Warts-mediated phosphorylation occurs, but find that this site is not sufficient to explain effects of Hippo signaling on Yki in vivo. These results identify modulation of subcellular localization as a mechanism of Yorkie regulation, and establish that this regulation occurs in vivo through multiple sites of Warts-dependent phosphorylation on Yorkie.
INTRODUCTION
Recent studies have linked the functions of several tumor suppressors to two interconnected signaling pathways, the Fat pathway and the Hippo pathway (reviewed in Pan, 2007; Saucedo and Edgar, 2007). These pathways act through Warts (Wts), a conserved Ser/Thr kinase, via two distinct mechanisms. The Fat signaling pathway influences the levels of Wts through a post-transcriptional process that requires the unconventional myosin Dachs (Cho et al., 2006). Thus, in fat mutant cells, levels of Wts are reduced. The Hippo pathway influences the activity of Wts without affecting its levels, through the kinase Hippo (Hpo), which phosphorylates Wts, and the co-factors Salvador (Sav) and Mob as Tumor Suppressor (Mats), which bind to Hpo and Wts (reviewed in Pan, 2007; Saucedo and Edgar, 2007). The tumor suppressors Expanded (Ex) and Merlin (Mer) act upstream of Hpo (Hamaratoglu et al., 2006). There is also crosstalk between these pathways, as Fat influences the levels of Ex at the subapical membrane (Bennett and Harvey, 2006; Feng and Irvine, 2007; Silva et al., 2006; Willecke et al., 2006).
Activated Wts phosphorylates, and thereby inhibits, a non-DNA binding transcriptional co-activator protein, Yorkie (Yki) (Huang et al., 2005). Yki promotes growth and inhibits apoptosis by enhancing the transcription of downstream genes, including Cyclin E, Cyclin B, DIAP1, and bantam (reviewed in Pan, 2007; Saucedo and Edgar, 2007). Mutation of yki inhibits growth and cell survival, whereas over-expression of yki promotes overgrowth, presumably because this overcomes negative regulatory mechanisms that normally limit its activity. Genetically, yki acts downstream of wts, and Wts-dependent phosphorylation can inhibit Yki’s transcriptional co-activator activity, as monitored by experiments in which a Yki:Gal4 fusion protein promotes transcription of a reporter gene in cultured Drosophila cells (Huang et al., 2005).
Most of the genes in the Fat and Hippo pathways are conserved in humans (reviewed in Pan, 2007; Saucedo and Edgar, 2007). Loss-of-function mutations in several mammalian homologues of Drosophila tumor suppressors have also been linked to cancers, supporting their conserved action as tumor suppressors. The mammalian homologue of Yki is YES-associated protein (YAP). YAP can rescue the lethality associated with Hpo pathway hyperactivation in Drosophila, implying functional conservation between Yki and YAP (Huang et al., 2005). The observation that over-expression of Yki promotes overgrowth would suggest that YAP might act as an oncogene in mammals, and recent studies have borne out this out expectation (Overholtzer et al., 2006; Zender et al., 2006).
The identification of Yorkie as the most downstream transcriptional regulator of the Fat and Hippo pathways provided an opportunity to elucidate molecular and cellular mechanisms through which tumor suppressors in these pathways affect growth control. Here we show that Wts phosphorylates Yki in vivo at multiple sites and regulates its nuclear localization. Phosphorylation and nuclear localization of Yki are also modulated by upstream tumor suppressors of the Fat and Hippo pathways. We also show that phosphorylation of Yki mediates binding to 14-3-3 proteins, acting via a conserved Ser that also contributes to normal regulation of Yki activity in vivo. Taken together, our studies indicate that Yki is regulated by Warts-mediated phosphorylation at multiple sites to influence its subcellular localization.
RESULTS
Yorkie is predominantly cytoplasmic
As studies of Yki’s mammalian homologue, YAP, suggested modulation of nuclear localization as a potential mode of YAP regulation (Basu et al., 2003), we investigated the subcellular localization of Yki in Drosophila. In one approach, we created a yki:GFP fusion gene, and expressed it under UAS-Gal4 control. Confirmation that this fusion protein is functional was provided by the observation that expression of yki:GFP under arm-Gal4 control was sufficient to rescue the lethality of a yki null allele, ykiB5. To specifically examine Yki:GFP function within the wing disc, we generated ykiB5 mutant clones in animals in which Yki:GFP was expressed in anterior cells under ci-Gal4 control. In posterior cells, ykiB5 clones were rare and small (Fig 1A). By contrast, in anterior cells, ykiB5 clones were readily recovered, indicating that Yki:GFP rescues the growth and viability of ykiB5 cells. Despite its presumed function as transcriptional co-activator, when the localization of Yki:GFP was examined in wing imaginal discs, it is predominantly cytoplasmic, although low levels appear in the nucleus (Fig. 1B).
Fig 1. Yki is predominantly cytoplasmic.

Panels show portions of wing imaginal discs; in this and subsequent figures panels marked prime show separate channels of the stain to the left. A) ykiB5 mutant clones (black arrows), marked by absence of lacZ (magenta), with UAS-yki:GFP (green) expressed under ci-Gal4 control. Twin clones (white arrows) are visible in both anterior and posterior compartments. B) Yki:GFP (green) expression in posterior cells (right) under en-Gal4 control; nuclei are marked by DAPI stain (blue). Nuclear Yki:GFP is barely above background. C) Yki-S168A:GFP (green) expression in posterior cells (right) under en-Gal4 control. Levels of nuclear Yki-S168A:GFP are higher than for Yki:GFP, but it is still predominantly cytoplasmic. D) ykiB5 mutant clones, marked by absence of lacZ (green), and stained for DNA (Dapi, blue) and Yki (red). E) Vertical section through the disc shown in D. The center of the wing disc forms a pseudostratified epithelium, with nuclei in different focal planes. Yki staining is detected throughout the apical-basal aspect of these cells, but is low in nuclei (e.g. as highlighted by asterisks to the right of nuclei).
In a second approach to investigating the subcellular localization of Yki, we generated antibodies against Yki. Confirmation of the specificity of anti-Yki staining was provided by examining clones of cells mutant for ykiB5, which encodes a protein null allele (Huang et al., 2005). To compensate for the poor growth and viability of yki clones, we employed the Minute technique, which gives clones a growth advantage over neighboring cells. This allowed recovery of yki mutant clones. The absence of staining in these clones confirmed that our antisera recognizes Yki (Fig. 1D, E). The wild-type localization profile of Yki is similar to that of Yki:GFP; Yki is predominantly cytoplasmic, although there are low levels in the nucleus. In vertical sections through the disc epithelium (Fig. 1E), Yki can be detected throughout the apical to basal aspect of these cells, but appears excluded from nuclei.
