

Abstract
Connective-tissue growth factor (CTGF) is a secreted protein implicated in multiple cellular events including angiogenesis, skeletogenesis and wound healing1. It is a member of the CCN family of secreted proteins, named after CTGF, cysteine-rich 61 (CYR61), and nephroblastoma overexpressed (NOV) proteins. The molecular mechanism by which CTGF or other CCN proteins regulate cell signalling is not known. CTGF contains a cysteine-rich domain (CR) similar to those found in chordin and other secreted proteins2, which in some cases have been reported to function as bone morphogenetic protein (BMP) and TGF-β binding domains3-6. Here we show that CTGF directly binds BMP4 and TGF-β1 through its CR domain. CTGF can antagonize BMP4 activity by preventing its binding to BMP receptors and has the opposite effect, enhancement of receptor binding, on TGF-β1. These results show that CTGF inhibits BMP and activates TGF-β signals by direct binding in the extracellular space.
By sequence comparison to Chordin we noticed that CTGF contains a CR module previously designated as von Willebrand type c domain7 (vwc). CTGF contains four distinct structural modules: an amino-terminal insulin-like growth-factor-binding domain (IGFB), followed by the CR/vwc domain, a thrombospondin type 1 repeat (TSP-1), and a carboxy-terminal cystine knot (CT) domain8 (Fig. 1a). The same modular architecture is shared by the other CCN family members. A comparison of the CR domain of CTGF with the CR domains of von Willebrand factor (vWF), thrombospondin (TSP), procollagens I and II and chordin showed conservation of ten regularly spaced cysteines and a few additional amino-acid residues characteristic of CR domains2 (Fig. 1b). Here we present functional studies on Xenopus laevis CTGF, a gene that is expressed in the floor plate, notochord, somites and other tissues known to be involved in early embryonic patterning (see Fig. S1 in the Supplementary Information).
Figure 1. CTGF encodes a CR-containing protein.

a, The connective tissue growth factor (CTGF) protein contains four structural modules following the signal peptide (SP): insulin-like growth-factor binding (IGFB) domain, chordin-like cysteinerich (CR) domain, thrombospondin type 1 repeat (TSP-1), and a C-terminal cystineknot (CT). b, Alignment of the CR domains of CTGF, von Willebrand Factor, thrombospondin, procollagen 1 and 2 and Chordin (GenBank accession numbers XP037056, NM000552, P35448, NP001835, P02452 and AAC42222, respectively). Black boxes represent identical amino acids and gray boxes similar amino acids; dashes indicate gaps introduced to optimize the alignment. Asterisks mark the ten identical cysteine residues characteristic of CR domains.
Synthetic CTGF mRNA injected into a single ventral blastomere at the four-cell stage was able to induce secondary axes9,10 in Xenopus embryos (66%, n = 115, Fig. 2b), which is consistent with inhibition of BMP signalling. The CTGF-induced axes were partial and never formed head structures with eyes, but included somites marked by the muscle-specific marker antibody 12-101 (data not shown). A construct encoding a secreted form of only the CR domain of CTGF (CTGF-CR, Fig. S2 in the Supplementary Information) was sufficient to induce ectopic axes, although at a lower frequency (30%, n = 78, Fig. 2c). Conversely, mRNA encoding CTGF lacking the CR domain (CTGF-ΔCR, Fig. S2) failed to induce ectopic axes (0%, n = 50, Fig. 2d). Reverse transcriptionpolymerase chain reaction (RT-PCR) analysis of ectodermal explants showed that microinjected CTGF mRNA strongly induced the anterior marker Otx-2 and the cement-gland marker XAG-1(Fig. 2e). In addition, CTGF caused a weaker induction of the panneural marker N-CAM and downregulation of the epidermal markers Msx-1 and Cytokeratin (Fig. 2e). Microinjection of CTGF mRNA into the animal pole resulted in embryos with enlarged heads, shortened trunk and tail structures (Fig. 2f, g), and expanded expression of the anterior cement-gland marker XAG-1 (Fig. 2h, i). In CTGF-injected embryos analysed at the neurula stage, the expression domain of the neural-plate marker Sox-2 was expanded (Fig. j, k) and, reciprocally, the expression domain of Msx-1, which demarcates the border between the neural plate and epidermis, was displaced laterally (Fig. 2m, n). Embryos injected with CTGF-CR also developed with expanded neural plates (Fig. 2l, o). We conclude from these in vivo results that CTGF can induce anti-BMP phenotypes in microinjected Xenopus embryos through its CR domain.
