

Abstract
Metastatic disease is the major cause of morbidity and mortality in cancer. Although surgery, chemotherapy, or radiation can often control primary tumor growth, successful eradication of disseminated metastases remains rare. We have now tested whether direct targeting tumor tissues to generate antitumor immune response before surgical excision produces sufficient CTL against micrometastases. One unsolved problem is whether such response allows coming CTL to be educated and then exit the tumor site. Another unsolved problem is whether these CTL can then patrol and effectively eliminate spontaneously metastasized tumor cells in the periphery. In this study, we have shown that adenovirus-expressing TNFSF14 [LIGHT (name derived from homologous to lymphotoxins, shows inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes); Ad-LIGHT] inoculated directly into primary 4T1 tumor, a highly aggressive, spontaneously metastasizing mammary carcinoma, followed by surgical removal of the primary tumor can eradicate established and disseminated metastatic tumor cells in the peripheral tissues. Furthermore, we clearly show with a fibrosarcoma model Ag104Ld that local treatment can generate plenty of tumor-specific CTL that exit the primary tumor and infiltrate distal tumors to completely eradicate distal tumors. Therefore, targeting the primary tumor with Ad-LIGHT before surgical excision is a new strategy to elicit better immune response for the eradication of spontaneous metastases.
Micrometastases can establish early in heterogeneous primary tumor development and seed into distal tissue sites before clinical detection (1). In many cases, metastases from primary cancer can occur when the primary tumor size is very small (2, 3). Therefore, at the time of diagnosis, many cancer patients already have microscopic metastases, an observation that has led to the development of postsurgical adjuvant therapy for patients with solid tumors. Despite advances in early detection and modifications to treatment regimens, success has been limited and optimal treatment of metastatic disease continues to pose a major challenge in cancer therapy.
A variety of human and murine cancers have been proven to be antigenic and recognizable by T cells (4 – 6). However, the naturally occurring T cell responses against malignancies in humans are often not sufficient to cause regression of tumors, primary or metastatic. Immunotherapy would potentially elicit tumor-reactive T cells that can seek and destroy disseminated tumor Ag-positive cancer cells while sparing the surrounding healthy tissues, but active vaccination for tumor-bearing hosts only shows limited benefit so far (7). Adoptive transfer of autologous antitumor T cells expanded ex vivo with defined Ags has shown promise, but only in a small subset of highly selected melanoma patients (7, 8). Furthermore, lack of well-defined Ags in most tumors limits either active vaccination or adoptive transfer therapy. Immunotherapy that is effective even without determination of specific tumor Ags would be more applicable and more therapeutically feasible. Current immunotherapy often follows conventional surgery, radiation, and chemotherapy. Surgery can reduce tumor burden but also remove the major source of tumor Ags, which may signal to retract the immune response while radiation and chemotherapy would further damage the existing immune response. It is still unclear when and how to boost active immune responses against tumor.
LIGHT (name derived from homologous to lymphotoxins, shows inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes; Genome Database designation, TN FSF14) is a TNF family member that interacts with lymphotoxin β receptor (LTβR)3 and herpesvirus entry mediator (HVEM) mainly expressed on stromal cells and T cells, respectively (9). LIGHT is able to interact with LTβR to regulate chemokine expression (10). In addition, LIGHT also exhibits potent, CD28-independent co-stimulatory activity for T cell priming and expansion leading to enhanced T cell immunity against tumors and/or increased auto-immunity (11–13). We have recently shown that the expression of LIGHT in the tumor environment induces LTβR-associated chemokines and adhesion molecules that attract and prime naive T cells leading to the rejection of established, highly progressive tumors in mice (14). It raises the possibility that we can target tumor tissues with LIGHT to generate more CTL that might be sufficient to clear metastases.
Recent studies have revealed that naive or effector-memory T cells can leave the periphery and enter the draining lymph nodes (DLNs) through an active process (15, 16). It is not yet known whether sufficient numbers of tumor-specific CTL recruited to the primary tumor can survive and exit the tumor environment to patrol peripheral tissues and eradicate disseminated metastases. In the current study, we were able to show that delivery of LIGHT, using a recombinant adenovirus, into the primary tumor can generate enough CTL to leave local tumor and patrol periphery tissues and eradicate spontaneous metastases. The simplicity of this strategy, aiming to use the tumor tissues as a source of training sites for the generation of CTL in situ, provides the means to effectively mount an antitumor-specific immune response against not only the primary tumor, but also disseminated metastases. As such, this model reveals therapeutic possibilities that may be attractive for future clinical translation.
Materials and Methods
Mice and cell line
Female C3H × C57BL/6F1 (C3B6F1), C57BL/6, BALB/c, and FVB mice, 4 – 8 wk old, were purchased from The Jackson Laboratory. 2C TCR-transgenic mice, OT-1 TCR-transgenic mice, both on RAG-1−/−/B6 background (2C, OT-1 mice), RAG-1−/−/B6, and P14 TCR-transgenic mice (P14 mice) were bred and maintained in the specific pathogen-free facility at the University of Chicago. Animal care and experiments were done in accordance with institutional and National Institutes of Health guidelines and were approved by an animal use committee at the University of Chicago. Fibrosarcoma Ag104Ld was described previously (14, 17). The B16-SIY melanoma cell line was generated as described (18). 4T1 is a 6-thioguanine-resistant cell line derived from a spontaneous mammary carcinoma (19). The MC38 colon cancer cell line was provided by Y. Liu (Ohio State University, Columbus, OH). Ag104Ld and 4T1 tumor cells were grown in DMEM (Invitrogen Life Technologies) supplemented with 10% FCS (HyClone). B16, B16-SIY, and MC38 tumor cells were grown in RPMI 1640 (Invitrogen Life Technologies) supplemented with 10% FCS.
The generation of adenovirus-expressing LIGHT (Ad-LIGHT)
The mutant murine LIGHT (mmLIGHT) was generated previously (14). To construct recombinant mmLIGHT-adenovirus, a BamHI/NotI fragment containing murine LIGHT cDNA cut from pcDNA3.1-mmLIGHT was cloned into the BamHI/NotI sites of the first parental plasmid, pLEP-ubp (left-end plasmid, Tetr) after human ubiquitin promoter (ubp). Subsequently, the pLEP-mmLIGHT was ligated to a second plasmid, pREP (right-end plasmid, Ampr) at a unique intron-encoded ClaI site. The ligation product was packaged with λ phage packaging extracts. The pLEP/pREP hybrid cosmids were selected grown on Amp/Tet LB agar plate. BglII digestion was used to further identify recombinant cosmid containing insert mmLIGHT. The Ad-mmLIGHT DNA fragment was liberated from its recombinant cosmid by I-CeuI digestion, and the mixture of I-CeuI digestion without further purification was transfected into 293 cells for recombinant adenovirus production. The Ad-mmLIGHT is referred to as Ad-LIGHT in this study.
