

Abstract
3,3′-Diindolylmethane (DIM) is a chemopreventive and chemotherapeutic phytochemical derived from the metabolism of indoles found at high concentrations in cruciferous vegetables. We have previously shown that DIM exhibits anti-angiogenic properties in cultured vascular endothelial cells and in Matrigel plug assays in rodents. In the present study, we demonstrate that DIM reduces the level of hypoxia-inducible factor (HIF)-1α in hypoxic tumor cell lines, as well as HIF-1 transcriptional activity as measured by a reporter assay. Moreover, DIM inhibited the expression of HIF-1-responsive endogenous genes, resulting in the reduced expression of key hypoxia responsive factors, VEGF, furin, enolase-1, glucose transporter-1 and phosphofructokinase. DIM reduced the level of HIF-1α in hypoxic cells by increasing the rate of the prolylhydroxylase- and proteosome-mediated degradation of HIF-1α, and by decreasing the rate of HIF-1α transcription. Using enzyme kinetics studies, we established that DIM interacts with the oligomycin-binding site on the F1 transmembrane component of mitochondrial F1F0-ATPase. The contributions of the resulting increases in levels of ROS and O2 in hypoxic cells to the inhibitory effects of DIM on HIF-1α expression are discussed. These studies are the first to show that DIM can decrease the accumulation and activity of the key angiogenesis regulatory factor, HIF-1α, in hypoxic tumor cells.
Keywords: 3,3′-Diindolylmethane; angiogenesis; hypoxia-inducible factor; cancer; ATPase; prolylhydroxylase
1. INTRODUCTION
Phytochemicals derived from cruciferous vegetables, including indole-3-carbinol (I3C) and its condensation product, 3,3′-diindolylmethane (DIM), are under study as promising anticancer agents [1–3]. DIM has chemopreventive properties that have been attributed partly to its interaction with aryl hydrocarbon receptor (AhR) and induction of Phase II detoxifying enzymes [4–8]. DIM can also inhibit the proliferation of breast cancer cells in vitro by inducing cell cycle arrest and by promoting apoptosis in both estrogen-dependent (MCF-7) and estrogen-independent (MDA-MB-231) breast cancer cell lines [9–14]. Oral treatments with I3C and DIM significantly reduce the incidence of 7,12-dimethylbenz(a)anthracene (DMBA)-induced mammary tumors in female rats and benzo(a)pyrene (BP)-induced neoplasia of the forestomach in female mice [2,9]. Long-term treatment with these indoles also has been shown to inhibit diethylnitrosamine (DEN)-initiated hepatocarcinogenesis in an infant mouse model [1]. DIM also inhibits the growth of established human mammary tumors in a xenograft model in mice [15]. Moreover, I3C and DIM have become widely used adjunct therapies for recurrent respiratory papillomatosis (RRP), caused by certain types of human papillomaviruses (HPVs) [16,17]. Thus, DIM has the potential to be a useful therapeutic agent against tumors and neoplasia in several tissues.
We have recently proposed that inhibition of tumor angiogenesis may be among the mechanisms by which DIM suppresses tumor growth [15]. We showed that DIM suppresses markers of angiogenesis in model systems, including inhibition of proliferation, migration and tube formation of cultured human vascular endothelial cells, and suppression of vascularization of Matrigel plugs in athymic mice [15].
Tumor angiogenesis plays a central role in primary tumor growth and metastasis [18]. Growth of a tumor beyond 2–3 mm3 requires development of a microvessel network to facilitate delivery of nutrients and oxygen to the tumor. Density of microvasculature has been used as an indicator of biological aggressiveness and metastatic potential in many primary tumors because neovascularization facilitates metastasis by allowing access of cancer cells to the circulation [19–22]. The abilities of primary breast, prostate and colorectal carcinomas to metastasize to the lymph nodes have been directly correlated to the degree of angiogenesis within the primary tumors [21–24].
The development of hypoxic conditions at the core of tumors reaching a critical size of a few millimeters in diameter is considered to be the initial stimulus that triggers tumor angiogenesis [20]. The hypoxia-induced factor (HIF)-1α accumulates rapidly in tumor cells exposed to hypoxic conditions and heterodimerizes with HIF-1β/ARNT to form HIF-1. HIF-1 is a transcription factor that regulates the expression of over 60 genes, including genes that encode several angiogenic factors and enzymes involved in energy metabolism [25,26].
Previous studies in our laboratory showed that DIM induced a G1 cell cycle arrest in human breast cancer MCF-7 cells by a mechanism that includes increased expression of the cell cycle inhibitor p21 [11]. We observed subsequently that DIM is a strong mitochondrial F1F0-ATPase inhibitor that can induce hyperpolarization of mitochondrial inner membrane, decrease cellular ATP level, and stimulate mitochondrial reactive oxygen species (ROS) production [27]. DIM-induced ROS production leads to the activation of stress-activated MAPK pathways involving p38 and JNK and the induction of expression of p21. Coincubation of cells with antioxidant vitamins significantly attenuated DIM-induced activation of p38 and JNK, and induction of p21 expression, indicating that oxidative stress is the major trigger of these events.
Since several studies have shown that inhibitors of mitochondrial respiration can inhibit the accumulation of HIF-1α in hypoxic cells, we examined whether DIM might function to inhibit angiogenesis by this means, as well. Thus, we further defined the inhibitory activity of DIM on F1F0-ATPase activity and examined whether this inhibition is associated with increased levels of ROS and O2 in hypoxic tumor cells. In addition, we determined the effects of this indole on the levels of hypoxia-induced HIF-1α accumulation in cultured tumor cells and on the transcriptional regulation of a HIF-1-responsive reporter gene and of several endogenous genes. Our results show that DIM strongly inhibited HIF-1α accumulation and HIF-1 activity in hypoxic tumor cells by a mechanism that involves reactivation of prolylhydroxyase activity and redistribution of intracellular oxygen.