Figure 2. CTGF mRNA injections induce anti-BMP phenotypes in Xenopusembryos.
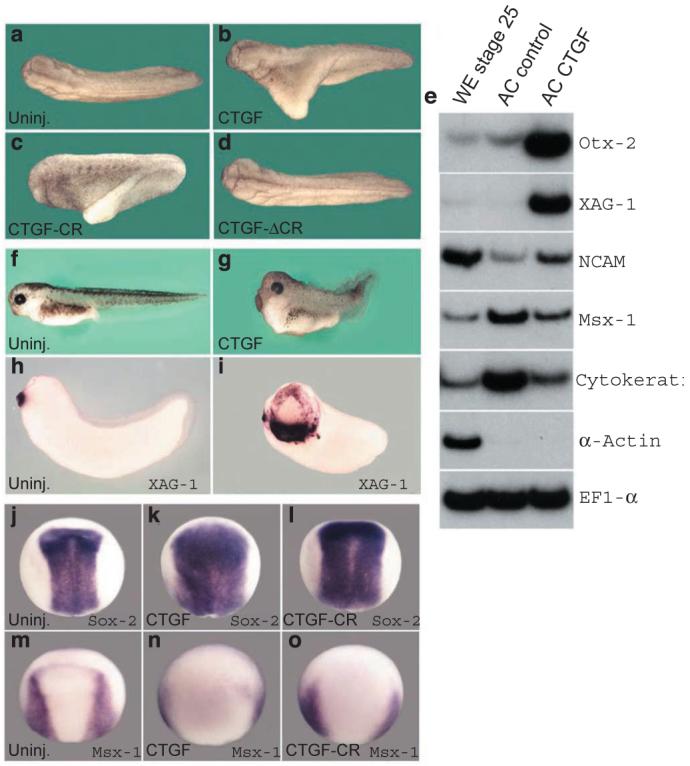
a-d, Dorsal views of tail-bud stage embryos. a, Uninjected; b, CTGF mRNA (150 pg); c, CTGF-CR mRNA (800 pg) and d, CTGF-ΔCR mRNA (500 pg) injected ventrally at 4-cell stage. e, RT-PCR analysis of whole embryo (WE), animal caps uninjected (AC control) or injected with 50 pg of CTGF mRNA per blastomere at the 4-cell stage (AC CTGF). Animal cap explants were dissected at stage 9 and cultured until sibling embryos reached stage 26. EF1-α was used as RNA loading control and α-Actin to monitor mesoderm in the ectodermal explants. Three-day tadpoles, f, uninjected, g, injected with 50 pg of CTGF mRNA into the animal region of each blastomere at 4-cell stage. Cement-gland marker XAG-1, h, uninjected, i, injected with 150 pg of CTGF mRNA per blastomere at 4-cell stage. j-o, in situ hybridization at neural plate stage for Sox-2 (j-l) and Msx-1 (m-o); j, m, Uninjected embryos; k, n, injected with 50 pg of CTGF; i, o, 150 pg of CTGF-CR mRNA injected per blastomere at 4-cell stage.
To test whether CTGF and BMP interacted biochemically, Flagepitope-tagged constructs of full-length CTGF, CTGF-CR, CTGF-ΔCR and the C terminus of CTGF (CTGF-CT) were generated and the proteins produced in cultured cells (Fig. S2). To produce CTGF in sufficient amounts for biochemical experiments we generated a Drosophila melanogaster S2 stable cell line secreting Flag-tagged Xenopus CTGF. Under these conditions the protein was full length and could be affinity-purified via its Flag tag (Fig. S2). Immunoprecipitation assays showed that full-length CTGF, like the positive control chordin, could bind BMP4 in solution (Fig. 3a, lanes 2, 3). Using a monoclonal antibody that is entirely specific for BMP4 (ref. 11), we showed in western blots that the binding of BMP4 to CTGF could be competed by an excess of BMP2 or TGF-β1 but not by IGF-1 (Fig. 3b, lanes 2-5). The interaction between CTGF and BMP4 was direct, because in chemical crosslinking experiments, full-length CTGF or the CR domain of CTGF (CTGFCR) could be crosslinked to BMP4, forming complexes of the expected molecular weights (Fig. 3c, lanes 2, 5). Conversely, CTGF constructs lacking the CR domain (CTGF-ΔCR and CTGF-CT) were unable to form complexes with BMP4 (Fig. 3c, lanes 3, 4). The observation that binding of BMP4 to CTGF could be partially competed by TGF-β1 (Fig. 3b, lane 4) suggested that CTGF might also bind TGF-β1. Chemical crosslinking experiments showed that TGF-β1 could also bind directly to CTGF or to CTGF-CR (Fig. 3d, e). The binding affinity of CTGF for BMP4 and TGF-β1 was determined by surface plasmon resonance (SPR) analysis, a method used previously to evaluate real-time interactions between BMP and follistatin12. Kinetic measurements using different concentrations of CTGF yielded dissociation constants (KD) of 5 nM for BMP4 and of 30 nM for TGF-β1 (Fig. 3f). We conclude from these biochemical studies that CTGF can directly bind BMP4 and TGF-β1 through its CR domain, and has a higher affinity for BMP4 than for TGF-β1.