Tumor injection, treatments, and evaluation of metastases by colonogenic assay
Ag104Ld, B16-SIY, MC38, and 4T1 tumors were inoculated s.c. into the area around the tail base on syngeneic C3B6F1, B6, or BALB/c mice. The tumor nodules were inoculated with indicated amount of Ad-LIGHT or Ad-control virus intratumorally in 50 μl of PBS. For surgical excision of primary 4T1 tumors, mice were anesthetized, and tumors were resected with sterilized instruments. Wounds were closed with metallic clips. All mice survived surgery. Mice in which primary tumors recurred at the site of the surgical excision were eliminated from the experiments. A colonogenic assay was used to evaluate metastases by 4T1 tumors as previously described (20). Briefly, lungs were collected and chopped before being dissociated in DMEM supplemented with 10% FCS containing 1.5 mg/ml collagenase type D (Sigma-Aldrich) (collagenase D solution) for 30 min in 37°C shaking incubator at 178 rpm speed. Organs were then plated at various dilutions in the DMEM supplemented with 10% FCS and 60 μM 6-thioguanine. Individual colonies representing micrometastases were counted after 5–10 days.
Measurement of cytokines in the spleen (SPL) and tumor
We prepared tumor and SPL homogenates as described (21). The amount of cytokines in the supernatants was quantified using the cytometric bead array kit (BD Biosciences) on a FACSCalibur cytometer equipped with CellQuestPro and CBA software (BD Biosciences) according to manufacturer’s instruction.
Adoptive transfer of 2C or P14 T cells and analysis of cells by FACS
LN cells and splenocytes were isolated from 2C or P14 mice and CD8+ T cells were negatively selected with a CD8+ T cell enrichment kit (Miltenyi Biotec). A total of 3 × 106 2C or P14 T cells were transferred into OT-1 mice. To transfer CFSE-labeled T cells, T cells were labeled with CFSE as described previously (14, 22), and 3 × 106 CFSE-labeled T cells were injected into mouse. Cells were isolated from the inguinal LNs (DLNs), other LNs (non-DLNs (NDLN)), SPL, or tumors at the time indicated. CFSE dilution was evaluated as described before (14, 21, 22).
Evaluation of the presence of Ag104Ld tumor cells in the lymphoid tissues after s.c. inoculation
Peripheral, mesenteric LNs, and SPL were collected 1, 6, 10, 14 days after s.c. inoculation of tumor cells, chopped, and dissociated in collagenase D solution for 20 min in a 37°C shaking incubator at 178 rpm speed and single-cell suspension was collected after 20 min and plated in DMEM supplemented with 10% FCS and 0.5 mg/ml G418. Plates were examined every week for 4 wk to detect colonies, which indicate the presence of live tumor cells. As a positive control, cultured tumor cells were added to the wild-type lymphoid organ before the procedure to obtain single-cell suspension begins. Only experiments that were sensitive enough to detect one tumor cell among at least 106 LN or SPL cells were counted.
In vivo cytotoxicity assay
The in vivo cytotoxicity assay was performed similarly as described by Barber et al. (23). In brief, splenocytes from Ld-positive BALB/c mice or control Ld-negative FVB mice were labeled with 10 μM cytosolic dye CFSE, referred to as highly labeled targets. And splenocytes from C3B6F1 mice were labeled with 1 μM CFSE, referred to as lowly labeled targets. The highly and lowly labeled cells were mixed with 1:1 ratio and transferred retro-orbitally (50 –100 million cells totally for each mouse, equal amount of cells transferred for each mouse in each individual experiment) into the indicated groups of recipient mice. After 18 h of in vivo killing, lymphocytes were isolated from the SPLs and analyzed by flow cytometry for target cell clearance. Target cells were differentiated from recipient cells based on CFSE staining and from each other based on different intensity of the staining. Gated on CFSE+ cells, the percentage of specific killing was calculated as the following: 100 − ((% CFSEhigh cells in tumor challenged mice/% CFSElow cells in tumor challenged mice)/(% CFSEhigh cells in naive mice/% CFSElow cells in naive mice)) × 100.
Statistical analysis
For analysis of the difference in tumor growth, the random effect models for longitudinal data were used to analyze such data because tumor growth was observed repeatedly over time on the same mouse. For each experiment, the tumor growth was assumed to depend on treatment and to follow a linear growth rate over time. The model gave an overall estimate of the intercept and slope of the linear growth for each group. We were interested mainly in comparing whether the slope, i.e., the growth rate, was different among different treatment groups. Both the intercept and slope were allowed to vary among individual mice. The actual tumor growth may not follow a linear growth trend over the entire follow-up period. The increase of tumor growth was slow at the early stage and became rapid at the later stage in some experiments. We also investigated this by adding a quadratic term of the follow-up time in the above random effect models. All other statistics were done using an unpaired Student two-tailed t test. Error bars represent SD.
Results
Ad-LIGHT controls spontaneous tumor metastases
The poorly immunogenic 4T1 mammary carcinoma closely mimics human breast cancer in its anatomical site, immunogenicity, growth characteristics, and more importantly, metastatic properties (19, 24, 25). 4T1 can metastasize to various organs, such as lung, as early as day 10 after inoculation of 105 4T1 tumor cells. To develop a clinically relevant therapeutic strategy to deliver LIGHT into established 4T1 to study its potential effect on metastasis, we generated a recombinant, replication-deficient Ad-LIGHT. We first infected the mammary carcinoma 4T1 tumor cells in vitro with Ad-LIGHT or adenovirus expressing LacZ as control reagent (Ad-control). The 4T1 tumor cells infected with Ad-LIGHT, but not Ad-control, expressed a high level of LIGHT 24 h after infection (Fig. 1). To determine whether Ad-LIGHT can induce expression of LIGHT in other tumor cells, we infected several tumor cell lines, including fibrosarcoma Ag104Ld and melanoma B16 in vitro for 24 h. Detection of LIGHT expression was evaluated using a soluble LTβR, one of the receptors for LIGHT. In fact, Ad-LIGHT was able to confer LIGHT expression on all three tumor cell lines we tested (Fig. 1). The 4T1 tumor cells were then harvested and 105 of LIGHT-expressing or Ad-control-infected 4T1 tumor cells were inoculated s.c. into BALB/c mice. Primary tumor growing s.c. was monitored for 35 days before the mice were sacrificed for the evaluation of metastases in the lung using a colonogenic assay. We found that the growth of the primary tumor was retarded but not completely rejected (Fig. 2A). Impressively, no colonies of metastatic cells were detected in the lungs of mice inoculated with LIGHT-expressing 4T1 tumor cells while a high number of metastases were detected in the lungs of mice bearing control tumors (Fig. 2B). These results indicated that LIGHT expression by the 4T1 tumor was sufficient to control metastases even in the presence of a growing primary tumor.
FIGURE 1.
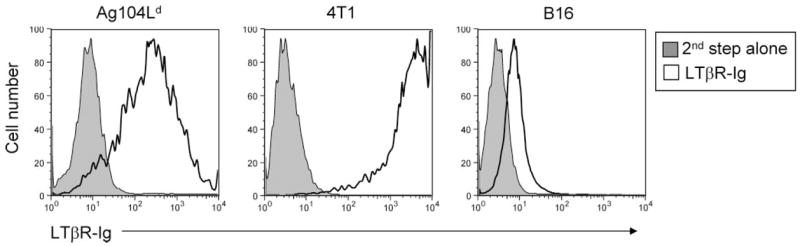
Expression of LIGHT by various tumor cell lines upon infection with Ad-LIGHT. A total of 1 × 106 tumor cells were plated in a 100-mm cell culture dish for 24 h then infected with Ad-LIGHT at 2 × 108 PFU/ml for 24 h. Cells were harvested and checked for LIGHT expression by FACS staining with LTβR-Ig at 0.02 mg/ml followed by second step Ab PE-coupled donkey anti-human IgG (white), or with second step Ab alone as control (gray). The experiments were repeated for three times. A representative graph was presented.