2. MATERIALS AND METHODS
2.1 Materials
Dulbecco’s modified Eagles’ medium (DMEM), Opti-MEM and lipofectamine were from Gibco/BRL (Grand Island, NY). Fetal bovine serum (FBS) was from Sigma Corp. (St. Louis, MO). MDA-MB-231 and HepG2 cells were from ATCC (Rockville, MD). Tri-reagent was from Molecular Research Center (Cincinnati, OH).
2.2. Cell Culture
The human breast tumor cell line, MDA-MB-231, and the human hepatoma HepG2 cell line were grown as adherent monolayers in DMEM, supplemented to 4.0 g/liter glucose, 3.7 g/L sodium bicarbonate and 10% FBS, in a humidified incubator at 37 °C and 5% CO2, and passaged at approximately 80% confluency. Cultures used in subsequent experiments were at less than 35 passages.
2.3. Western Blot Analysis
Cells were cultured in 6-well plates to near confluency, treated as indicated in the figure legends and placed in normoxic (20% O2) or hypoxic (1% O2) incubators for 4 hours. Cells were immediately harvested in 200 μL of loading buffer (10% glycerol, 5% 2-mercaptoethanol, 10% SDS, 0.125 M Tris-HCl pH 6.7, 0.15% bromophenol blue), sonicated for 12 seconds, heated to 99.9 °C for 5 min and fractionated by electrophoresis on 10% polyacrylamide, 0.1% SDS resolving gels. Proteins were electrically transferred to Immobilon-II membranes (Millipore, Billerica, MA), and blocked at room temperature for 1 hour with 5% non-fat dry milk in wash buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20). Blots were subsequently incubated with antibodies against human HIF-1α (BD Pharmigen, San Diego, CA). The antibody concentration was 0.25 μg/mL in blocking buffer. Immunoreactive proteins were detected after 1 h of incubation at room temperature with horseradish peroxidase-conjugated secondary antibodies. Goat anti-mouse antibodies were used as secondary antibodies (Bio-Rad) after being diluted 1:3000 in wash buffer. Blots were treated with ECL reagents (PerkinElmer Life Sciences), and chemoluminescence was detected using BioMax MR film (Kodak) or quantitated directly by densitometry using an AlphaImager HP imaging system (Alpha Innotech, San Leandro, CA). Equal protein loading was ascertained by Ponceau-S-staining of the membranes after transfer and by stripping the membranes and reprobing with antibodies against human β-tubulin.
2.4. Determination of mRNA by RT-PCR
Cells were lysed by addition of Tri-reagent (Molecular Research Center, Inc., Cincinnati, OH) and chloroform was used for phase separation. After centrifugation, the aqueous upper phase was collected and total RNA was precipitated by isopropanol, washed with 75% ethanol, and dissolved in DEPC treated water. Levels of specific mRNAs were determined by reverse transcription and polymerase chain reaction (RT-PCR) using Gibco enzymes and reagents. Reverse transcription was done using a 15-mer oligo-dT primer to generate cDNAs from all mRNAs. Levels of specific mRNA for the genes indicated in the Results section were determined by PCR using the specific 20-mer primer pairs below on a RoboCycler G96 (Stratagene, Cedar Creek, TX): Furin-f, 5′-TGTGGTGTAGGTGTGGCCTA-3′, Furin-r, 5′GCTGATGGACAGCGTGTAGA-3′. VEGF-f, 5′-GTCATTGGCAGAAAACCA-3′, VEGF-r, 5′AGTTTTGCCAA-TTCACACT-3′. Enolase-1-f, 5′-GCTCCGGGACAATGATAAGA-3′, Enolase-1-r, 5′-CGCCATTGATGACATTGAAC-3′. Phosphofructokinase-f, 5′-CAAAAGCTACCTG-GCGAAAG-3′, Phosphofructokinase-r, 5′-TAGTACACGATGCGGCTCTG-3′. Glut-1-f, 5′-ACAGGCAGCTGGATGAGACT-3′, Glut-1-r, 5′-CATAGCCACCTCCTGGGAT-A-3′. GAPDH-f, 5′-GAAGGTGAAGGTCGGAGTC-3′, GAPDH-r, 5′-GAAGATGGT-GATGGATTTC-3′.
All primers were designed to have an annealing temperature of 58 °C. After the following number of PCR cycles (94 °C, 45 sec, 58°C, 90 sec, 72 °C 60 sec) 20 for furin, enolase-1 and glut-1, 30 for VEGF and 18 for GAPDH, the PCR products were dyed with ethidium bromide and separated on a 15% agarose gel and photographed under UV light. For the purpose of illustration, the results are shown as an inverted image (black and white negative).
2.5. Reporter assay
The HIF-1 inducible luciferase reporter pH3SVL containing a total of six HIF-1 DNA-binding sites derived from the transferrin gene, was a gift from Dr. Katschinski (28). The reporter plasmid was transiently transfected in MDA-MB-231 cells by lipofection (Lipofectamine, Gibco) and after treatments as indicated in the Results section, luciferase activity in cell lysates was measured as described previously [29].
2.6. Oxygen availability assay
The mit-R-Luc-pCDNA3 expressing the mitochondrial targeting sequence of MnSOD fused to the N-terminus of the Renilla luciferase was a gift from Dr. Thilo Hagen (30). Transient transfection in MDA-MB-231 cells by lipofection was followed by a 48 hour recovery period to allow sufficient expression of the luciferase. The entire oxygen assay procedure was carried out in a hypoxic glove box filled with 1% O2/5% CO2 at 37 °C. Transfected cells were trypsinized and resuspended in medium that had been equilibrated with the hypoxic gas mixture for 2 hours, and aliquoted into luminometer cuvettes and allowed to equilibrate for 10 minutes. Treatments as indicated in the “Results” section were for 1 hour. Coelenterazine (5 μg/mL) was added and luminescence recorded for 10 seconds.