Figure 3. CTGF binds BMP4 and TGF-β1 through the CR domain.

a, Westernblot analysis of BMP4 (5 nM) bound to CTGF (10 nM, lane 3) or chordin (5 nM, lane 2) after immunoprecipitation (IP) as described previously4. To determine IP effectiveness, the same blots were subsequently probed with anti-Flag antibody for CTGF and anti-myc antibody for chordin. b, Competition of BMP4 (5 nM) bound to CTGF (10 nM) by IP in the absence (lane 2) or presence of 20-fold molar excess of BMP2 (lane 3), TGF-β1 (lane 4) or IGF-1 (lane 5). CTGF IP efficiency was monitored with anti-Flag antibody. c, Chemical crosslinking of CTGF and CTGF mutant constructs to BMP4. Membrane was probed with anti-BMP4 antibody. Bands corresponding to BMP4 dimer (B), BMP4-CTGF complex (B-C) and BMP4:CTGF-CR complex (B-CR) are indicated. d, e, Western blot probed with anti-Flag antibody after chemical crosslinking of CTGF and CTGF-CR to TGF-β1. Bands corresponding to CTGF monomer (C), CTGF-TGF-β1 (C-T) complex, and to CTGF-CR monomer (CR), to the dimer of CTGF-CR (CR2) and to the complex of a dimer of CTGF-CR with TGF-β1 (CR2-T) are indicated. The TGF-β1 was added at 25 nM and 50 nM. f, Sensorgrams of SPR analyses showing the binding of purified CTGF to BMP4 or to TGF-β1. CTGF protein was run (black arrowhead) over BMP4 or TGF-β1 sensor chips. After CTGF flow ended (white arrowhead), dissociation was monitored by a decrease in the resonance units. Kinetic experiments were performed using CTGF concentrations ranging between 6.5 nM and 210 nM of full-length CTGF.
Purified CTGF inhibited BMP signalling in 10T1/2 cells13,14 (Fig. 4a, b). Increasing amounts of CTGF inhibited the alkaline phosphatase (AP) activity induced by 2.5 nM BMP4 in a dose-dependent manner (Fig. 4b) with 50% inhibition (IC50) at approximately 12 nM. The phosphorylation of Smad1 induced by 0.5 nM BMP4 was inhibited by the addition of 20 nM CTGF (Fig. 4c, lanes 2 and 3) to human 293T cell cultures15. Interestingly, an excess of BMP4 (10 nM) was able to quench the antagonistic effect of CTGF on BMP4 action (Fig. 4c, lanes 4 and 5). To address the molecular mechanism by which CTGF inhibited BMP4 signalling we next examined the effect of CTGF on the binding of BMP4 to its cognate receptor16. Increasing amounts of CTGF antagonized BMP4 binding to a recombinant type Ia BMP-receptor-Fc fusion protein, as shown by western blot analysis with anti-BMP4 antibody (Fig. 4d, lanes 2-5). We conclude that binding of BMP4 to CTGF inhibits BMP signalling by preventing the binding of BMP4 to its cognate receptor. This molecular mechanism of action is analogous to that of the BMP antagonist chordin13.
Figure 4. CTGF antagonizes BMP4 signalling by inhibiting receptor binding.