Yki is phosphorylated by Warts in vivo
Prior studies of Yki phosphorylation have all relied on experiments in cultured cells. To investigate whether the phosphorylation status of Yki is detectably influenced in vivo by Wts or other upstream tumor suppressors, we examined endogenous Yki in lysates of wing imaginal discs by Western blotting. Because yki mutations are lethal, the specificity of anti-Yki sera on Western blots was characterized by RNAi treatment of cultured cells; Yki is the predominant band detected (Fig. 2C). Western blotting of wing imaginal discs revealed a prominent smeared band of the expected mobility (~50 kD) (Fig. 2). In a wts mutant that permits survival of animals until the third larval instar (wtsP2) the Yki band appears sharper, consistent with the possibility that Yki phosphorylation is reduced, but the difference in mobility is subtle (Fig. 2A). Thus, to further evaluate the in vivo phosphorylation status of Yki, we employed a new technique for separating phosphorylated isoforms, Phos-tag gels (Kinoshita et al., 2006). The Phos-tag is a phosphate binding compound, which, when incorporated into polyacrylamide gels, can result in an exaggerated mobility shift for phosphorylated proteins, dependent upon the degree of phosphorylation (Kinoshita et al., 2006). When wing disc lysates are run on Phos-tag gels, several prominent Yki bands are detected, implying that several distinct phophophorylated isoforms are present in wild-type tissue (Fig. 2B). Most of these isoforms are absent in wts mutants (Fig. 2B), implying that the bulk of Yki phosphorylation in vivo is Wts-dependent. Confirmation that the mobility shifts observed are a consequence of phosphorylation was provided by treatment of lysates with calf intestinal alkaline phosphatase (CIP). CIP treatment of lysates resulted in sharper bands, with slightly faster mobility, on conventional gels, and collapsed the bands observed on Phos-tag gels into a single major band, resulting in a mobility profile similar to that observed in wtsP2 (Fig. 2). Thus, endogenous Yki is phosphorylated in vivo at multiple sites in a Wts-dependent process.
Fig 2. Yki is phosphorylated by Wts in vivo.
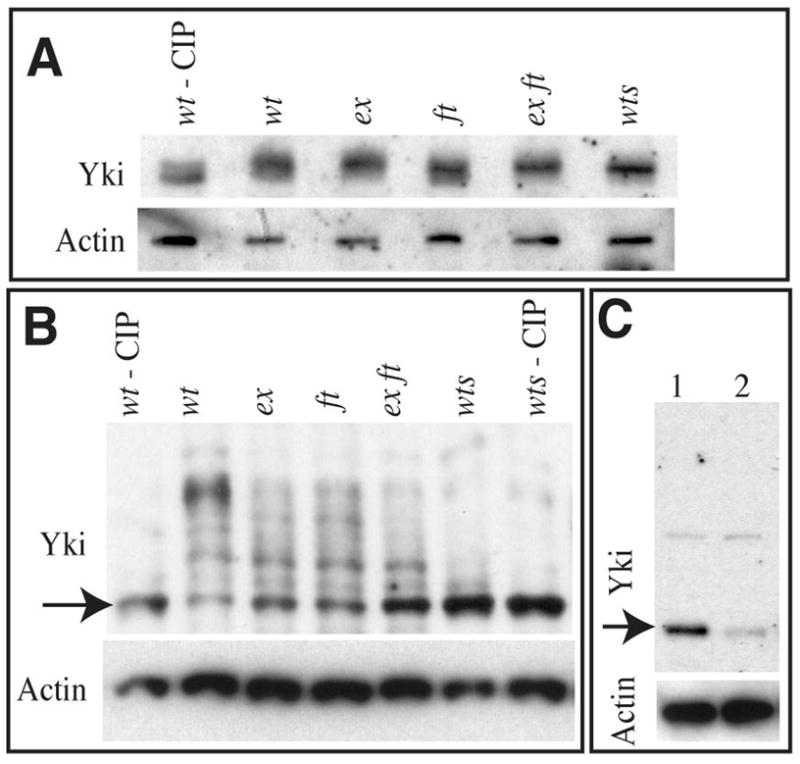
A) Western blots of lysates of wing imaginal discs from animals of wild type, treated with CIP (wt – CIP), wild type (wt), exe1, ftG-rv, exe1 ftG-rv, wtsP2, as indicated, run on a conventional 7.5% PAGE gel, probed with anti-Yki, and, as a loading control, anti-α-Actin. B) Western blot of lysates of wing imaginal discs from animals of wild type, treated with CIP (wt – CIP), wild type (wt), exe1, ftG-rv, exe1 ftG-rv, wtsP2, wtsP2 treated with CIP, as indicated, run on a Phos-Tag gel (25 μM Phos-Tag), probed with anti-Yki, and, as a loading control, anti-Actin. Arrow points to band corresponding to unphosphorylated Yki. C) Conventional 4–15% PAGE gel, probed with anti-Yki (upper panel), and, as a loading control (lower panel) anti-α-Actin. Lane 1 Shows S2 cell lysate treated with ds RNA corresponding to GFP (control), Lane 2 shows lysate from S2 cells treated with ds RNA corresponding to yki. Yki protein (arrow) is reduced, but a faint background band (above) is unaffected.
Wts is regulated by upstream tumor suppressors of the Fat and Hippo pathways. Many of these are lethal, but it is possible to recover third instar larvae homozygous for fat or ex null mutants. In fat mutant discs, Yki appears as smeared band on conventional gels, with a mobility similar to that in wild-type (Fig. 2A). However, on Phos-tag gels it is clear that Yki phosphorylation is reduced but not absent in fat mutants, as slower mobility isoforms are decreased, and the fastest mobility (and presumably unphosphorylated) isoform is increased. (Fig. 2B). These results imply that Wts function is partially compromised, consistent with the observation that Wts levels are reduced but not absent in fat mutants (Cho et al., 2006). Ex acts within the Hippo pathway, and regulates the activity of Wts (Hamaratoglu et al., 2006). Yki mobility in lysates of ex mutants was similar to that in fat mutants: phosphoryated Yki isoforms appear reduced but not eliminated, while unphosphorylated Yki appears increased (Fig. 2B). Although it has been reported that ex fat double mutants have the same phenotype as fat or ex single mutants (Bennett and Harvey, 2006; Silva et al., 2006), a more recent study revealed that they have actually act in parallel and have additive phenotypes (Feng and Irvine, 2007). Consistent with this, we observed an additive effect on Yki dephosphorylation in lysates from ex fat double mutants (Fig. 2B).
Wts regulates the nuclear localization of Yki
The results described above indicate that most Yki is subject to Wts-dependent phosphorylation in vivo. To investigate how Yki is influenced by phosphorylation at a cellular level, we created clones of cells mutant for wts, and examined Yki staining. In contrast to the cytoplasmic localization of Yki within wild-type cells, Yki appears distributed throughout the nucleus and cytoplasm of wts mutant cells (Fig. 3A). Thus, Wts influences the subcellular localization of Yki: Wts excludes Yki from the nucleus, presumably via Wts-dependent phosphorylation, but in the absence of Wts, Yki is free to travel to the nucleus.