FIGURE 2.
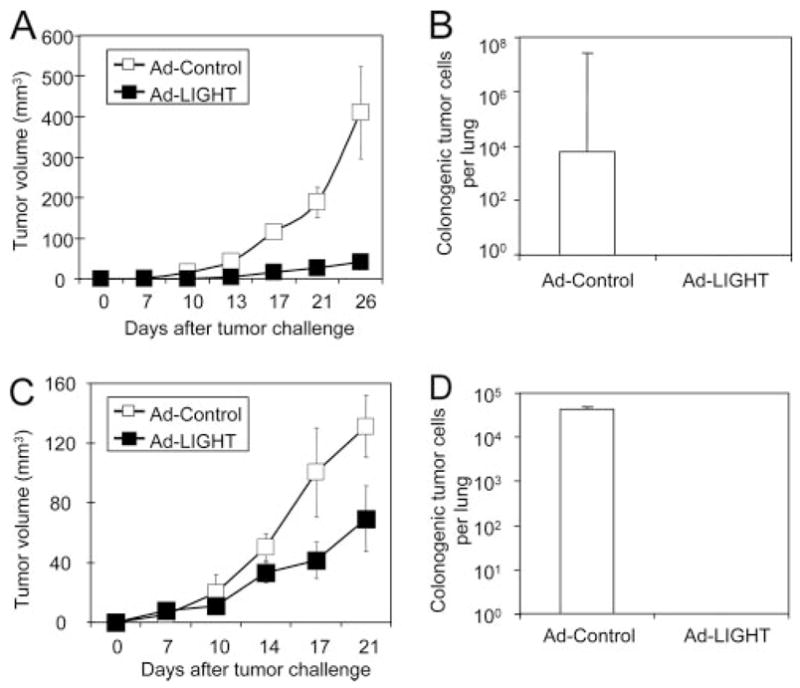
Ad-LIGHT treatment controls spontaneous metastases. A and B, Expression of LIGHT on 4T1 tumor cells prevents the development of spontaneous metastases. In vitro-cultured 4T1 mammary carcinoma (1 × 106 cells) were infected with Ad-LIGHT or Ad-control (2 × 108 PFU/ml) for 24 h and then 1 × 105 cells were injected s.c. into the flank of BALB/c mice. Tumor growth was monitored (A) until mice were sacrificed on day 35 posttumor inoculation for analysis of lung metastases with colonogenic assay (B). C and D, The effect of Ad-LIGHT on tumor growth and metastases when given 7 days after tumor implantation. 4T1 tumor cells (1 × 105) were injected s.c. into the flank of BALB/C mice on day 0. Seven days later, 2.5 × 109 PFU of Ad-LIGHT (■) or Ad-control (□) was intratumorally administered and tumor growth was continually monitored (C). Data are given as tumor volume (mean ± SD). D, Using the same protocol, primary tumor was surgically removed on day 18 and mice were sacrificed on day 35 posttumor inoculation for analysis of lung metastases with colonogenic assay. Data are a representative of two independent experiments and five mice in each group.
We next wanted to test the therapeutic effects of Ad-LIGHT on the established primary tumor. We investigated whether Ad-LIGHT delivered directly into an established tumor would control cancer metastases. The s.c. growth of 105 4T1 tumor cells in BALB/c mice was established for 7 days, followed by the intra-tumor injection of 2.5 × 109 PFU of Ad-LIGHT or Ad-control. We observed that the growth of the primary tumor was somewhat suppressed after Ad-LIGHT treatment but continued to grow (Fig. 2C). To mimic the clinical therapeutic setting, the primary tumor was surgically removed 18 days after the tumor inoculation. Mice were then sacrificed to evaluate the metastases in the lung using the colonogenic assay 35 days postprimary tumor inoculation. Interestingly, we detected no metastases in the lung of the mice that had been given Ad-LIGHT treatment while large numbers of metastatic cancer cells were found in the lung of the control mice (Fig. 2D). These results demonstrate that LIGHT delivered by adenoviral gene transfer into an established primary tumor induced significant antitumor effects to control the occurrence of spontaneous metastases.
Ad-LIGHT mediates the rejection of established spontaneous lung metastases
There are two possible general mechanisms by which Ad-LIGHT treatment could inhibit the number of metastatic cancer cells present in the lung. One is that Ad-LIGHT-induced antitumor immunity suppresses the growth of the primary tumor which then prevents the tumor cells to metastasize to other sites. The other is that Ad-LIGHT triggers a potent antitumor immunity to cause rejection of already seeded distal tumors. Given the potent antimetastatic activity of Ad-LIGHT treatment, we wanted to examine whether Ad-LIGHT treatment would possibly be effective to treat hosts already bearing detectable metastases. Because s.c. injection of 105 4T1 tumor cells consistently results in detectable metastases in the lung 11 days posttumor inoculation, Ad-LIGHT treatment after the 11-day time point would indicate the effectiveness of the treatment on already seeded metastatic cancer cells. We inoculated 105 4T1 tumor cells s.c. to BALB/c mice, provided intratumoral injection of 1 × 109 PFU of Ad-LIGHT or Ad-control 14 and 17 days later, and then surgically removed the primary tumor 24 days after initial tumor inoculation. The mice were sacrificed and the metastases in the lung were analyzed 35 days postinoculation of primary tumor. Although we found a large number of metastatic colonies in the lung of the Ad-control-treated mice, a dramatic decrease in the number of metastatic cancer cells was detected in Ad-LIGHT-treated mice (Fig. 3A). More importantly, treatments with Ad-LIGHT in combination with surgery significantly reduced lung metastases compared with surgery alone even in the event that surgery was performed early on day 14 when Ad-LIGHT treatment started (Fig. 3A and Table I). The antimetastatic effect of Ad-LIGHT was dependent on CD8+ T cells as depletion of CD8+ T cells starting at the time of Ad-LIGHT treatment completely abolished the protective effects (Fig. 3A and Table I). Furthermore, the combination treatment of surgery and Ad-LIGHT, starting on day 14 after primary tumor inoculation resulted in a substantial increase of mice free of any detectable cancer cells in the lung, from 13.6% to 51.6%, while surgery alone showed 100% metastases (Table I). Together, these results strongly suggest that local treatment of cancer with Ad-LIGHT is effective at eradicating pre-existing metastases. With this combination therapy of Ad-LIGHT and surgery, 50% of mice consistently survived long term while 100% of mice in the control group died of metastases shortly after surgery (Fig. 3B). Therefore, local treatment with Ad-LIGHT generated a strong antitumor immunity that was dependent on CD8+ T cells to eradicate pre-existing metastases in the periphery, conferring long-term protection and eventual cure.
FIGURE 3.