2.7. Measurement of ROS production in whole cells
Intracellular ROS were detected with the cell permeable fluorescent probe CM-H2DCFDA by flow cytometry as described previously (31). Cells were seeded into six-well culture plates at a density of 5 × 105/well, and 24 hours later, they were placed in the hypoxic chamber and the medium was replaced with medium that had been pre-equilibrated with the hypoxic gas mixture for 2 hours and treated with different concentrations of DIM or 1 mM DTT for 1 hour. An identical set of wells was kept in normoxic conditions for comparison. During the last 30 minutes of treatment, cells were dyed with 1 μg/mL CM-H2DCFDA for 30 minutes in the dark at 37 °C. Cells were then trypsinized and resuspended in hypoxic medium, transferred to flow cytometer tubes that were sealed to prevent reoxygenation and analyzed on a Beckman-Coulter EPICS XL flow cytometer.
2.8. PK/LDH–coupled ATPase assay
The F1F0-ATPase activity of sub-mitochondrial particles was measured spectrophotometrically at 340 nm by coupling the production of ADP to the oxidation of NADH via pyruvate kinase and lactate dehydrogenase described previously (36). The reaction mixture contained a final volume of 400 μL at 25 °C and included the following: Tris-HCl (pH 8), 2 mM ATP, 2 mM MgCl2, 50 mM KCl, 0.2 mM EDTA, 0.2 mM NADH, 1 mM PEP, 5 units pyruvate kinase, 8 units lactate dehydrogenase, and 25 to 50 μg mitochondrial proteins. These assay conditions minimized the contribution of other transport ATPases, such as Na,K-ATPase. Test compounds were added before the reaction was started by addition of 25 μg of sub-mitochondrial particles at 25 °C.
3. RESULTS
3.1. DIM inhibited hypoxia induced accumulation of HIF-1α in tumor-derived cells
Accumulation of HIF-1α in hypoxic cells is achieved by inhibition of its normally rapid degradation through proline hydroxylation, ubiquitination and proteasomal processing. We examined the effects of DIM on the levels of HIF-1α in two human tumor cell lines, MBA-MB-231 and HepG2. Cells were treated with a range of concentrations of DIM and after 4 hours of exposure to 1% O2, levels of HIF-1α in cell lysates were analyzed by Western blot. As shown in Figure 1A, the level of HIF-1α protein was highly elevated in hypoxic, compared to normoxic cells. Under normoxic conditions DIM did not affect the low level of HIF-1α expression. Under hypoxic conditions increasing concentrations of DIM caused a progressive decrease in HIF-1α expression in both cell lines. Concentrations of DIM as low as 1 μM were effective and complete inhibition of expression was found with 50 μM DIM in both cell lines. Inhibitors of prolyl hydroxylase, i.e. CPX, DPL and DMOG, acted as hypoxia mimics in normoxic conditions and reversed the inhibitory effect of DIM in hypoxic conditions (Figure 1B). The proteosome inhibitor MG132 caused a very strong increase of HIF-1α in normoxic conditions and cotreatment with DIM 50 μM prevented this accumulation, suggesting that, at high concentration, DIM might inhibit HIF-1α synthesis. In hypoxic conditions, MG132 partially countered the inhibitory effect of DIM, suggesting that proteasomic degradation contributes to the effects of DIM on the levels of HIF-1α. We found, also, that the antioxidants, DTT and NAC, did not reverse the effects of DIM on HIF-1α accumulation (Figure 1C).
Figure 1. A. DIM inhibits hypoxia-induced increased level of HIF-1α.

MDA-MB-231 and HepG2 cells grown to near confluent density in 6-well plates were treated with various concentrations of DIM in serum free medium and incubated in normoxic or hypoxic conditions for 4 hours. Levels of HIF-1α were measured by Western blot. β-Tubulin was used as a loading control. B. Effects of PHD inhibitors on the level of HIF-1α protein. MDA-MB-231 cells were treated with DIM and the iron chelators ciclopirox olamine (CPX) and 2–2′dipyridyl (DPL) and a metabolic prolyl hydroxylase inhibitor, dimethyloxaloylglycine (DMOG) and incubated in normoxic or hypoxic conditions for 4 hours. Levels of HIF-1αprotein were measured by Western blot. C. Effects of a proteosome inhibitor and of antioxidants on the level of HIF-1a protein. MDA-MB-231 cells were treated with DIM and the proteosome inhibitor, MG132 (20 mM), the PHD inhibitor 2-2′dipyridyl (DPL) (100 mM), and the antioxidants dithiothreitol (DTT) (1 mM) or N-acetyl-cysteine (NAC) (2 mM), and incubated in normoxic or hypoxic conditions for 4 hours. Levels of HIF-1α protein were measured by Western blot. Representative blots are shown. Experiments were reproduced at least three times with identical results.
3.2. DIM reduced the transcriptional activity of HIF-1
To determine whether the observed DIM-mediated decrease of HIF-1α protein level was reflected in a subsequent decrease in HIF-1 transcriptional activity, we measured luciferase expression in cells transfected with a HIF-1-responsive reporter construct. As shown in Figure 2, after 8 hours of exposure to hypoxia, there was an approximately 4.5-fold increase in HIF-1-responsive luciferase activity compared with normoxic cells. Increasing concentrations of DIM progressively decreased this activity to the same level as found in normoxic cells. Inhibitors of prolyl hydroxylase increased HIF-1 activity in normoxic cells and reversed the effects of DIM in hypoxic cells. DIM had no effect on the low level of HIF-1 activity in normoxic conditions. Cotreatment with the antioxidants, DTT or NAC, did not reverse the inhibitory effect of DIM in hypoxic cells.
Figure 2. Reporter Assay of HIF-1 transcriptional activity.

MDA-MB-231 cells grown to 60% confluency in 6-well plates were transfected with the HIF-1 reporter construct, pH3-SVL, using the lipofectamine transfection reagent. Cells were allowed to recover overnight, given the indicated concentrations of DIM and the PHD inhibitors 2-2′dipyridyl (DPL) and dimethyloxaloylglycine (DMOG), and the antioxidants dithiothreitol (DTT) or N-acetyl-cysteine (NAC) and incubated in normoxic or hypoxic conditions for 8 hours. Activity of luciferase in cell lysates was measured by chemiluminescence. Protein concentration in cell lysates was measured using the Bradford assay and results expressed as relative light units (RLU) per mg protein as the average +/− SD for three replicates.