Fig 3. Fat and Hippo signaling regulate the nuclear localization of Yki.
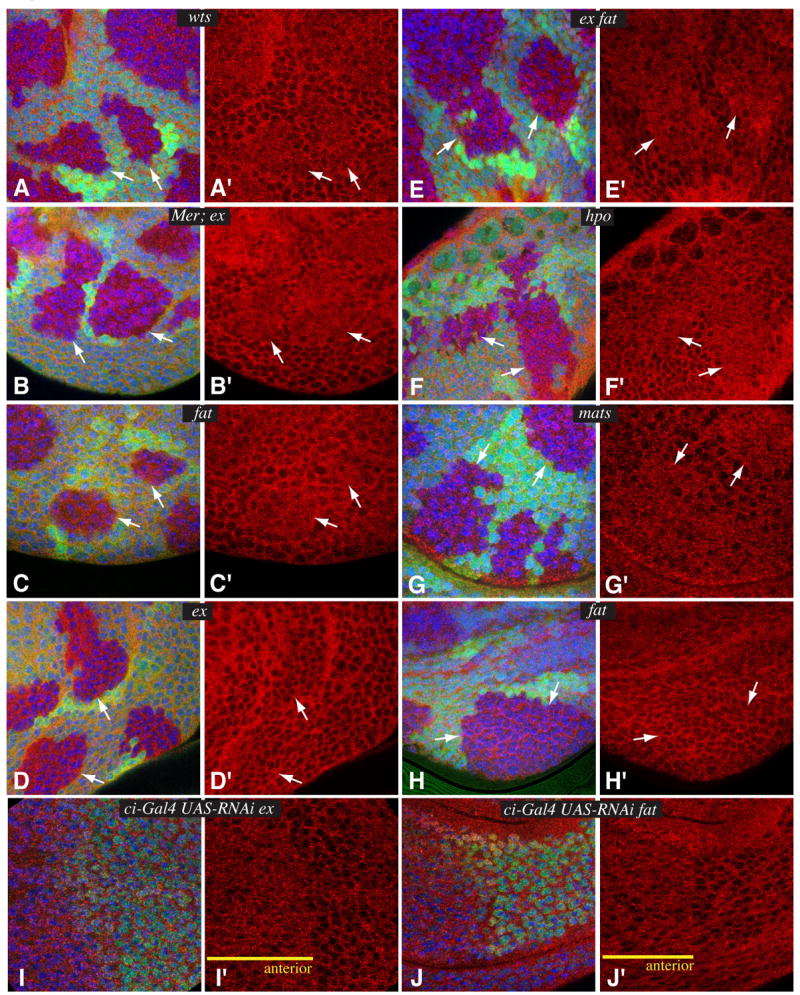
Panels A-H show close-ups of imaginal discs, stained for Yki (red), nuclei (DAPI, blue), and containing mutant clones marked by absence of GFP (green). A) wtsX1 B) Mer4; exe1 C) fatG-rv D) exe1 E) exe1 fatG-rv F) hpo42-47 G) matse235 H) fatG-rv. Arrows point to clone edges. I, J Show closeups of imaginal discs expressing RNAi lines under ci-Gal4 control, stained for Yki (red), nuclei (DAPI, blue), and Engrailed (green). ci-Gal4 drives expression in anterior cells (yellow line), partially complementary to Engrailed. I) ci-Gal4 UAS-ex RNAi UAS-dcr2. J) ci-Gal4 UAS-fat RNAi UAS-dcr2.
We next investigated the influence of tumor suppressors that act upstream of wts. Increased nuclear Yki was clearly observed within clones of cells mutant for the Wts kinase hpo (Fig. 3F), and the Wts co-factor mats (Fig. 3G). Clones of cells doubly mutant for two more upstream regulators of the Hippo pathway, ex and Mer, also exhibited Yki staining throughout the cytoplasm and nucleus (Fig. 3B), comparable to that observed in wts. Thus, the action of these tumor suppressors within the Hippo pathway can be directly visualized in vivo by their effect on Yki localization. ex single mutant clones did not detectably increase nuclear Yki (Fig. 3D), presumably due to the partial redundancy of ex with Mer. fat mutants have effects on growth and Yki phosphorylation comparable to ex mutants, and in most cases fat mutant clones also failed to exhibit a noticeable increase in the levels of nuclear Yki (Fig. 3H), although we did identify two exceptional clones (out of over 100 examined) in which an effect on Yki localization could be clearly observed (Fig. 3C). ex fat double mutants exhibit more severe phenotypes and effects on Yki dephosphorylation than single mutants (Fig. 2B), reflective of their parallel action in the Fat and Hippo pathways (Cho et al., 2006; Feng and Irvine, 2007). ex fat double mutant clones also consistently exhibited nuclear Yki staining (Fig. 3E). This observation suggests that both fat and ex can modulate the subcellular localization of Yki, but only in double mutant clones is the effect strong enough to be clearly detected. To further investigate the influence of ex and fat, we examined Yki staining in wing discs in which their levels were reduced in anterior cells throughout development by RNAi. When double stranded RNAs corresponding to fat or ex were expressed under ci-Gal4 control, together with the RNAi enhancer dicer2 (Dietzl et al., 2007), a modest but consistent increase in nuclear Yki was observed (Fig. 3I,J), confirming that these genes can each individually influence Yki localization. Expression of dicer2 alone had no effect on Yki (not shown). As the ex and fat mutations we employed are thought to be null alleles, we surmise that the apparently stronger RNAi phenotype results from the perdurance of gene products within mutant clones. Altogether, our observations establish a correlation among the overgrowth phenotypes of Fat and Hippo pathway mutants, their effects on Yki phosphorylation, and their effects on Yki nuclear localization.
Wts-dependent interaction of Yki with 14-3-3
It has been proposed that in cultured mammalian cells, YAP can be phosphorylated by Akt, creating an interaction site for 14-3-3 proteins, which then prevents YAP from localizing to the nucleus (Basu et al., 2003). To investigate whether the influence of Wts on Yki nuclear localization might involve modulating binding to 14-3-3 proteins, we performed co-immunoprecipitation assays on proteins expressed in cultured Drosophila S2 cells. Epitope-tagged forms of Yki and the two Drosophila 14-3-3 proteins (ε and ξ), were transfected into cells in the presence or absence of Wts and the Wts activators Hpo and Sav. As the ability of Akt to modulate association of YAP with 14-3-3 proteins has been mapped to a specific, conserved Ser residue (Basu et al., 2003), corresponding to Ser168 of Drosophila Yki, we also expressed a mutant form of Yki in which this was changed to Ala (Yki-S168A:FLAG). In the absence of exogenous kinases, weak association between 14-3-3 proteins and Yki could be detected (Fig. 4A). This interaction presumably depends on phosphorylation of Ser168 by endogenous kinases, as it was eliminated by the S168A mutation. Notably, the interaction between 14-3-3 and Yki was enhanced by expression and activation of Wts (Fig 4A). This enhanced interaction was also abolished by the S168A mutation (Fig. 4A). These observations imply that Wts-mediated phosphorylation of Ser168 promotes binding of Yki to 14-3-3 proteins.