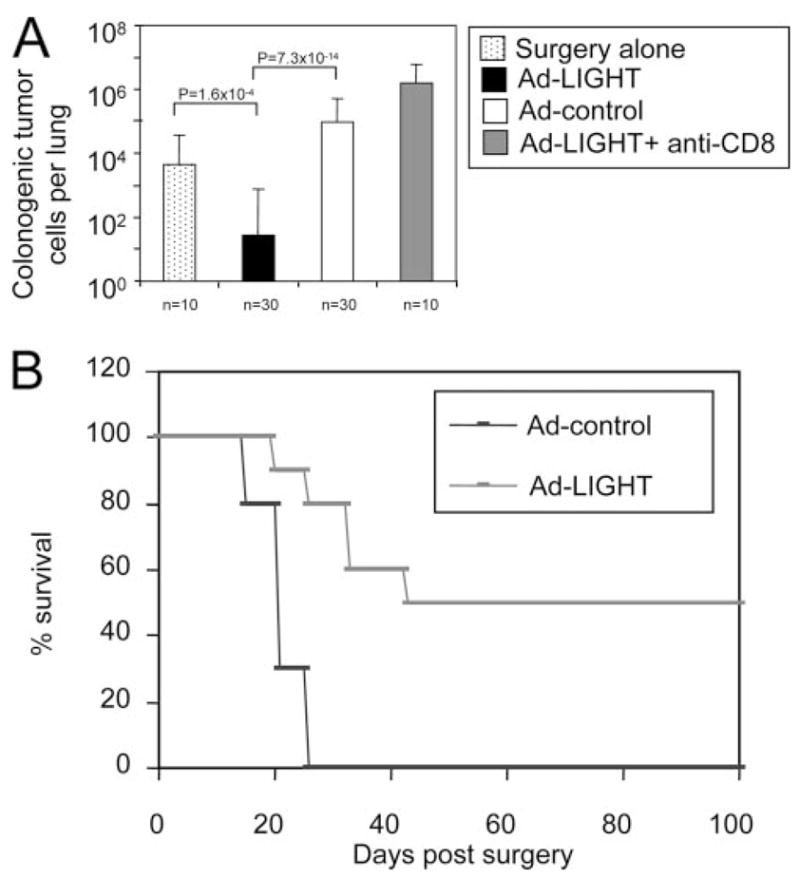
Ad-LIGHT treatment eradicates established metastases. A, 4T1 mammary carcinoma cells injected s.c. into the flank of BALB/c mice were treated intratumorally with 1 × 109 PFU Ad-LIGHT (black) or Ad-control (white) on days 14 and 17 posttumor inoculation. One group of mice was treated with surgery alone on day 14 posttumor challenge (dotted). Other 4T1 tumor-bearing mice were treated with Ad-LIGHT in the same way, with the addition of CD8 depletion by anti-CD8 Ab (YTS. 169.4.2), starting day 14 after primary tumor inoculation (gray). Anti-CD8 Ab was given to mice i.p., 125 μg/mouse once every week until the mice were sacrificed for analysis. More than 90% of CD8+ T cells were depleted by this regimen, as confirmed by FACS staining of peripheral blood. Except for the mice that were treated with surgery alone, the primary tumors (~150 mm3) on other mice were surgically resected on day 24 and mice were sacrificed for analysis of lung metastases with colonogenic assay on day 35. Data are a pool of multiple independent experiments. B, Fifty percent of mice treated with Ad-LIGHT in combination with surgery survive long term. 4T1 mammary carcinoma cells injected s.c. into the flank of BALB/c mice were treated intratumorally with 1 × 109 PFU Ad-LIGHT (pink), Ad-control (blue) on days 14 and 17 posttumor inoculation. Primary tumors on mice were surgically resected on day 24. The mice were checked for survival over 100 days postsurgery. Similar data have been generated in two independent facilities (University of Chicago and Biogen). A representative of two experiments, 10 mice in each group, was shown.
Table I.
Ad-LIGHT eradicates metastases and promotes long-term survival
| Treatments and Timea | Time of Sacrifice, In Daysa | Number of Mice Free of Tumor Cells in the Lung/All Mice (%)b |
|---|---|---|
| None | 14 | 3/22 (13.6%) |
| Surgery on day 14 | 35 | 0/10 (0%) |
| Ad-controlc on day 14+ Surgery on day 24 | 35 | 0/35 (0%) |
| Ad-LIGHTc on day 14+ Surgery on day 24 | 35 | 18/35 (51.4%) |
| Ad-LIGHT and CD8 depletiond on day 14+ Surgery on day 24 | 35 | 0/35 (0%) |
Days after primary tumor inoculation.
Pooled from several independent experiments.
Total of 2.5 × 109 PFU Ad-control (LacZ) or Ad-LIGHT was injected intratumorly per mouse.
A total of 125 mg of depleting anti-CD8 Ab was injected on day 14 and once every week.
LIGHT delivered by adenovirus mediates host resistance to various tumor lines
Next, to demonstrate the versatility of the Ad-LIGHT treatment in other tumor models also defined as aggressive and/or poorly immunogenic, we inoculated a fibrosarcoma line, Ag104Ld; a melanoma line B16-SIY; and a colon cancer line MC38. Ag104Ld tumor cells grow aggressively in vivo and extremely difficult to treat. Inoculation of only 104 tumor cells is sufficient to kill an immunocompetent host in 30 days, without a detectable immune response (14, 17). High dose of Ag104Ld tumor cells (106) was injected s.c. into C3B6F1 mice. Ad-LIGHT (2.5 × 109 PFU) or Ad-control was injected intratumorly 14 days after tumor challenge when the tumor mass was well-established. Following Ad-LIGHT treatment, the Ag104Ld tumors persisted for a few days before being completely rejected, while those treated with Ad-control continued to grow progressively (Fig. 4A). Tumor rejection mediated by Ad-LIGHT was CD8+ T cell-dependent and led to strong memory protection against the rechallenge of Ag104Ld at doses as high as 107 tumor cells (data not shown). The therapeutic effect of Ad-LIGHT is not limited to 4T1 and Ag104Ld. Melanoma B16s and colon cancer MC38 are known to be aggressive and poorly responsive to various treatments (26, 27). The growth of B16-SIY and MC38 was also inhibited by intratumor injection of Ad-LIGHT (Fig. 4, B and C). These results demonstrate that local treatment of Ad-LIGHT can mediate partial or complete control of various large established tumors.
FIGURE 4.
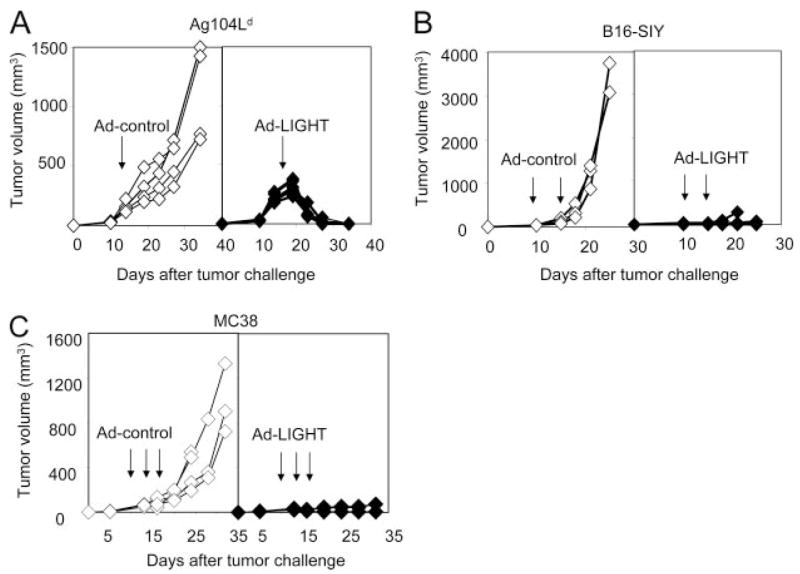
Intratumoral Ad-LIGHT treatment inhibits the growth of various established tumors. A, Antitumor effects of Ad-LIGHT on Ag104Ld tumors. A total of 106 Ag104Ld tumor cells were injected s.c. into the C3B6F1 mice. A total of 2.5 × 109 PFU Ad-LIGHT (◆) or Ad-control (◇) was injected intratumorly 14 days after tumor challenge. Tumor growth was recorded. One representative of three experiments was shown. B, Antitumor effects of Ad-LIGHT on B16-SIY tumors. B6 wild-type (WT) mice were injected s.c. with 1 × 106 cells near the base of the tail. Ten days later, 1 × 109 PFU of either Ad-LIGHT (◆) or Ad-control (◇) was injected intratumorly. The treatment was repeated 4 days later followed by continued monitoring for tumor growth. One representative of two experiments is shown. C, Antitumor effects of Ad-LIGHT on MC38 tumors. B6 WT mice were injected subcutaneously with 1 × 105 cells near the base of the tail. Ten days later, 1 × 109 PFU of either Ad-LIGHT (◆) or Ad-control (◇) was injected intratumorly. The treatment was repeated twice, 3 days apart, followed by continued monitoring for tumor growth. One representative of three experiments was shown. Arrow indicates the time of injection. Tumor volume was calculated as length × width × height/2.