3.3. DIM decreased the expression of endogenous HIF-1-responsive genes
We next examined the effect of DIM on expression of several HIF-responsive endogenous genes in human liver (HepG2) and breast tumor (MDA-MB-231) derived cells. RT-PCR analyses indicated that the levels of expression of VEGF, enolase-1 (Eno-1), glucose transporter-1 (GLUT-1), phosphofructokinase (PFK-1), and furin, in hypoxic cells were strongly reduced by DIM in concentration-dependent manners, whereas the lower levels of expression of these genes found in normoxic cells were not affected by DIM (Figure 3). Eno-1, GLUT-1 and PFK-1 are part of the glycolytic pathway. VEGF regulates vascular development in hypoxic tissues. Furin is a pro-protein convertase that mediates the proteolytic activation of a large number of polypeptides involved in the establishment of new blood vessels such as TGFβ1, PDGF, IGF-2 and.IGF-1 receptor. Although the data indicate a maximum and very strong inhibition of expression of the HIF-1-responsive genes at 25 μM DIM in both cell lines, pronounced inhibition was seen for expression of furin, Eno-1 and GLUT-1 following exposure to only 1 μM DIM.
Figure 3. DIM inhibits hypoxia-induced increased transcription of endogenous HIF-1-responsive genes.

MDA-MB-231 (A) and HepG2 (B) cells grown to near confluent density in 6-well plates were given the indicated concentrations of DIM in serum free medium and incubated in normoxic or hypoxic conditions for 24 hours. Total RNA was isolated and levels of mRNA of HIF-1 responsive genes were measured by semi-quantitative RT-PCR. Representative blots are shown. Experiments were reproduced at least three times with identical results.
3.4. Antioxidants quench DIM-induced ROS in hypoxic cells
Our previous studies have shown that DIM induces the release of ROS in cultured tumor cells and in primary murine macrophage cultures. Since some studies have reported that antioxidants can oblate HIF-1α accumulation [32] and DIM is a chemical antioxidant [33], we examined the effects of DIM on ROS production in hypoxic cells. The intracellular levels of ROS were quantified by flow cytometry using the cell permeable fluorescent probe CM-H2DCFDA. Hypoxic cells had a basal level of ROS approximately twice that of normoxic cells and treatment with DIM caused a five-fold increase in ROS levels, whereas in normoxic cells DIM caused a smaller increase. Treatment with the antioxidant DTT countered the effect of hypoxia on ROS production and in cotreatments with DIM, DTT also decreased the levels of ROS substantially. (Figure 4). These results indicate that DIM is a potent pro-oxidant in hypoxic cells and that the standard antioxidant, DTT, can efficiently quench the ROS.
Figure 4. The pro-oxidant effects of DIM in hypoxic cells are quenched by DTT.
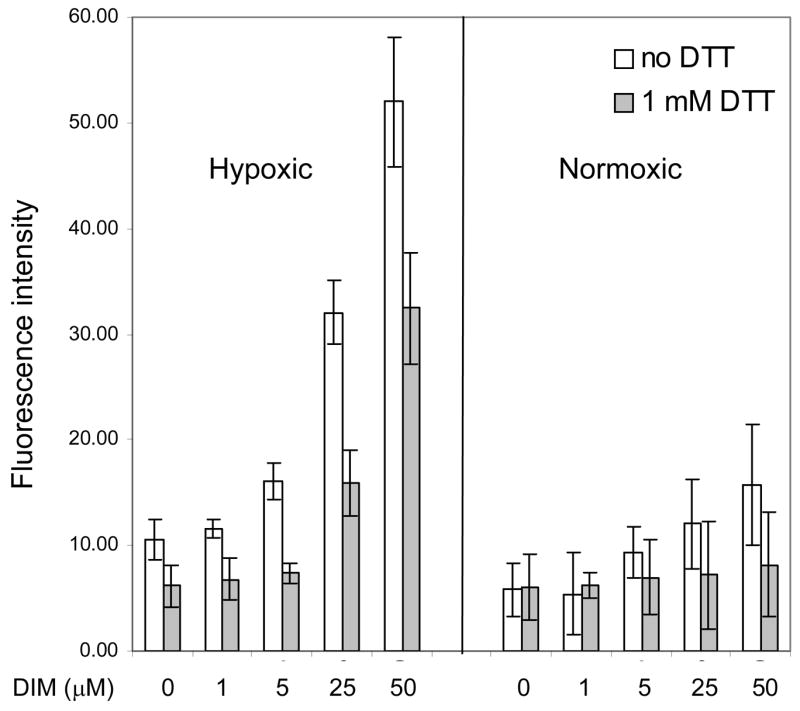
Intracellular ROS were detected with the cell permeable fluorescent probe CM-H2DCFDA by flow cytometry as described in the Methods section. Cells were treated with the indicated range of concentrations of DIM and co-treated with DTT. Results shown are the average +/− SD of at least three replicates.
Taken together, these results indicate that DIM can strongly inhibit the accumulation and transcriptional activity of HIF-1α by a mechanism that involves the activation of prolyl hydroxylase and appears to be independent of DIM-induced release of ROS in tumor cells.
3.5. DIM affects the rates of synthesis and degradation of HIF-1α
The effect of DIM on the steady state levels of HIF-1α in hypoxic conditions shown in Figure 1A could result from a reduced rate of synthesis, an increased rate of degradation or a combination of both and therefore, we measured these two components separately. First, the rate of synthesis was determined by measuring accumulation of HIF-1α protein over a period of 60 min following inhibiton of degradation by the proteosome inhibitor MG132 in normoxic conditions (Figure 5A). With complete inhibition of degradation, the protein accumulation correlates directly with the rate of synthesis Relative abundance of HIF-1α protein was quantified by densitometric analysis of the Western blots by measuring the chemiluminescence directly with a digital imager. The rapid accumulation of HIF-1α protein in cells treated with DMSO as the control, from none at time 0 to 140 density units at 60 min, was not significantly affected by DIM at 10 and 25 μM, but higher concentrations strongly reduced the rate of synthesis, with 50 μM DIM having an effect similar to that of the cycloheximide (CHX) control.