Fig 4. Yki phosphorylation and 14-3-3 binding.
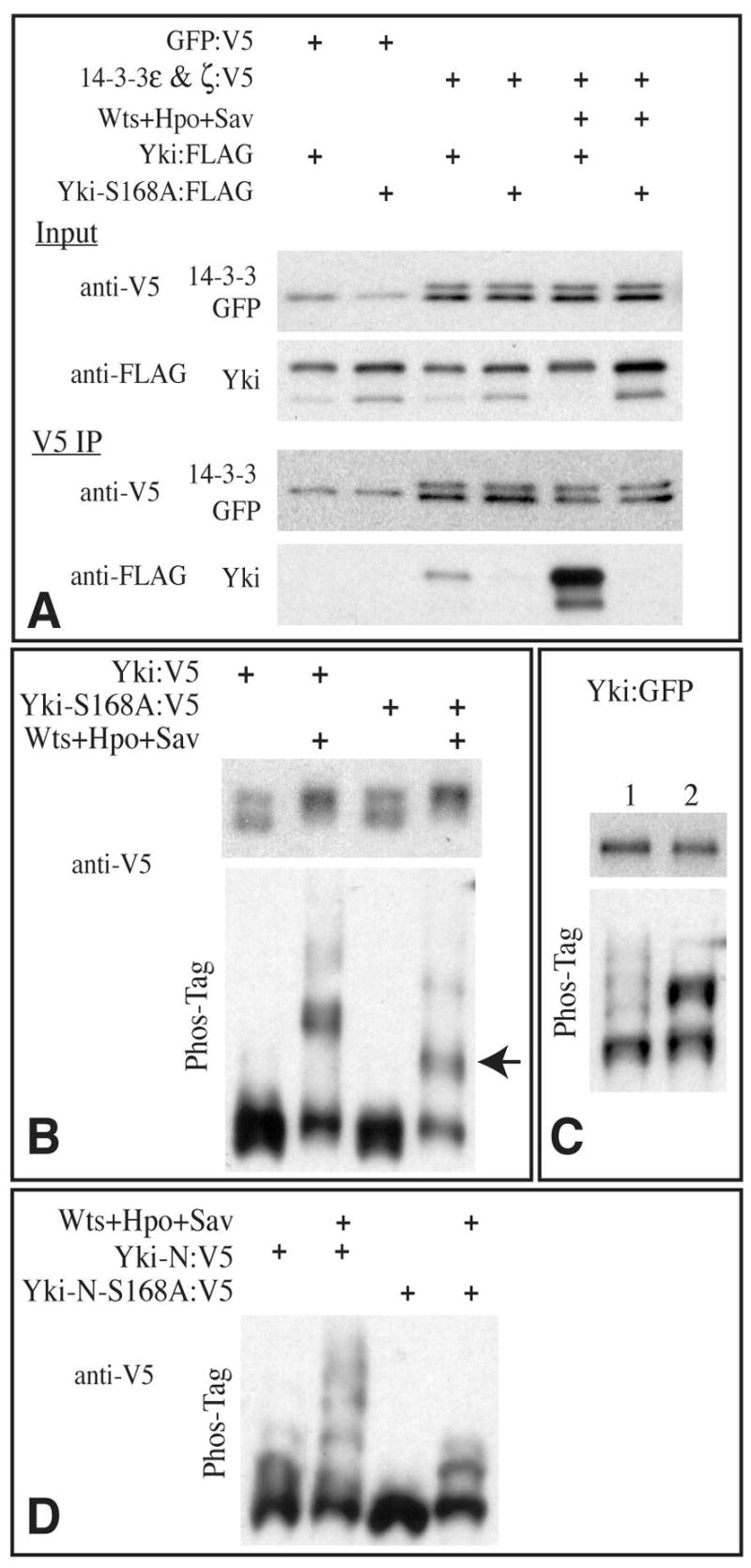
A) Western blots (4–15% PAGE) of samples from S2 cells co-transfected with the indicated proteins. Bottom two panels show blot on material precipitated with anti-V5 beads, upper two panel shows input; GFP:V5 (control protein) and 14-3-3:V5 proteins have similar mobility. B) Western blots (using anti-V5 epitope) on lysates of S2 cells co-transfected to express the indicated proteins. Transfection of Wts, Hpo, and Sav promotes phosphorylation of Yki and Yki-S168A, as revealed both on conventional SDS-PAGE (top panels) and Phos-Tag gels (30 μM Phos-Tag, bottom panels), however a distinct mobility isoform (arrow), representing partially phosphorylated Yki, is observed with Yki-S168A mutant but not wild-type Yki on Phos-tag gels. Transfer of heavily phosphorylated proteins from Phos-Tag gels can be poor, hence they may be underrepresented on these blots. C) Western blots of lysates of wing imaginal discs from animals expressing Yki:GFP (lane 1) or Yki-S168A:GFP (lane 2) under sd-Gal4 control. Upper panel shows conventional SDS-PAGE, lower panel shows a Phos-tag gel (60 μM Phos-Tag) D) Western blot on lysates of S2 cells co-transfected to express the indicated proteins, run on a Phos-tag gel (60 μM Phos-Tag). Yki-N is an N terminal fragment of Yki, comprising the first 240 aa.
To compare the effects of Wts on 14-3-3 binding with its effects on Yki phosphorylation, we examined Yki mobility on Western blots of cultured cell lysates. On conventional gels, activated Wts effected a substantial shift in Yki mobility, implying that Yki is heavily phosphorylated when co-transfected with activated Wts (Fig. 4B). This shift was not detectably influenced by the S168A mutation, implying that Ser168 is just one of multiple Wts-dependent phosphorylation sites on Yki. A shift was also observed on Phos-tag gels for both wild type and S168A mutant Yki (Fig. 4B). These experiments also illustrated one limitation of the Phos-tag gels, as there was often a decrease in the amount of Yki detected, which, as overall levels of Yki were not decreased on conventional gels, presumably resulted from the relatively poor transfer efficiency of heavily phosphorylated proteins out of Phos-tag gels (Kinoshita et al., 2006). Interestingly, on Phos-Tag gels S168A mutant Yki also exhibited a distinct, modestly shifted isoform that was not observed in experiments with wild-type Yki. This was observed both in cultured cells (Fig. 4B), and in wing disc lysates (Fig. 4C). This suggests that mutation of Ser168 can impair further phosphorylation of Yki. However, as interpretation of this result was potentially complicated by a reduced ability to detect heavily phosphorylated isoforms of Yki on Phos-tag gels, we also examined the mobility of smaller fragments of Yki.