Ad-LIGHT treatment at local site enhances T cell-mediated responses at local and distal site
We have demonstrated that Ad-LIGHT-mediated rejection of metastases was CD8+ T cell dependent in 4T1 tumor model. Next, we would like to trace Ag-specific T cell responses and visualize T cell trafficking in the process of local Ad-LIGHT treatment leading to the eradication of a distal tumor. 4T1 tumor model lacks a traceable tumor Ag. We decided to use Ag104Ld tumor system to examine the antitumor T cell responses because it bears a defined dominant model tumor Ag Ld, which can be recognized by 2C TCR-transgenic T cells through direct Ag-presentation pathway (28). We inoculated C3B6F1 mice with two s.c. Ag104Ld tumors 6 days apart to mimic a s.c. metastatic model. The intratumoral Ad-LIGHT treatment of the primary tumor was given 5 days after the secondary tumor inoculation. We found that both the primary and secondary tumors were rejected in the mice treated with Ad-LIGHT only at the primary tumor, but not in the mice treated with Ad-control (Fig. 5A). Because the effect of Ad-LIGHT is CD8-mediated, we examined the percentage of CD8+ T cells among the Ly5.2+ leukocytes, and IFN-γ-producing effectors among all the CD8+ T cells in the DLNs, SPL, primary and secondary tumors. The number of CD8+ T cells and IFN-γ-producing effector CD8+ T cells did not change significantly in the lymphoid organs, yet it increased dramatically in both the primary and the secondary tumors after Ad-LIGHT treatment (Fig. 5, B and C). The development of antitumor immunity can be associated with a change of the cytokine environment (21). To examine the cytokine milieu in the lymphoid organs, and inside the tumor itself, we harvested the SPL and tumor tissues 7 days after Ad-LIGHT or Ad-control treatment for cytokine measurement. Interestingly, we found that the levels of proinflammatory cytokines, TNF-α and IFN-γ, were increased considerably in the primary and the secondary tumors of Ad-LIGHT-treated mice comparing to the Ad-control-treated ones (Fig. 5D). TNF-α was also found increased in the SPL of Ad-LIGHT-treated mice (Fig. 5D). Other cytokines such as IL-2, IL-4, IL-5, or IL-6 were not significantly different between the two groups of mice in the lymphoid organ or inside the tumor microenvironments. Thus, tumor rejection mediated by Ad-LIGHT was accompanied by an obvious increase of CD8+ T cells, IFN-γ producing effector T cells, and the augmentation of inflammatory cytokines, TNF-α and IFN-γ, inside both the primary and secondary tumors.
FIGURE 5.
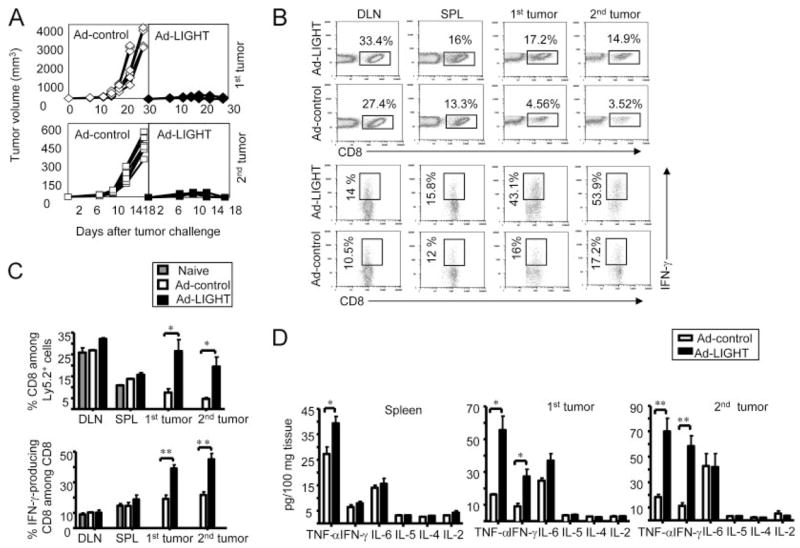
Ad-LIGHT treatment on the primary tumor induces strong antitumor immune responses in the secondary tumor. Each of the C3B6F1 mice was inoculated with 105 Ag104Ld and 105 Ag104Ld tumor cells 6 days after primary tumor challenge. The intratumor Ad-LIGHT treatment (2.5 × 109 PFU) on the primary tumor was given 5 days after the secondary tumor inoculation. The growth of both of the primary and secondary tumors was monitored (A). The percentage of CD8+ T cells among Ly5.2+ cells, and IFN-γ-producing CD8+ T cells among CD8+ T cells in the DLN, SPL, primary and secondary tumors were examined (B) and the statistics was calculated (C). The SPL and tumor tissues were harvested 7 days after Ad-LIGHT or Ad-control treatment, ground the tissues and collected the supernatant for cytokine measurement (D). Representative or pooled data from two experiments, five mice each group for each, were shown.
Ad-LIGHT targeting the primary tumor generates more CTL
We next performed adoptive transfer experiments to test whether Ad-LIGHT can drive the primary tumor to generate more antitumor CTL, which can subsequently circulate systemically and facilitate rejection of the distal tumor. To investigate whether the T cells from lymphoid organs of treated mice are capable of mediating the rejection of established tumors, we inoculated 105 Ag104Ld tumor cells into C3B6F1 mice, then treated with 2.5 × 109 PFU of Ad-LIGHT or Ad-control 14 days posttumor inoculation. At 21 days, we collected the SPL and LNs from the treated mice and purified the T cells and adoptively transferred 107 of these T cells to C3B6F1 mice bearing an Ag104Ld tumor that had been established for 7 days. We found the mice that received T cells from Ad-LIGHT-treated mice all rejected their tumors while the ones that received T cells from Ad-control-treated mice died of tumor burden the same as the mice received no treatments (Fig. 6A). This result suggests that activated Ag-specific T cells present in circulation after Ad-LIGHT treatment, but not control virus, are sufficient to reject established tumors. Next, we would like to directly visualize in vivo whether Ad-LIGHT treatment at the primary tumor would generate more CTL that are able to kill targets systemically in an Ag-specific manner. We performed in vivo killing assays to compare the abilities of CTL from Ad-LIGHT or Ad-control-treated Ag104Ld tumor-bearing mice to kill the Ld-positive vs Ld-negative targets. Our results showed that CFSE-labeled BALB/c (Ld-positive) splenocytes as targets were killed significantly more in the Ad-LIGHT-treated tumor-bearing hosts than in the Ad-control-treated mice, which killed Ld-positive targets similarly as Ld-negative Ag104 tumor-bearing mice (Fig. 6, B and C). The CTL functions were specific against Ag Ld because FVB (Ld-negative) splenocytes as targets were not killed in both groups of tumor-bearing mice comparing to the naive mice (Fig. 6, B and C). Therefore, it is possible that targeting primary tumor tissue may generate tumor Ag-specific CTL that exit to circulation, which then patrol peripheral tissues for eradication of distal tumors.