Figure 5. Rates of synthesis and degradation of HIF-1α.

A, Rate of synthesis was ascertained from the accumulation of HIF-1α protein over time measured by Western blots, following inhibiton of degradation by the proteasome inhibitor, MG132 in normoxic conditions. Relative abundance of HIF-1α protein was quantified by densitometric analysis of the Western blots. The intensity of the chemiluminescence was measured by a digital imaging system and expressed as relative density units. The protein synthesis inhibitor, cycloheximide (CHX) was used as control. B, Degradation rates were determined by the reduction of HIF-1α protein level at 30 minutes after addition of CHX to cells previously placed in hypoxic conditions for 4 hours. For each concentration of DIM, a control treatment without CHX was used to account for the change in steady state caused by the effect of DIM on the rate of synthesis. C, The relative rate of degradation is calculated as the ratio of the level of HIF-1α protein without CHX over that with CHX for each concentration of DIM and expressed relative to the rate for the DMSO control. The rate of synthesis as measured by the accumulation of HIF-1α at 60 minutes after proteosome inhibition is also shown, relative to that of the DMSO control. Results shown are the average +/− SD of at least three replicates.
The relative rate of degradation of HIF-1α was subsequently determined by the decrease of HIF-1α protein level at 30 minutes after addition of CHX to cells previously placed in hypoxic conditions for 4 hours (Figure 5B). For each concentration of DIM a control treatment without CHX was used to account for the change in steady state caused by the effect of DIM on the rate of synthesis. The relative rate of degradation was calculated as the ratio of the level of HIF-1α protein without CHX over that with CHX for each concentration of DIM and expressed relative to the rate for the DMSO control (Figure 5C). The rate of synthesis as measured by the accumulation of HIF-1α at 60 minutes after proteosome inhibition is also included in Figure 5C relative to that of the DMSO control. Taken together, the results show that at the lower concentrations of 10 and 25 μM, the effect of DIM on the steady state levels of HIF-1α resulted primarily from an increase in degradation, whereas the higher concentrations of 35 and 50 μM DIM strongly reduced synthesis without further increasing degradation.
3.6. DIM affects oxygen availability in hypoxic cells
Inhibitors of mitochondrial respiration such as nitric oxide (NO), myxothiazol and sodium azide, have been reported to increase intracellular O2 availability in hypoxic cells. To examine whether DIM functons in this manner to reactivate prolyl hydroxylases, we employed a Renilla luciferase reporter that is solely dependent on available O2 to oxidize its substrate, coelenterazine. Cells were transfected with an expression vector encoding Renilla luciferase fused to the mitochondrial targeting sequence of MnSOD. Two days after transfection, cells were trypsinized and resuspended in serum-free DMEM that had been equilibrated with 1% O2 for two hours. Transfected cells were treated under hypoxic conditions with DIM and the NO donor, NOC12 (3-(2-hydroxy-1-(1-methylethyl)-2-nitrosohydrazino)-1-propanamine), as the positive control, and subjected to the luciferase activity assay as described in the Methods and Materials section. Samples were then allowed to re-equilibrate to 21% O2 for 10 minutes and luminescence was measured again. The results shown in Figure 6 indicate that oxygen availability increases gradually with DIM concentration to reach 2.3-fold over control at 50 μM DIM. The NO donor positive control produced a 2.8-fold change.
Figure 6. Effect of DIM on intracellular oxygen availability.
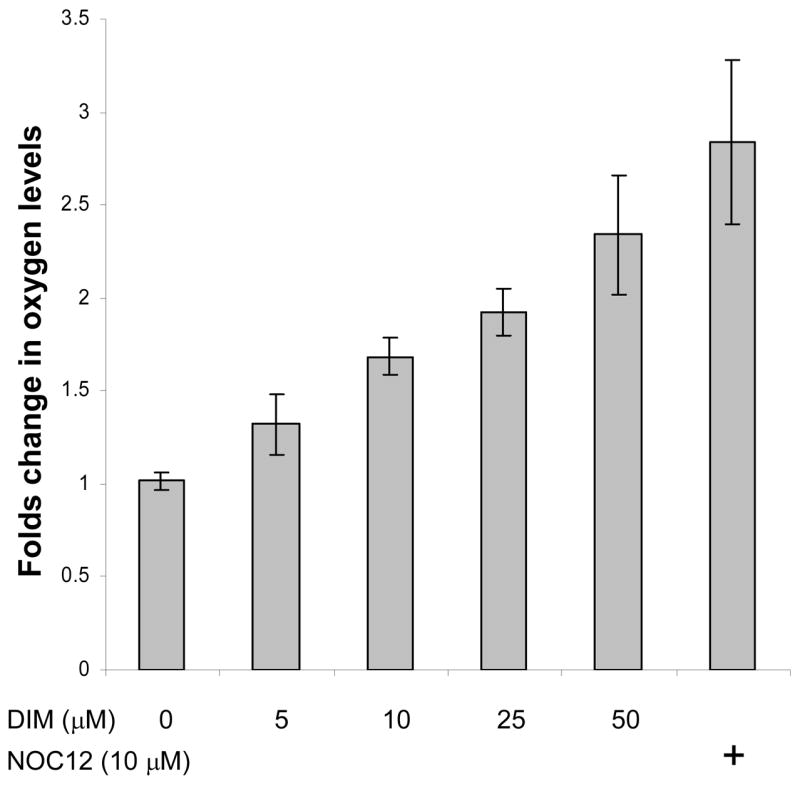
Two days after transfection with the expression vector encoding Renilla luciferase, cells were trypsinized and resuspended in serum-free DMEM that had been pre-equilibrated with 1% O2 for two hours. The treatments were with DIM and the NO donor, NOC12, as the positive control, and the luciferase activity assays were all done in an hypoxic glove box filled with 1% O2/5% CO2 at 37 °C. Results shown are the average +/− SD of at least three replicates.