An N-terminal fragment of Yki (Yki-N:V5), comprising the first 240 amino acids, was shifted into several bands on Phos-tag gels by co-expression of activated Wts (Fig. 4D). Conversely, the same fragment with the S168A mutation was shifted into only three bands. These results confirm that Ser168 can be a site of Warts-mediated phosphorylation. Since multiple phosphorylated isoforms appear to be affected by Ser168 mutation, they also suggest that phosphorylation of Yki at Ser168 might promote phosphorylation of Yki at additional sites, in other words, that it might act as a priming site. The influence of S168A mutation on Wts phosphorylation does not appear to be due to an effect on Wts-Yki binding, as physical interaction between them, as assayed by co-immunoprecipitation from S2 cells, was not detectably affected (not shown).
S168A mutant Yki is hyperactivated
The influence of mutation of Ser168 on 14-3-3 binding and Yki phosphorylation in S2 cells suggested that Yki might be regulated in vivo via this site. This was investigated by introducing the S168A mutation into a Yki:GFP transgene expressed under UAS control in Drosophila. As different chromosomal insertions of UAS transgenes are associated with different levels of expression, we examined more than ten insertions each for Yki:GFP and Yki-S168A:GFP. We also directly examined their relative expression levels by Western blotting, and for comparison, that of a published UAS-yki transgene (Huang et al., 2005). Expression of UAS-yki under GMR-Gal4 control resulted in overgrown eyes (Fig. 5B), whereas expression of UAS-yki:GFP didn’t cause any obvious eye overgrowth (Fig. 5C), even though UAS-yki:GFP was active in rescue experiments (Fig. 1A). However, examination of Yki levels by Western blotting and by confocal microscopy (Fig. 5E and data not shown), revealed that UAS-yki directs higher expression levels of Yki than any of our UAS-yki:GFP transgene insertions. Thus, Yki:GFP might be less active simply because its expression levels are lower.
Fig 5. Yki-S168A is hyperactivated.
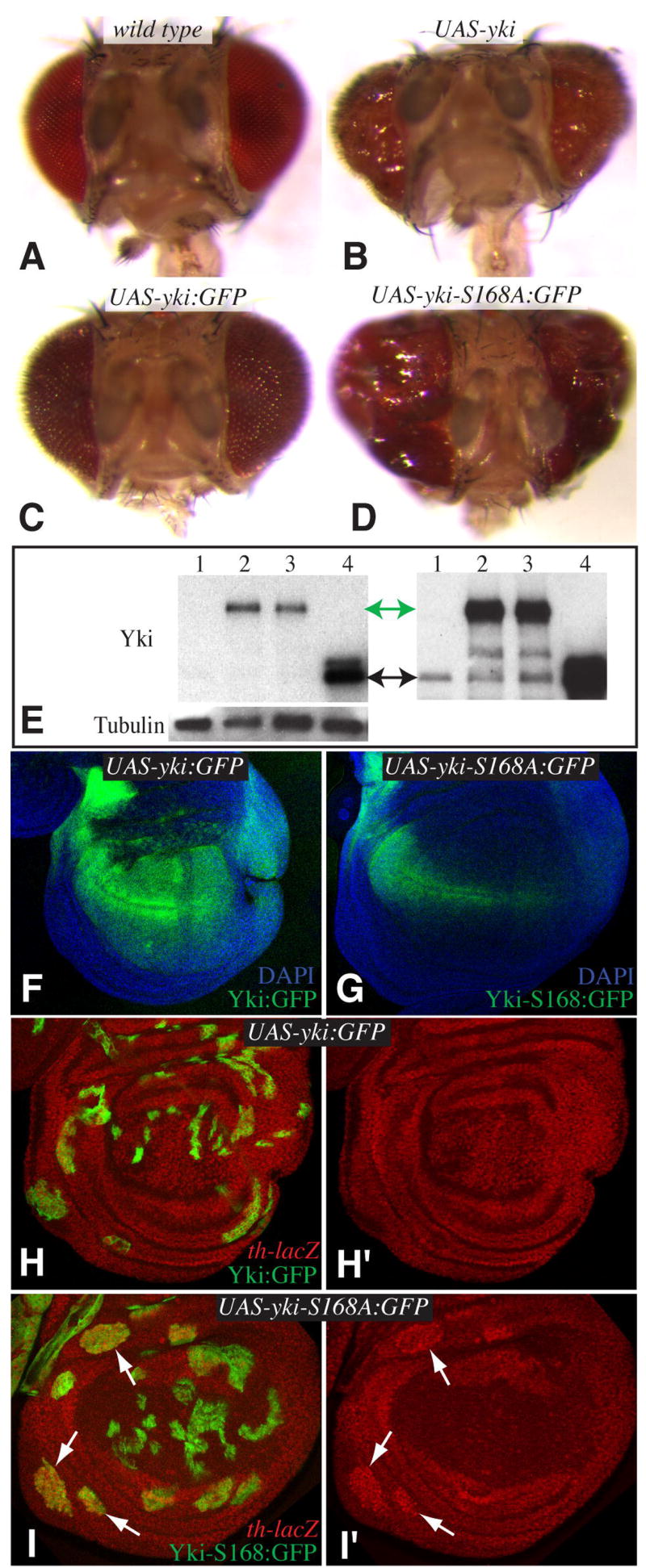
A–D) Heads of adult flies from wild type (A), UAS-yki GMR-Gal4 (B), UAS-yki:GFP GMR-Gal4 (C), UAS-yki-S168A:GFP GMR-Gal4 (D). E) Western blot (4-15% gradient gel) with anti-Yki on lysates from wing imaginal discs of 1. Wild-type 2. sd-Gal4 UAS-yki:GFP 3. sd-Gal4 UAS-yki-S168:GFP. 4. sd-Gal4 UAS-yki. Upper left panel shows a lower exposure, endogenous Yki is barely visible; upper right panel shows a longer exposure of the same gel, endogenous Yki is now visible. Green arrow marks Yki:GFP, black arrow marks Yki. Lower left panel shows anti-α-Tubulin as a loading control. F,G) Wing imaginal discs form sd-Gal4 UAS-yki:GFP (F) and sd-Gal4 UAS-yki-S168:GFP. Images were collected in parallel with same confocal settings, note that Yki-S168A:GFP expression (green) is weaker than Yki:GFP expression but the disc is overgrown. H,I) Wing imaginal discs containing clones of cells expressing UAS-yki:GFP (H) or UAS-yki-S168:GFP (I) (green), and stained for expression of a th-lacZ reporter (red). th expression is induced by yki-S168:GFP (arrows), but not by yki:GFP. In I) th-lacZ expression in the center of the disc is out of the plane of focus.