FIGURE 6.
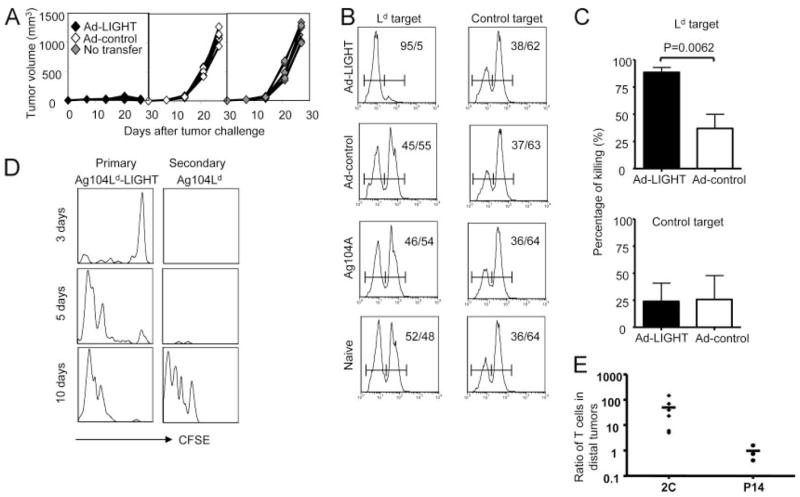
Ag-specific T cells stimulated by LIGHT inside the primary tumor traffic to the distal sites. A, T cells from the lymphoid organs of Ad-LIGHT-treated mice mediate the rejection of established tumors upon adoptive transfer. A total of 105 Ag104Ld tumor cells were inoculated into the C3B6F1 mice. The mice were then treated with 2.5 × 109 PFU of Ad-LIGHT or Ad-control 14 days posttumor challenge. Seven days after the treatment, we collected the SPL and LNs from the treated mice and purified the T cells and adoptively transferred 107 of these T cells to C3B6F1 mice bearing an Ag104Ld tumor that has been established for 7 days. The tumor growth on these mice was monitored. B and C, Intratumor Ad-LIGHT treatment promotes Ag-specific CTL. A total of 105 Ag104Ld or Ag104 tumor cells were inoculated into the C3B6F1 mice. The mice bearing Ag104Ld tumor were then treated with 2.5 × 109 PFU of Ad-LIGHT or Ad-control 14 days posttumor challenge. Seven days after treatment, in vivo killings were tested in the mice as described in Materials and Methods. Gated on CFSE+ cells, the representative percentage of highly labeled targets, BALB/c (Ld target) or FVB (control target) splenocytes, and lowly labeled C3B6F1 splenocytes in dedicated groups of recipient mice were shown (B). The statistics of percentage of specific killing from pooled experiments including total five mice from Ad-LIGHT treated group and eight mice from Ad control-treated group were shown (C). D and E, Each of the OT-1 mice was inoculated with two tumors. The primary tumor was either 106 Ag104Ld or Ag104Ld-LIGHT, while the secondary one was 105 Ag104Ld tumor cells. Ten and 14 days posttumor challenge 3 × 106 CFSE-labeled 2C T cells, or 3 × 106 2C T cells and equal numbers of P14 T cells were infused into mice. Presence and CFSE dilution of the 2C T cells were evaluated at days 3, 5, and 10 after adoptive transfer inside the primary Ag104Ld-LIGHT tumor and the secondary Ag104Ld tumors, one representative of three mice for each time points were shown (B). The percentage of 2C T cells identified by Vβ8-positive and anti-2C-specific TCR Ab 1B2-positive cells, and P14 T cells identified by Vβ8-positive and 1B2-negative cells in the DLN, non-DLN, SPL, primary and secondary tumors in the hosts bearing a LIGHT-expressing Ag104Ld or parental Ag104Ld was monitored and the ratios of 2C or P14 T cells in the secondary tumors with a LIGHT-positive Ag104Ld primary tumor vs a parental Ag104Ld primary tumor were compared (C).
Activated tumor Ag-specific T cells generated from the primary tumor move to the distal tumor
Although tumor-specific effector cells have been detected in tumors of mice and humans (4 – 6), it remains to be shown whether these T cells possess the ability to leave the tumor site and home to distant metastases. To demonstrate that the tumor Ag-specific effector T cells generated in the LIGHT-expressing primary tumor could patrol the peripheral tissues to infiltrate a distal secondary tumor mass that is not expressing LIGHT, we used a tumor cell line, Ag104Ld transfected with LIGHT. It is difficult to confirm whether the population of activated tumor-specific T cells detected in DLN originated from the tumor or were generated in lymphoid tissue, because cross-presentation of tumor Ags can occur in both compartments. To circumvent this caveat, we used a system in which reactive T cells recognize Ag only through direct presentation on tumor cells. We have previously reported that in OT-1 hosts (H-2b), adoptively transferred 2C TCR-transgenic T cells recognizing Ag104Ld tumors, cannot be activated by indirect presentation, but can only be activated by direct presentation of Ld by the tumor cells (14, 22). These host mice lack endogenous T cells reactive to the Ag104Ld tumor, and 2C T cells are the only T cells mediating the antitumor responses. We found no homeostatic expansion of these 2C cells upon transfer into these mice in the presence of CD8+ OT-1 T cells. To demonstrate whether effector T cells leave the primary tumor to traffic to the distal tumor, we used this model system in which 2C T cells were adoptively transferred to tumor-bearing OT-1 mice. The primary tumor was either 106 Ag104Ld or Ag104Ld-LIGHT, while the secondary tumor was 105 Ag104Ld. CFSE-labeled 2C T cells were transferred 10–14 days posttumor challenge. Proliferation of the 2C T cells inside the tumor environment was evaluated on days 3, 5, and 10 after the adoptive transfer. Even only 3 days after transfer, CFSEhigh non-proliferating 2C cells were present in the LIGHT-expressing Ag104Ld primary tumor and lymphoid tissues. Subsequent proliferation of these 2C T cells was well-observed inside tumor but not DLN 5 days after transfer, as indicated by the in situ dilution of CFSE. By day 10, a larger number of proliferated 2C T cells were found in Ag104Ld-LIGHT tumors. In the secondary Ag104Ld tumor, the presence of diluted CFSE-labeled 2C T cells was detected in mice bearing Ag104Ld-LIGHT tumor (Fig. 6, D and E). 2C T cells were barely detectable in either the primary or the secondary tumors in mice bearing Ag104Ld as primary tumor (data not shown).