a, Dose-dependent induction of alkaline phosphatase (AP) by BMP4 in 10T1/2 cells is inhibited by CTGF. b, Adding increasing amounts of CTGF inhibits AP induced by 2.5 nM of BMP4. We estimate that CTGF can block BMP4 activity with an IC50 of about 12 nM. Error bars represent standard deviations of triplicate samples. Experiments were independently performed three times. c, Inhibition of Smad1 phosphorylation in 293T cells by CTGF at low, but not high, BMP4 concentrations. d, CTGF inhibits binding of BMP4 to its cognate receptor. BMP4 (1 nM) was preincubated 1 h at room temperature with 10, 25 and 50 nM (black triangle) of affinity-purified CTGF (lanes 3-5), and BMPRIa-Fc was added at 1 nM for 2 h. Lane 1 corresponds to the loading of 50% of the total BMP4 used in each reaction. Lane 6 shows negative control of BMP4 binding to protein A beads in the absence of the BMPRIa-Fc.
CTGF did not inhibit binding of TGF-β1 to a TGFβ-receptor II-Fc (TβRII-Fc) recombinant protein. On the contrary, preincubation with CTGF increased the crosslinking of [125I]TGF-β1 to recombinant TβRII-Fc in solution (Fig. 5a). CTGF also enhanced the binding of [125I]TGF-β1 to endogenous receptors on the surface of cultured cells. Using the method of Massague17, it was found that crosslinking of TGF-β to all three TGF-β interacting proteins — betaglycan, TβRII and TβRI — increased in the presence of CTGF (Fig. 5b). Because CTGF binds TGF-β1 with relatively low affinity (with a KD value of only 30 nM, Fig. 3f), it is conceivable that CTGF may function as a chaperone to modify the conformation or solubility of TGF-β1 and facilitate presentation to its cognate receptors, which have affinities in the picomolar range17. Signalling by TGF-β1 was also increased by CTGF. At low TGF-β1 concentrations, CTGF potentiated the phosphorylation of Smad2 induced by TGF-β1 (Fig. 5c, lanes 2 and 3) in fetal mink lung (Mv1Lu) cell cultures. At higher concentrations of TGF-β1, CTGF did not increase Smad2 phosphorylation, indicating that CTGF serves to potentiate the effects of limiting amounts of TGF-β1 and has no detectable effects of its own on these cells (Fig. 5c, lanes 4-6). In addition, a TGF-β reporter gene18 was induced 6-10-fold by as little as 3 nM CTGF (Fig 5d). TGF-β1 was required, as CTGF alone did not induce reporter-gene expression, even at 45 nM (Fig. 5d).
Figure 5. CTGF potentiates TGF-β1 receptor binding and signalling.

a, Crosslinking of [125I]-TGF-β1 to 1 nM TGF-β receptor Fc fusion protein (TβRII-Fc) in the presence (+) or absence (-) of 10 nM purified CTGF. The TβRII-Fc-TGF-β1 complex is seen in the upper part of the gel. Different concentrations of [125I]-TGF-β1, ranging from 10 pM to 0.5 nM, were used. b, Labelling of cell-surface proteins by [125I]-TGF-β1 in NRK cells after chemical crosslinking with DSS. 125I-TGF-β1 (100 pM) was pre-incubated 1 h with (+) or without (-) 6 nM CTGF before adding for receptor binding17. Specific binding was shown by competition with 50-fold unlabelled TGF-β1 (lane 3). Lane 4 shows negative control crosslinking of CTGF, and [I125]-TGF-β1 in the absence of cells. The three major cell surface TGF-β receptors17 are indicated: betaglycan (βG, 250 kDa), TGF-β-receptor II (TβRII, 80 kDa) and TGF-β-receptor I (TβRI, 68 kDa). c, Two hours after addition of proteins, Smad2 phosphorylation in Mv1Lu cells was stimulated by 6 nM CTGF in the presence of 10 pM TGF-β1. This effect was not seen when TGF-β1 was increased fivefold. Lane 6 shows that CTGF has no effect on its own. Note that Smad2 phosphorylation was induced at lower concentrations of TGF-β1 than those required in reporter-gene or cell-differentiation assays (see below), perhaps reflecting the shorter treatment times involved. d, Luciferase activity 36 h after transfection of Mv1Lu cells with the TGF-β inducible reporter p3TP-lux. Constant TGF-β1 and different concentrations of CTGF were used. Note that CTGF protein stimulates TGF-β1 signalling even when as little as 3 nM is used. e, Number of spherical cell aggregates per square centimeter observed in Mv1Lu cells after 12 h of treatment with different growth factors as described above. The concentrations of CTGF and TGF-β1 were 3 nM and 200 pM, respectively. Note that the BMP inhibitors Chordin and BMPRIa-Fc could not replace CTGF, and that soluble TGFRII-Fc inhibited spherical aggregate formation. f, Immunofluorescence showing the formation of spherical cell aggregates in P19 embryonal carcinoma cells treated with 5 nM CTGF. P19 cell aggregates were positive for PECAM-1 and vWF.