In contrast to the mild affects of Yki:GFP expression under GMR-Gal4 control, expression of Yki-S168A:GFP under GMR-Gal4 control generated substantial eye overgrowths (Fig. 5D). In this case, transgene insertions that generated strong overgrowths were associated with Yki-S168A:GFP levels at or below those of wild-type Yki:GFP insertions that did not generate detectable effects on growth (Fig. 5E–G and data not shown). Thus, the stronger phenotypes of Yki-S168A:GFP in comparison to Yki:GFP imply that the S168A mutation hyperactivates Yki, presumably by making it resistant to an inhibitory mechanism that acts via Ser168 phosphorylation. Examination of Yki:GFP-expressing clones in imaginal discs further supported this conclusion. Clones expressing Yki:GFP exhibited irregular shapes and normal sizes (Figs 5H,6A). By contrast, clones expressing Yki-S168A:GFP were larger, and were also rounder (Fig. 5I,6B), which is characteristic of mutations in Hippo and Fat pathway tumor suppressors. Mutation of Fat or Hippo pathway tumor suppressors is associated with the induction of downstream target genes, including thread (th, Diap1) and ex. Expression of th-lacZ (Fig. 5H,I) and ex-lacZ reporters (not shown) was elevated in association with Yki-S168A:GFP-expressing clones, but was not detectably affected by Yki:GFP-expressing clones. Altogether, these results indicate that S168A mutant Yki is hyperactivated.
Fig 6. Influence of Warts on clone growth and survival.
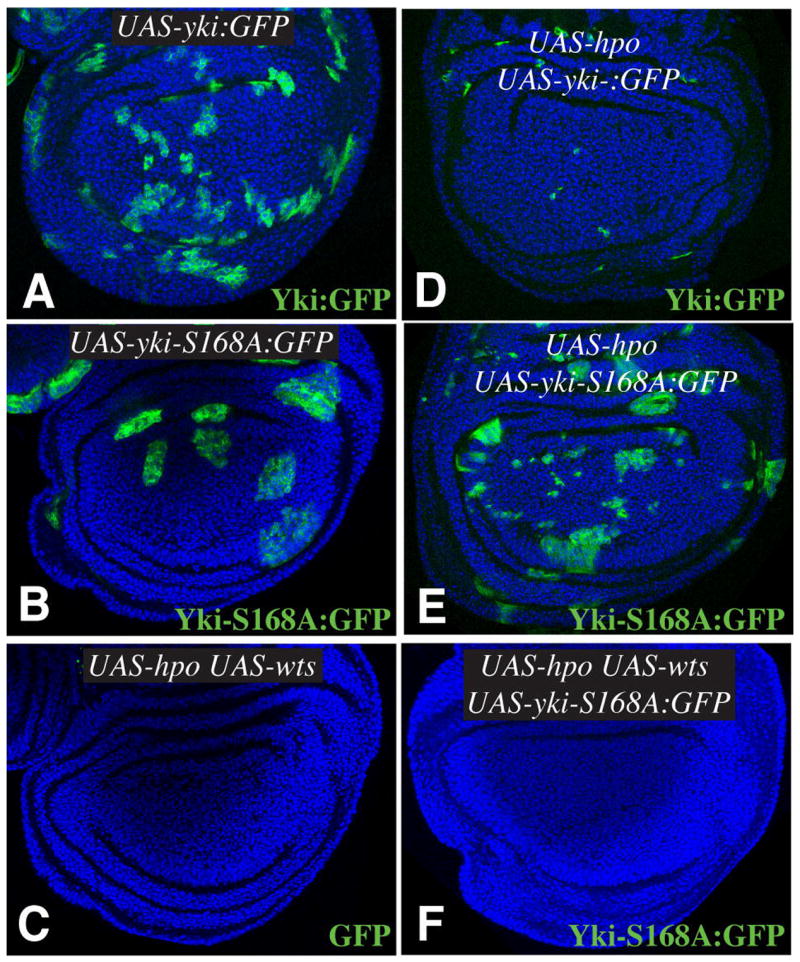
A–F show wing imaginal discs from animals in which clones of cells expressing UAS transgenes under control of actin-Gal4 have been induced by Flip-out: A) UAS-yki:GFP B) UAS-yki-S168A:GFP C) UAS-hpo UAS-wts UAS-GFP D) UAS-yki:GFP UAS-hpo E) UAS-hpo UAS-yki-S168A:GFP F) UAS-hpo UAS-wts UAS-yki-S168A:GFP. GFP expression is green, nuclei are blue.
To investigate whether this correlates with an effect on nuclear localization, we compared the subcellular localization of Yki-S168A:GFP to that of Yki:GFP. Yki:GFP and Yki-S168A:GFP are both predominantly localized to the cytoplasm (Fig. 1B,C). However, in the case of Yki-S168A:GFP there is a shift in the relative distribution, as slightly higher levels are observed in the nucleus than for Yki:GFP (Quantitation of average pixel intensities in the nucleus versus the cytoplasm yielded a ratio of 0.43 for Yki:GFP, and 0.59 for Yki-S168A:GFP). Thus, Ser168 contributes to, but does not fully explain, the exclusion of Yki from the nucleus.
S168A mutant Yki exhibits reduced sensitivity to Wts in vivo
To further investigate how Ser168 influences the regulation of Yki by Wts in vivo, we examined the consequences of Wts activation on Yki-S168A activity in imaginal discs. Over-expression of Hpo activates Wts, which inhibits Yki and thereby promotes apoptosis. Thus, Hpo-expressing clones are not normally recovered in imaginal discs (Fig. 6C and data not shown). When Yki:GFP was co-expressed with Hpo, clones were tiny and rarely detected (Fig 6D), indicating that expression of over-expression of Yki:GFP was not sufficient to block the effects of Hpo expression. However, when Yki-S168A:GFP was co-expressed with Hpo, clones were larger and more numerous (Fig. 6E). Thus, the S168A mutation reduces the sensitivity of cells to the pro-apoptotic effects of Hpo over-expression. To achieve even higher levels of Wts activation, we co-expressed Wts together with Hpo. In this case, the size and frequency of clones was greatly reduced even for Yki-S168A:GFP-expressing clones, and in most cases no clones were recovered (Fig. 6F). Thus, Yki-S168A:GFP can still be affected by high level Wts activation in vivo. However, at lower Wts activation levels (i.e. expression of Hpo alone) mutation of Ser168 confers partial resistance to Wts activation.