To determine whether this influx of 2C T cells into the Ag104Ld tumor of these mice was due to Ag-specific infiltration or by passive circulation, we transferred P14 T cells, which are lymphocytic choriomeningitis virus gp33-specific TCR transgenic T cells and are nonresponsive to Ag104Ld tumors, in equivalent numbers with our Ag-specific 2C T cells. We found that 2C and P14 T cells were present at a comparable level in the DLN, NDLN, SPL as well as tumors on the mice bearing two Ag104Ld tumors (data not shown). In contrast, there were much more 2C than P14 T cells in the secondary Ag104Ld tumor in the mice bearing a LIGHT-expressing Ag104Ld primary tumor (Fig. 6E). Although the number of tumor Ag-irrelevant P14 T cells in the secondary tumors was comparable, 2C T cells were 5–100 times more abundant in the secondary tumor in the presence of a LIGHT-positive primary tumor than a parental primary tumor (Fig. 6E). We failed to detect any live tumor cells in the LNs 1, 6, 10, and 14 days after tumor challenge using a sensitive colonogenic assay that is capable of identifying one live tumor cell among 106 cells from lymphoid organs after s.c. tumor challenge (data not shown). This ruled out that s.c. Ag104Ld tumor cells migrate in a significant amount to the lymphoid tissues to prime 2C T cells. Thus, priming by the Ag104Ld tumors is dependent primarily on the direct presentation to 2C T cells inside the tumor. The result of these experiments strongly suggests that some activated Ag-specific T cells are capable of migrating out of the tumor and into systemic circulation following activation and expansion by LIGHT inside the tumor. Using Ag104ALd and 2C system allows us to conclude this while other systems with potential cross-priming could not address this issue clearly. Taken together, these results demonstrated that LIGHT expression on tumor cells mediated by the Ad-LIGHT treatment can generate large numbers of effector cells from the primary tumor to efficiently survey peripheral tissues and direct proinflammatory changes within distal tumors, leading to its regression.
Discussion
Metastasis is frequently a fatal step in the progression of solid malignancies. Disseminated metastatic tumor cells can remain dormant and clinically undetectable for months or even years following surgical resection of the primary tumor, leading to subsequent clinical disease recurrence. Metastases are difficult to be completely eradicated by surgery, chemotherapy, and radiation. In this study, we have demonstrated that local delivery of LIGHT into the primary tumor not only prevents the formation of metastases, but also eradicates established metastases in peripheral tissues. Conventional treatments that surgically remove tumor first to be followed by chemotherapy, or radiation often fail to clear metastases. Our study shows that primary tumor can be used as a major site to generate better CTL that then leave the primary tumor, patrol, detect, and eradicate established metastatic tumors. Therefore, strategically planned immunotherapy, using primary tumor tissue as the site for generating and sustaining a tumor-specific immune response, can in fact bypass current therapeutic limitations that rely on determination of specific tumor Ags and often fail to clear metastastic tumor.
An advantage of intratumor treatment with LIGHT is that anti-tumor CTL can be effectively and rapidly generated inside tumor without the knowledge of tumor Ags. The following four experiments we conducted support the notion that the clearance of the distal metastases may largely be attributable to the effects of LIGHT in the primary tumor, rather than a general systemic immune stimulation by the adenovirus expressing LIGHT: 1) highly progressive tumor cells stably transfected with LIGHT mediated rejection of distal tumors completely. 2) 4T1 tumor cells infected with Ad-LIGHT in vitro resulted in no metastasis. In this model, any remaining viral excess was washed out before inoculation of LIGHT-infected tumor cells, thus rendering systemic stimulation by Ad-LIGHT impossible. 3) 4T1 tumor cells infected with Ad-LIGHT as a therapeutic vaccine led to the eradication of metastases (data not shown). 4) Our preliminary data showed that inoculation of Ad-LIGHT outside of the primary tumor failed to prevent lung metastasis. There are additional advantages of intratumor treatment over systemic treatment, one of which is to limit auto-immunity: 1) there are more tumor-specific T cells and fewer autoreactive T cells inside the tumor tissues; 2) there are more unique tumor Ags inside tumor; 3) suppressive environment inside the tumor prevents nonspecific activation or bystander activation of autoreactive T cells inside tumor; 4) T cells will be more readily activated when first signal (tumor Ags) and second single (LIGHT) are closely provided. Intratumor injection of LIGHT provides second signal inside tumor tissues so that the tumor-specific T cells have more chance to be activated by tumor Ags in the same environment. We have not observed any signs of autoimmunity on mice that were treated with Ad-LIGHT and survived long term (over 100 days postsurgery) so far. This suggested that intratumor Ad-LIGHT treatment mainly promotes immune responses against unique tumor Ag(s) rather than shared ones, at least in Ag104Ld and 4T1, the two models we have observed for long term. Taken together, our data strongly suggest that targeting the primary tumor with Ad-LIGHT effectively breaks tolerance of T cells and promotes Ag-specific CTL to clear metastases or distal tumors.
4T1 mouse mammary carcinoma is a poorly immunogenic tumor model that shares many characteristics with human breast cancers, and is an established model for metastatic cancers (19, 24, 25). When 105 tumor cells are inoculated s.c., the tumor begins to metastasize to the DLN, lung, liver, and other organs only 10 days postinoculation. Mice succumb to lung metastases within 5–7 wk. Our study demonstrates that local treatment of 4T1 with Ad-LIGHT on day 14 can eradicate established metastatic cells in the peripheral tissues. With surgical excision of the primary tumor a week subsequent to dissemination of cancer cells, treatment with Ad-LIGHT becomes even more effective. Although the generation of CTL by intratumor treatment with Ad-LIGHT could not eradicate the primary tumor, it was sufficient to eradicate metastatic tumors, which is more difficult to manage by conventional treatments. In fact, immunotherapies have greater potential for treating micrometastases while conventional treatments using surgery and local radiation may be more effective in removing primary tumors. A combination of conventional treatment and immunotherapy is likely to be necessary for a cure of malignancies for most patients. It remains to be determined whether Ad-LIGHT in combination with other treatments can augment the immune response to completely eradicate more established metastases in the periphery.
Before our study, it had not been clearly demonstrated whether effector T cells that had infiltrated the tumor exit and enter the draining lymphoid organs for systemic circulation to eradicate distal tumors. This has been in part due to the lack of an appropriate model system to trace the path of tumor-specific CTL from the tumor to lymphoid structures and to distinguish the site of T cell activation due to the possible Ag cross-presentation in both compartments. To this end, we used a unique tumor model, the 2C-Ag104Ld system, to trace Ag-specific T cells inside the primary tumor, the DLN, and secondary tumor sites. The uniqueness of the model system is that the Ag Ld expressed by the tumor cells can only be seen by 2C T cells directly because the Ld processed and presented by the APCs from a H-2b host cannot be recognized by 2C T cells (14, 17). Because earlier kinetic and genetic studies have clearly demonstrated that T cell priming normally initiates in the lymphoid organ regardless of direct or cross-priming (29, 30), it is possible that tumor cells injected s.c. migrate to the lymphoid organ to prime T cells there via direct presentation. To exclude this possibility, we have also verified the lack of any Ag104Ld tumor cells inside DLN that could directly prime 2C T cells in the lymphoid tissues. Therefore, no naive 2C T cells are activated in the DLNs in this setting. Rather, 2C T cells must move into the tumor site for direct encounter with the Ld-expressing tumor cells for initial activation, after which they circulate back to the lymphoid tissues. In the presence of LIGHT inside the tumor, our study clearly shows that CTL are efficiently primed and subsequently circulate to infiltrate LIGHT-negative distal tumors. Without LIGHT expression in the primary tumor, few activated T cells were detected in DLNs or at a secondary tumor site. It is likely that these effector/memory T cells generated in the local tumor site in the presence of LIGHT are able to leave the tumor and patrol the periphery and target the metastatic tumor cells.