3.7. DIM is a non-competitive inhibitor of mitochondrial F1F0-ATPase
We have reported previously that DIM can inhibit mitochondrial F1F0-ATPase activity, and thereby inhibit cellular respiration. To extend these studies we compared the binding kinetics of DIM with certain established ATPase inhibitors in an effort to more precisely define the binding site of DIM on this important enzyme. ATPase activities were measured in vitro using sub-mitochondrial particles prepared from mouse liver. The results presented as double inverse plots (1/V versus I/S) in Figure 7A show that DIM is a non-competitive inhibitor of ATP synthase, i.e. DIM caused a reduction in velocity but no change in affinity for the ATP substrate. The Dixon plot in Figure 7B (1/V versus [I]) gives a Ki value of 25 μM for DIM. The combined inhibition kinetics of DIM with oligomycin (Figure 7C) or with resveratrol (Figure 7D) indicate that DIM binds to the same site on the transmembrane proton channel (F0) as oligomycin (parallel lines), but does not bind to the same site as resveratrol (convergent lines) on the catalytic domain (F1). Also, DIM did not inhibit activity of solubilized F1- ATPase (data not shown), which provides further evidence that the indole did not interact with the catalytic domain.
Figure 7. Kinetics of inhibition of F1F0-ATPase activity by DIM.

A, Double inverse plot (1/V versus I/S). B, Dixon plot (1/V versus [I]) gives a Ki value of 25 μM for DIM. C and D, Combined inhibition kinetics of DIM with oligomycin or with resveratrol.
4. DISCUSSION
We established that DIM inhibited HIF-1α accumulation and HIF-1 activity in hypoxic cells and that the inhibitors of prolylhydroxylase, CPX, DPL and DMOG, and of the proteasome, MG132, could reverse this inhibition. DIM inhibited the expression of major HIF-1-regulated endogenous genes, most notably the pro-angiogenic factors, furin and VEGF, in a concentration-dependent manner in hypoxic tumor cells. We confirmed that DIM is a strong inhibitor of mitochondrial F1F0-ATPase and determined that DIM binds to the oligomycin-binding site in the transmembrane F0 domain. This inhibitory effect on respiration was accompanied by an increase in intracellular levels of O2 and very strong increases in ROS production with exposure to the higher concentrations of DIM. Although the ROS were efficiently quenched by co-treatment with antioxidants, including DTT, as well as NAC, ascorbate, GSH, vitamin E and vitamin C (data not shown), the antioxidant treatments did not reverse the effects of DIM on HIF-1α accumulation or HIF-1 activities. In addition, inhibitors of the stress kinases, PI3K and p38, as well as catalase over-expression (data not shown), did not counter the effects of DIM. These results suggest that the effects of DIM, especially on HIF-1α degradation, are not likely to result from ROS release and related oxidative stress induced signaling.
The more likely mechanism for the lower concentrations of DIM is the reactivation of the prolyl hydroxylation degradation pathway as a result of an increase in levels of cellular O2. Indeed, the inhibitory effects of DIM were accompanied by an approximately 2-fold increase in O2 levels in hypoxic cells. Furthermore, previous studies have shown that several established inhibitors of mitochondrial electron transport, including myxothiazol, sodium azide, antimycin A, rotenone, nitric oxide and oligomycin, all can inhibit HIF-1α accumulation in hypoxic cells and that this effect is not countered by co-treatments with any of several antioxidants [30,31]. These published studies further showed that cotreatments with prolyl hydroxylase inhibitors (DMO and DFO) or the proteasome inhibitor (MG-132) largely reversed the stabilizing effects of the respiratory inhibitors on HIF-1α levels. Thus, the results we present for DIM are consistent with these previous reports on the effects of other respiration inhibitors and provide strong support for the hypothesis that the inhibitory effects of the lower concentrations of DIM on F1F0-ATPase with resultant accumulation of cellular O2 are responsible for the reactivation of the prolyl hydroxylase-dependent degradation pathway of HIF-1α.
Our results show further that exposure of cells to the higher concentrations of DIM (>25 μM) results in a near total oblation of HIF-1α expression. In this concentration range, the rate of HIF-1α synthesis continues to decline, while the rate of HIF-1α degradation shows no further increase, (c.f. Figure 5.c.) Although the mechanism of this aspect of DIM activity is yet to be fully understood, this effect is consistent with reports of activities of other inhibitors of mitochondrial respiration. Indeed, studies with mitochondrial thioredoxin (Trx2) have shown that over expression of this protein can inhibit translation of HIF-1α by a mechanism that involves the attenuation of activities of Akt, p70S6K and eIF-4E, which together mediate HIF-1α translation [34] It is suggested that the observed down-regulation of Akt signaling is a result of ROS production arising from a disruption of mitochondrial membrane potential by over-expressed Trx2. We have shown that DIM can produce an increase in mitochondrial membrane protential as reported for Trx2 [27]. In addition, we and others have observed that DIM can inhibit Akt signaling as does Trx2 over expression, which is consistent with similar modes of action of high concentrations of DIM and Trx2 on HIF-1α translation [35]. Although our results indicated a strong increase in ROS production along with a strong decrease in HIF-1α synthesis with the higher concentrations of DIM, the specific role of ROS in this inhibition is yet to be determined. Although we observed no effect of soluble antioxidants on HIF-1α oblation by high concentrations of DIM, the possibility remains that the effects might arise from a compartmentalized production of ROS in mitochondria that might not be quenched by the soluble antioxidants, as was suggested for the Trx2 studies [34].