DISCUSSION
Yki was first identified as a candidate transcription factor of the Hippo pathway based on genetic studies. A molecular link between Yki and Hippo signaling was then suggested by over-expression experiments in cultured cell assays. By establishing that Yki phosphorylation and localization are influenced in imaginal discs by upstream components of the Fat and Hippo pathways, our results confirm that Yki is directly regulated by these pathways in vivo. Since upstream regulators of Yki activity, including Fat, Ex and Mer, are specifically localized at the subapical membrane, whereas Yki is much more broadly distributed, there must exist some mechanism to transmit the signaling status of upstream components like Fat and Ex to Yki, or Yki itself must diffuse throughout the cell.
The observation that nuclear localization of Yki is regulated by Wts defines a cellular mechanism by which Wts-dependent phosphorylation inhibits Yki activity. In general there is a correlation between the phosphorylation of Yki and its localization; i.e. genotypes that most strongly reduce Yki phosphorylation (e.g. wts, ex fat) most strongly affect Yki localization, while genotypes with weaker effects on phosphorylation (e.g. fat, ex) have weaker effects on Yki localization. Although it is formally possible that inhibition of nuclear localization is not the only mechanism by which phosphorylation inhibits Yki activity, we think it more likely that an increase in nuclear Yki accounts for all biological effects, but that in some cases (e.g. ex or fat mutant clones), the increase was below our sensitivity of detection. In favor of this we note the additive effects of ex and fat mutants on Yki localization, which parallels their additive effects on Yki phosphorylation (Fig. 2B), and their additive effects on growth and gene expression (Feng and Irvine, 2007). This would also be consistent with the observation that Yki-S168A:GFP is hyperactivated, but its subcellular localization is only slightly shifted. Additionally, yki is essential for normal growth and viability, presumably via its influence on downstream gene expression, yet endogenous Yki is barely detectable in the nucleus. In this respect, Yki could be comparable to transcriptional co-activators of other signaling pathways, such as the Notch intracellular domain, which can have substantial biological activity even under conditions where its levels in the nucleus are below detection using conventional antibody staining techniques (Struhl and Adachi, 1998). Since even very low levels of endogenous Yki have significant biological effects, then even a slight increase might be sufficient to substantially influence downstream gene expression and growth.
Several transcription factors have previously been reported to be regulated by phosphorylation-dependent effects on nuclear localization, and a molecular mechanism for this involving binding to 14-3-3 proteins has been described (reviewed in Mackintosh, 2004). 14-3-3 binding also appears to contribute to the regulation of Yki localization, as phosphorylation of Yki promotes binding to 14-3-3 proteins in cultured cells. Moreover, this binding is dependent upon Ser168, which conforms to a consensus site for 14-3-3 binding, and which influences Yki activity and localization in vivo. However, our results suggest that there may be other cytoplasmic tethers that contribute to the retention of phosphorylated Yki in the cytoplasm. Analysis on Phos-tag gels established that Yki is phosphorylated on multiple sites. When Ser168 is mutated, binding to 14-3-3 proteins was eliminated, consistent with the observation that this is the only Ser within Yki that conforms to a 14-3-3 binding site consensus sequence. However, the bulk of Yki-S168A:GFP was retained in the cytoplasm. By contrast, when wts was mutant, Yki was equally distributed between cytoplasm and nucleus. Additionally, the observation that the growth and survival of cells expressing Yki-S168A:GFP could be blocked when Wts and Hpo were co-expressed implies that nuclear localization of this mutant form of Yki can still be blocked by Wts-mediated phosphorylation at other sites. Moreover, the ability of Wts to promote phosphorylation of Yki was reduced but not eliminated by mutation of Ser168. Altogether, these results suggest that Ser168 can only account for part of the regulation of Yki by Wts, which, if it is the only 14-3-3 interaction site, implies the existence of other cytoplasmic tethers.
While this manuscript was under review, the observation that Yki and YAP localization can be influenced by Warts-mediated phosphorylation was independently reported (Dong et al., 2007; Zhao et al., 2007). Our results overlap with these studies in describing the influence of Yki phosphorylation on Yki localization, and the binding to 14-3-3 proteins mediated by phosphorylation of Yki Ser168 (or its mammalian equivalent, YAP Ser127). Our observations extend these published findings by reporting, for the first time, the phosphorylation status of Yki in vivo. This has allowed us to characterize the effects of upstream regulators under endogenous expression conditions (as opposed to the extreme levels of expression and phosphorylation associated with co-transfection experiments in cultured cells). Importantly, our results imply that even subtle changes in phosphorylation and nuclear localization, such as are associated with fat mutants, can have dramatic effects on downstream gene expression and growth. Our results also clearly indicate that multiple sites can contribute to Yki phosphorylation and localization, in contrast to the inference that Ser 168 of Yki is the only relevant site (Dong et al., 2007) but in agreement with the inference that there are multiple Lats phosphorylation sites on YAP (Zhao et al., 2007).
MATERIALS AND METHODS
Fly stocks
Mutant clones were generated by FLP-FRT-mediated recombination using w; ykiB5 FRT42D/CyO-GFP, w; wtsX1 FRT82B/TM6b, w; exe1 FRT40A/CyO-GFP, w; ft8 FRT40A/CyO-GFP, w; ftGrv FRT40A/CyO-GFP, w; exe1 ftGrv FRT40A/CyO, yw hs-FLP; arm-lacZ M FRT42D/CyO-GFP, yw hs-FLP; Ubi-GFP FRT40A/CyO-GFP, yw hs-FLP; FRT82B Ubi-GFP/TM3b, yw hs-FLP; FRT42D Ubi-GFP/CyO-GFP, yw Mer4/FM7-GFP; Ubi-GFP, Ubi-Mer+ FRT40A/CyO-GFP and w; exe1 FRT40A/CyO-GFP; hs-FLP, Sb/TM6b.
Ectopic expression clones were created by Flip-out using yw hs-FLP[122]; act> y+>gal4 (AyGal4) or yw hs-FLP [122]; act> y+>gal4 UAS-GFP (AyGal4-GFP), UAS-yki, UAS-yki:GFP, UAS-ykiS168A:GFP, UAS-hpo, and UAS-wts. Ectopic expression of UAS-yki, UAS-yki:GFP, and UAS-yki-S168A:GFP was also induced with GMR-gal4, sd-gal4, dpp-gal4, ptc-Gal4, act-Gal4, tub-Gal4 ey-Gal4 and ci-gal4.
For rescue experiments in the wing imaginal discs, hs-FLP; ykiB5 FRT42D/CyO;Ci-gal4/TM6B and hs-FLP; arm-lacZ FRT42D; UAS-yki:GFP/TM6b were used. Among more than ten insertions each, we have retained four UAS-yki:GFP insertions, 4-4-Y and 4-12-1 (2nd chromosome), 4-3-Y and 4-9-Y (3rd chromosome) and four UAS-yki-S168A:GFP insertions, 10-2-1 and 10-12-1 (2nd chromosome) and 10-7-Y and 10-9-Y (3rd chromosome).