It has been shown that LIGHT can mediate tumor cell apoptosis via signaling through tumor-expressed HVEM and LTβR, leading to suppression of tumor growth in Rag1−/− mice (31, 32). Although in the presence of a large amount of IFN-γ, LTβR signaling may directly lead to tumor cell apoptosis in the absence of T cells (33), our data here indicate that CD8+ T cells may be essential for LIGHT-mediated rejection of distal tumors or metastases in the Ag104Ld and 4T1 tumor models. It will be interesting to investigate whether intratumor Ad-LIGHT induces LTβR or HVEM-expressing tumor cell apoptosis which also contributes to an enhanced immune response for rejecting distal or metastatic tumor cells.
Overall, our study has addressed a few key issues in treating metastatic tumors. The uniqueness of our model system allows us to clearly distinguish between tumor Ag-specific T cells that are activated in lymphoid tissues before tumor encounter, vs those activated directly within the tumor microenvironment. Our model system allows us to be among the very few who carefully examine the trafficking of tumor Ag-specific CD8+ T cells within the primary tumor after local Ad-LIGHT immunotherapy, and to demonstrate that the exiting CTL can circulate in the periphery while maintaining effector function. Furthermore, such CTL could accumulate in the distal tumor, leading to eradication of preexisting metastases. Therefore, this strategy has the potential for translation to clinical application toward the treatment of systemic metastases.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH) (AI062026, CA115540, and CA09296). P.Y. is a recipient of an NIH Training Grant (5T32DK07074) and Digestive Diseases Research Core Center Seeding Grant P30-DK42086.
Abbreviations used in this paper: LTβR, lymphotoxin β receptor; HVEM, herpes-virus entry mediatior; Ad-LIGHT, adenovirus-expressing LIGHT; mmLIGHT, mutant murine LIGHT; LN, lymph node; DLN, draining LN; NDLN, nondraining LN; SPL, spleen.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Fidler IJ. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 2.Koscielny S, Tubiana M, Le MG, Valleron AJ, Mouriesse H, Contesso G, Sarrazin D. Breast cancer: relationship between the size of the primary tumour and the probability of metastatic dissemination. Br J Cancer. 1984;49:709–715. doi: 10.1038/bjc.1984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koscielny S, Tubiana M, Valleron AJ. A simulation model of the natural history of human breast cancer. Br J Cancer. 1985;52:515–524. doi: 10.1038/bjc.1985.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber H. Tumor immunology. In: Paul WE, editor. Fundamental Immunology. Lippincott Raven Press; New York: 2003. pp. 1557–1592. [Google Scholar]
- 7.Rosenberg SA. The emergence of modern cancer immunotherapy. Ann Surg Oncol. 2005;12:344–346. doi: 10.1245/ASO.2005.01.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Foster A, Chin R, Yu P, Sun Y, Wang Y, Pfeffer K, Fu YX. The complementation of lymphotoxin deficiency with LIGHT, a newly discovered TNF family member, for the restoration of secondary lymphoid structure and function. Eur J Immunol. 2002;32:1969–1979. doi: 10.1002/1521-4141(200207)32:7<1969::AID-IMMU1969>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 11.Tamada K, Shimozaki K, Chapoval AI, Zhu G, Sica G, Flies D, Boone T, Hsu H, Fu YX, Nagata S, et al. Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nat Med. 2000;6:283–289. doi: 10.1038/73136. [DOI] [PubMed] [Google Scholar]
- 12.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Anders RA, Wang Y, Turner JR, Abraham C, Pfeffer K, Fu YX. The critical role of LIGHT in promoting intestinal inflammation and Crohn’s disease. J Immunol. 2005;174:8173–8182. doi: 10.4049/jimmunol.174.12.8173. [DOI] [PubMed] [Google Scholar]
- 14.Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, Fu YX. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. 2004;5:141–149. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- 15.Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, Butcher EC. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. 2005;6:895–901. doi: 10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 17.Wick M, Dubey P, Koeppen H, Siegel CT, Fields PE, Chen L, Bluestone JA, Schreiber H. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med. 1997;186:229–238. doi: 10.1084/jem.186.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 19.Miller FR, Miller BE, Heppner GH. Characterization of metastatic heterogeneity among subpopulations of a single mouse mammary tumor: heterogeneity in phenotypic stability. Invasion Metastasis. 1983;3:22–31. [PubMed] [Google Scholar]
- 20.Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W. Current Protocols in Immunology. John Wiley and Sons; 1991. pp. 20.2.1–20.2.11. [Google Scholar]
- 21.Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu YX. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779–791. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu P, Spiotto MT, Lee Y, Schreiber H, Fu YX. Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J Exp Med. 2003;197:985–995. doi: 10.1084/jem.20021804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003;171:27–31. doi: 10.4049/jimmunol.171.1.27. [DOI] [PubMed] [Google Scholar]
- 24.Parviz M, Chin CS, Graham LJ, Miller C, Lee C, George K, Bear HD. Successful adoptive immunotherapy with vaccine-sensitized T cells, despite no effect with vaccination alone in a weakly immunogenic tumor model. Cancer Immunol Immunother. 2003;52:739–750. doi: 10.1007/s00262-003-0405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998;58:1486–1493. [PubMed] [Google Scholar]
- 26.Kocak E, Lute K, Chang X, May KF, Jr, Exten KR, Zhang H, Abdessalam SF, Lehman AM, Jarjoura D, Zheng P, Liu Y. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 2006;66:7276–7284. doi: 10.1158/0008-5472.CAN-05-2128. [DOI] [PubMed] [Google Scholar]
- 27.Wilcox RA, Flies DB, Zhu G, Johnson AJ, Tamada K, Chapoval AI, Strome SE, Pease LR, Chen L. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest. 2002;109:651–659. doi: 10.1172/JCI14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Udaka K, Tsomides TJ, Eisen HN. A naturally occurring peptide recognized by alloreactive CD8+ cytotoxic T lymphocytes in association with a class I MHC protein. Cell. 1992;69:989–998. doi: 10.1016/0092-8674(92)90617-l. [DOI] [PubMed] [Google Scholar]
- 29.Bai XF, Gao JX, Liu J, Wen J, Zheng P, Liu Y. On the site and mode of antigen presentation for the initiation of clonal expansion of CD8 T cells specific for a natural tumor antigen. Cancer Res. 2001;61:6860–6867. [PubMed] [Google Scholar]
- 30.Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, Hemmi S, Hengartner H, Zinkernagel RM. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058–1064. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 31.Harrop JA, McDonnell PC, Brigham-Burke M, Lyn SD, Minton J, Tan KB, Dede K, Spampanato J, Silverman C, Hensley P, et al. Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J Biol Chem. 1998;273:27548–27556. doi: 10.1074/jbc.273.42.27548. [DOI] [PubMed] [Google Scholar]
- 32.Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, Jiang GW, Lu J, Tan J, Ugustus M, et al. LIGHT, a novel ligand for lymphotoxin β receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest. 1998;102:1142–1151. doi: 10.1172/JCI3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Browning JL, Miatkowski K, Sizing I, Griffiths D, Zafari M, Benjamin CD, Meier W, Mackay F. Signaling through the lymphotoxin β receptor induces the death of some adenocarcinoma tumor lines. J Exp Med. 1996;183:867–878. doi: 10.1084/jem.183.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]