The oxygenation effect of DIM may have consequences for tumor therapy beyond those resulting from a decrease in HIF-1α accumulation in hypoxic tumor tissue. Oxygenation of tumors ranges from 30% to less than 10% the oxygenation level of normal tissues [36]. A major result of these low oxygen levels is a resistance of tumor tissue to radiation therapy [37]. Although there is considerable interest in developing means of increasing tumor sensitivity to radiation, only a few substances are reported to be successful in this regard. Recent studies have shown that one such substance, nelfinavir, exhibits antiangiogenic activity by a mechanism that involves inhibition Akt signaling and VEGF expression, and down-regulation of HIF-1α expression in hypoxic tumor cells [36]. In addition, this drug was shown to increase oxygenation of xenograft tumor tissue and increase the sensitivity of the tumors to radiation. In light of the similarities in the effects of DIM and nelfinavir on Akt and HIF-1 signaling and angiogenesis, it is reasonable to suggest that DIM could increase tumor oxygenation and sensitivity to radiation, as well.
HIF-1 has become an attractive target for the development of anti-cancer drugs [38]. Studies in many laboratories have shown that potential therapeutic agents can disrupt the HIF-1 signaling pathway through a variety of mechanisms, including the inhibition of HIF-1α protein synthesis, nuclear translocation, HIF-1 transactivation of target genes, and stabilization [39]. The inhibitory effects of DIM on mitochondrial respiration through inhibition of F1F0-ATPase appear to be primarily responsible for the increase in HIF-1α degradation and decrease in HIF-1α transcription that we observed. It is of interest to note that a group of cancer protective phytochemicals, including resveratrol, have been shown to both down-regulate HIF-1α in hypoxic cells and to inhibit mitochondrial F1F0-ATPase [40–42]. Thus, F1F0-ATPase and other components of the mitochondrial respiratory chain, appear to be important molecular targets for a relatively large group of potentially useful antiangiogenic agents from plants.
Acknowledgments
Financial support provided by Department of Defense, Army Breast Cancer Research Program Grant DAMDI 7-96-1-6159 and grants CA69056 and CA102360 from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wattenberg LW, Loub WD. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer research. 1978;38(5):1410–3. [PubMed] [Google Scholar]
- 2.Grubbs CJ, Steele VE, Casebolt T, Juliana MM, Eto I, Whitaker LM, Dragnev KH, Kelloff GJ, Lubet RL. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer research. 1995;15(3):709–16. [PubMed] [Google Scholar]
- 3.Latxague L, Gardrat C, Coustille JL, Viaud MC, Rollin P. Identification of enzymatic degradation products from synthesized glucobrassicin by gas chromatography-mass spectrometry. Journal of Chromatography. 1991;586(1):166–70. [Google Scholar]
- 4.Wortelboer HM, de Kruif CA, van Iersel AA, Falke HE, Noordhoek J, Blaauboer BJ. Acid reaction products of indole-3-carbinol and their effects on cytochrome P450 and phase II enzymes in rat and monkey hepatocytes. Biochemical pharmacology. 1992;43(7):1439–47. doi: 10.1016/0006-2952(92)90200-3. [DOI] [PubMed] [Google Scholar]
- 5.Bradfield CA, Bjeldanes LF. Dietary modification of xenobiotic metabolism contribution of indolylic compounds present in brassica-oleracea. Journal of Agricultural & Food Chemistry. 1987;35(6):896–900. [Google Scholar]
- 6.Bradfield CA, Bjeldanes LF. Structure-activity relationships of dietary indoles: a proposed mechanism of action as modifiers of xenobiotic metabolism. Journal of toxicology and environmental health. 1987;21(3):311–23. doi: 10.1080/15287398709531021. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi N, Dashwood RH, Bjeldanes LF, Bailey GS, Williams DE. Regulation of hepatic cytochrome P4501A by indole-3-carbinol: transient induction with continuous feeding in rainbow trout. Food Chem Toxicol. 1995;33(2):111–20. doi: 10.1016/0278-6915(94)00117-7. [DOI] [PubMed] [Google Scholar]
- 8.Chen I, Safe S, Bjeldanes L. Indole-3-carbinol and diindolylmethane as aryl hydrocarbon (Ah) receptor agonists and antagonists in T47D human breast cancer cells. Biochemical pharmacology. 1996;51(8):1069–76. doi: 10.1016/0006-2952(96)00060-3. [DOI] [PubMed] [Google Scholar]
- 9.Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19(9):1631–9. doi: 10.1093/carcin/19.9.1631. [DOI] [PubMed] [Google Scholar]
- 10.Hong C, Kim HA, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane (DIM) induces a G(1) cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21(WAF1/CIP1) expression. Carcinogenesis. 2002;23(8):1297–305. doi: 10.1093/carcin/23.8.1297. [DOI] [PubMed] [Google Scholar]
- 11.Hong C, Firestone GL, Bjeldanes LF. Bcl-2 family-mediated apoptotic effects of 3,3′-diindolylmethane (DIM) in human breast cancer cells. Biochemical pharmacology. 2002;63(6):1085–97. doi: 10.1016/s0006-2952(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 12.Firestone GL, Bjeldanes LF. Indole-3-carbinol and 3-3′-diindolylmethane antiproliferative signaling pathways control cell-cycle gene transcription in human breast cancer cells by regulating promoter-Sp1 transcription factor interactions. The Journal of nutrition. 2003;133(7 Suppl):2448S–55S. doi: 10.1093/jn/133.7.2448S. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Hsu BAJ, Kinseth BAM, Bjeldanes LF, Firestone GL. Indole-3-carbinol induces a G1 cell cycle arrest and inhibits prostate-specific antigen production in human LNCaP prostate carcinoma cells. Cancer. 2003;98(11):2511–20. doi: 10.1002/cncr.11844. [DOI] [PubMed] [Google Scholar]
- 14.Ge X, Fares FA, Yannai S. Induction of apoptosis in MCF-7 cells by indole-3-carbinol is independent of p53 and bax. Anticancer research. 1999;19(4B):3199–203. [PubMed] [Google Scholar]