For reduction of ex or fat levels by RNAi, we used UAS-IR constructs 9396 (fat) and 22994 (ex), together with UAS-dicer2 (Vienna Drosophila RNAi Center) (Dietzl et al., 2007).
Yorkie antibody preparation
Full length Yki was cloned into pGEX-3X (Amersham Biosciences) at a SmaI site. GST:Yki was in BL21(DE3) E. coli (Invitrogen) by induction with 1mM IPTG. Insoluble inclusion bodies contained the majority of GST-Yki, and were used to immunize rabbits (Cocalico Biologicals).
Histology and imagaing
Imaginal discs were fixed and stained as described previously(Cho and Irvine, 2004), using as primary antibodies rabbit anti-Yki (1:400), anti-En (Developmental Studies Hybridoma Bank) and goat anti-β-gal (1:1000, Biogenesis). Fluorescent stains were captured on a Leica TCS SP5 confocal microscope. For some panels, maximum projection through multiple horizontal sections was performed to allow visualization of staining in different focal planes.
Generation of transgenic flies
The S168A mutation was introduced into Yki by primer-mediated site-directed mutagenesis. Yki and Yki-S168A with C terminal HA-tags (Huang et al., 2005) were amplified by PCR and cloned into pEGFP-N1 (Clontech) to construct yki:GFP and yki-S168A:GFP; these were then cloned into pUAST for P-element mediated transformation (Duke University Model System Genomics).
Protein blotting
Homozygous mutant wing discs were obtained from wandering third instar larvae using exe1/CyO-GFP, ftGrv/L14, exe1 ftGrv/CyO-GFP, and wtsP2 FRT82B/TM6b stocks; Oregon-R was used as wild type. Larvae were collected and dissected in PBS (10mM Sodium Phosphate, 2.7 mM KCl, 137 mM NaCl, pH 7.4), and wing discs were collected and lysed in CIP buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.9, 10 mM MgCl2, 1 mM DTT), 1 mM PMSF, 0.1% NP40, protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktail (Calbiochem). For CIP treatment phosphatase inhibitor cocktail was omitted, and lysate was incubated 37°C 30 min in 20 units CIP (NEB) per 100 μL reaction. Lysates were cleared by centrifugation and subjected to SDS-PAGE. To detect phosphorylated Yorkie in SDS-PAGE, we used Phos-tag AAL-107 (FMS Laboratory) according to the manufacturer’s instruction. Western blotting was performed using rabbit anti-Yki (1:4000), mouse anti-Actin (1:5000, CalBiochem), rat anti-HA:Hrp (1:2000, Roche), mouse anti-α-Tubulin (1:5000, Sigma), mouse anti-V5:Hrp (1:5000, Invitrogen).
Plasmid constructs
For immunoprecipitation assays, 14-3-3ε and 14-3-3ξ were amplified by PCR using total cDNA of S2 cells as a template and then cloned into pAc5.1/V5-HisA (Iinvitrogen). For assays of phosphorylation-dependent mobility shifts, Yki:V5, Yki-S168A:V5, Yki-N:V5, and Yki-N-S168A:V5 were constructed in pAc5.1/V5-HisA vector (Invitrogen). Flag-tagged Hpo, Myc-tagged Wts and HA-tagged Sav expressing constructs were from J. Jiang (Jia et al., 2003). pUAST-Yki:FLAG(3x) and pUAST-Yki:FLAG:FLAG(3x) were constructed by cloning Yki:FLAG and Yki:FLAG into pUAST-FLAG(3x) (Y. Mao).
S2 cell assays and co-immunoprecipitation
S2 cells were cultured with Schneider’s Drosophila medium (Invitrogen) and 10% FBS (Sigma). Transfections were performed with Cellfectin (Invitrogen) according to manufacture’s protocol. Co-immunoprecipitation assays were performed as described previously (Chen et al., 2003). To make yki double stranded RNA, the first 500 bp of yki coding region was amplified by PCR and subcloned into pCR2.1-TOPO vector (Invitrogen) and then both forward and reverse clones of yki were amplified using M13 primers for use as templates for T7 polymerase in vitro transcription. Generation of double strand RNA and RNAi in S2 cells was then performed as described (Clemens et al., 2000).
Acknowledgments
We thank J. Jiang, E. Hafen, B. Hay, Z.C. Lai, D. Pan, the Vienna Drosophila RNAi Center, the Developmental Studies Hybridoma Bank, and the Bloomington Drosophila Stock Ceter for reagents, Y. Feng for advice on Phos-Tag gels, R. Pan for photographing fly heads, and C. Rauskolb for comments on the manuscript. This research was supported by the Howard Hughes Medical Institute and NIH post-doctoral fellowship 1F32GM079817 to H.O.
References
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11–23. doi: 10.1016/s1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- Bennett FC, Harvey KF. Fat Cadherin Modulates Organ Size in Drosophila via the Salvador/Warts/Hippo Signaling Pathway. Curr Biol. 2006;16:2101–10. doi: 10.1016/j.cub.2006.09.045. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–68. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD. Delineation of a Fat tumor suppressor pathway. Nat Genet. 2006;38:1142–50. doi: 10.1038/ng1887. [DOI] [PubMed] [Google Scholar]
- Cho E, Irvine KD. Action of fat, four-jointed, dachsous and dachs in distal-to-proximal wing signaling. Development. 2004;131:4489–500. doi: 10.1242/dev.01315. [DOI] [PubMed] [Google Scholar]
- Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, Hemmings BA, Dixon JE. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci U S A. 2000;97:6499–503. doi: 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–6. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–33. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Irvine KD. Fat and Expanded act in parallel to regulate growth through Warts. Proc Natl Acad Sci U S A. 2007;104:20362–7. doi: 10.1073/pnas.0706722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–34. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Jia J, Zhang W, Wang B, Trinko R, Jiang J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 2003;17:2514–9. doi: 10.1101/gad.1134003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–57. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Mackintosh C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem J. 2004;381:329–42. doi: 10.1042/BJ20031332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103:12405–10. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21:886–97. doi: 10.1101/gad.1536007. [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Edgar BA. Filling out the Hippo pathway. Nat Rev Mol Cell Biol. 2007;8:613–621. doi: 10.1038/nrm2221. [DOI] [PubMed] [Google Scholar]
- Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H. The Tumor-Suppressor Gene fat Controls Tissue Growth Upstream of Expanded in the Hippo Signaling Pathway. Curr Biol. 2006;16:2081–9. doi: 10.1016/j.cub.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell. 1998;93:649–60. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Willecke M, Hamaratoglu F, Kango-Singh M, Udan R, Chen CL, Tao C, Zhang X, Halder G. The Fat Cadherin Acts through the Hippo Tumor-Suppressor Pathway to Regulate Tissue Size. Curr Biol. 2006;16:2090–100. doi: 10.1016/j.cub.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–61. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]