- 15.Chang X, Tou JC, Hong C, Kim HA, Riby JE, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane inhibits angiogenesis and the growth of transplantable human breast carcinoma in athymic mice. Carcinogenesis. 2005;26(4):771–8. doi: 10.1093/carcin/bgi018. [DOI] [PubMed] [Google Scholar]
- 16.Auborn KJ. Therapy for recurrent respiratory papillomatosis. Antivir Ther. 2002;7:1–9. [PubMed] [Google Scholar]
- 17.Wiatrak BJ. Overview of recurrent respiratory papillomatosis. Curr Opin Otolaryngol Head Neck Surg. 2003;11:433–441. doi: 10.1097/00020840-200312000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Folkman J. Tumor angiogenesis: therapeutic implications. The New England journal of medicine. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 19.Liotta LA, Kleinerman J, Saidel GM. Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer research. 1974;34(5):997–1004. [PubMed] [Google Scholar]
- 20.Folkman J, Shing Y. Angiogenesis. The Journal of biological chemistry. 1992;267(16):10931–4. [PubMed] [Google Scholar]
- 21.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. The American journal of pathology. 1993;143(2):401–9. [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi Y, Bucana CD, Liu W, Yoneda J, Kitadai Y, Cleary KR, Ellis LM. Platelet-derived endothelial cell growth factor in human colon cancer angiogenesis: role of infiltrating cells. Journal of the National Cancer Institute. 1996;88(16):1146–51. doi: 10.1093/jnci/88.16.1146. [DOI] [PubMed] [Google Scholar]
- 23.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. The New England journal of medicine. 1991;324(1):1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer research. 1995;55(18):3964–8. [PubMed] [Google Scholar]
- 25.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annual review of cell and developmental biology. 1999;15:551–78. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochemical pharmacology. 2000;59(1):47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- 27.Gong Y, Sohn H, Xue L, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane is a novel mitochondrial H(+)-ATP synthase inhibitor that can induce p21(Cip1/Waf1) expression by induction of oxidative stress in human breast cancer cells. Cancer research. 2006;66(9):4880–7. doi: 10.1158/0008-5472.CAN-05-4162. [DOI] [PubMed] [Google Scholar]
- 28.Ibrahim NO, Hahn T, Franke C, Stiehl DP, Wirthner R, Wenger RH, Katschinski DM. Induction of the hypoxia-inducible factor system by low levels of heat shock protein 90 inhibitors. Cancer research. 2005;65(23):11094–100. doi: 10.1158/0008-5472.CAN-05-1877. [DOI] [PubMed] [Google Scholar]
- 29.Riby JE, Chang GH, Firestone GL, Bjeldanes LF. Ligand-independent activation of estrogen receptor function by 3, 3′-diindolylmethane in human breast cancer cells. Biochemical pharmacology. 2000;60(2):167–77. doi: 10.1016/s0006-2952(00)00307-5. [DOI] [PubMed] [Google Scholar]
- 30.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302(5652):1975–8. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 31.Gong Y, Agani FH. Oligomycin inhibits HIF-1alpha expression in hypoxic tumor cells. American journal of physiology. 2005;288(5):C1023–9. doi: 10.1152/ajpcell.00443.2004. [DOI] [PubMed] [Google Scholar]
- 32.Kohl R, Zhou J, Brune B. Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1alpha stabilization. Free Radic Biol Med. 2006 Apr 15;40(8):1430–42. doi: 10.1016/j.freeradbiomed.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Benabadji SH, Wen R, Zheng JB, Dong XC, Yuan SG. Anticarcinogenic and antioxidant activity of diindolylmethane derivatives. Acta Pharmacol Sin. 2004;25:666–71. [PubMed] [Google Scholar]
- 34.Zhou J, Damdimopoulos AE, Spyrou G, Brune B. Thioredoxin 1 and thioredoxin 2 have opposed regulatory functions on hypoxia-inducible factor-1alpha. J Biol Chem. 2007 Mar 9;282(10):7482–90. doi: 10.1074/jbc.M608289200. [DOI] [PubMed] [Google Scholar]
- 35.Rahman KW, Sarkar FH. Inhibition of nuclear translocation of nuclear factor-{kappa}B contributes to 3,3′-diindolylmethane-induced apoptosis in breast cancer cells. Cancer Res. 2005 Jan 1;65(1):364–71. [PubMed] [Google Scholar]
- 36.Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM, Koch CJ, Hahn SM, Maity A. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. 2006 Sep 15;66(18):9252–9. doi: 10.1158/0008-5472.CAN-06-1239. [DOI] [PubMed] [Google Scholar]
- 37.Gatenby RA, Kessler HB, Rosenblum JS, Coia LR, Moldofsky PJ, Hartz WH, Broder GJ. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int J Radiat Oncol Biol Phys. 1988 May;14(5):831–8. doi: 10.1016/0360-3016(88)90002-8. [DOI] [PubMed] [Google Scholar]
- 38.Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev. 2007 Jun;26(2):341–52. doi: 10.1007/s10555-007-9059-x. [DOI] [PubMed] [Google Scholar]
- 39.Belozerov VE, Van Meir EG. Hypoxia inducible factor-1: a novel target for cancer therapy. Anti-cancer drugs. 2005;16(9):901–9. doi: 10.1097/01.cad.0000180116.85912.69. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Q, Tang X, Lu QY, Zhang ZF, Brown J, Le AD. Resveratrol inhibits hypoxia-induced accumulation of hypoxia-inducible factor-1alpha and VEGF expression in human tongue squamous cell carcinoma and hepatoma cells. Molecular cancer therapeutics. 2005;4(10):1465–74. doi: 10.1158/1535-7163.MCT-05-0198. [DOI] [PubMed] [Google Scholar]
- 41.Wang B, Li H, Yan H, Xiao JG. Genistein inhibited hypoxia-inducible factor-1alpha expression induced by hypoxia and cobalt chloride in human retinal pigment epithelium cells. Methods and findings in experimental and clinical pharmacology. 2005;27(3):179–84. doi: 10.1358/mf.2005.27.3.890875. [DOI] [PubMed] [Google Scholar]
- 42.Zheng J, Ramirez VD. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. British journal of pharmacology. 2000;130(5):1115–23. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]