The synergy between CTGF and TGF-β1 was accompanied by a striking and unexpected morphological change in the cultures. Spherical aggregates of cells were observed in Mv1Lu cells treated with TGF-β1 and purified CTGF, but not when treated with either factor on its own (Fig. 5e and Fig. S3 in the Supplementary Information). These effects of CTGF were not caused by a blockade of endogenous BMP signals, because addition of 10 nM chordin or BMPRIa-Fc (which are sufficient to inhibit BMP4 signalling in cell culture, data not shown) together with TGF-β1 did not induce spherical structures in Mv1Lu cells (Fig. 5e). In embryonal carcinoma P19 cells similar structures were induced by CTGF alone (Fig. 5f), presumably because these cells produce endogenous TGF-β (see the legend to Fig. S3). The cell aggregates were positive for platelet-endothelial cell-adhesion molecule (PECAM-1) and for vWF (Fig. 5f and Fig. S3). Although these molecules are markers for endothelial cells, it has recently been reported that PECAM-1 is not entirely specific for blood vessels19 and therefore the determination of whether the spherical structures reported here correspond to blood-vessel-like structures must await further experimentation. We conclude that CTGF protein enhances TGF-β1 signalling by several criteria, including receptor and cell-surface binding, Smad2 phosphorylation, activation of gene expression, and differentiation of cultured cell lines.
Although multiple physiological roles have been proposed for CTGF, its signalling pathway remains unknown. CTGF can bind to low-density lipoprotein receptor-related protein and integrins20,21, but the consequences of these interactions on down-stream signaltransduction pathways are not known. The results reported here indicate that CTGF signals in part through the binding of BMP4/2 and TGF-β1 in the extracellular space. We find that CTGF inhibits BMP but activates TGF-β1 signalling. TGF-β is a well-known inducer of extracellular matrix (ECM) components such as collagen and fibronectin22,23. The CTGF gene may participate in this event, as its promoter contains a TGF-β response element24,25. In support of this view, collagen deposition and anchorage-independent growth induced by TGF-β in fibrob-lasts can be inhibited by adding neutralizing antibodies against CTGF23,26, indicating a synergistic relationship between CTGF and TGF-β. The function of the other domains in CTGF remains to be investigated: the IGFB domain has been shown to bind IGF although this interaction is of low affinity (ref. 27 and our unpublished observations), TSP-1 domains have been implicated in extracellular matrix and vascular endothelial growth factor (VEGF) binding28,29, and the cystine-knot module may contribute a dimerization motif or additional signalling capabilities30. In conclusion, the results presented here suggest a molecular mechanism for CTGF function by which this protein can activate TGF-β signalling and inhibit BMP activity by directly binding to these growth factors in the extracellular space through its Chordin-like domain.
Methods
DNA constructs and embryo manipulations
The complete open reading frame of Xenopus CTGF was amplified by PCR from cDNA of stage 25 embryos using the following primers: forward 5′-AAA CTC GAG GAA CTA ATC TGC TGC TGT-3′ and reverse 5′-AAA TCT AGA TGT GTT TCT CTG CCT CTA-3′, based on the sequence from GenBank accession number U43523 entered by M. L. King and Z. Ying in 1997. The PCR product was cloned in the expression vector pCS2+. Epitope-tag subclones were generated by amplifying CTGF lacking the signal peptide and inserting the fragment in-frame into pCS2+ containing a chordin signal peptide until Ala41, followed by the Flag-tag sequence (pchd-Flag). The primers used for Flag-tagged fulllength CTGF or deletion mutants were as follows: CTGF 5′-AAA CTC GAG CAG GAG TGT AAT GGG GAA T-3′ and 5′-AAA TCT AGA TGT GTT TCT CTG CCT CTA-3′; CTGF-CR 5′-AAA CTC GAG GCC CCT TGT TGT GTT CGG-3′ and 5′-AAA TCT AGA TAG CGC GGA TTA GGG ATG GA-3′; CTGF-CT, 5′-AAA CTC GAG GGT CCT GAT CCA TCC CTA-3′ and T7 primer. CTGF-ΔCR was generated by deletion of sequences between Ala95 and Ala191 of CTGF by PCR mutagenesis. Detailed protocols for embryo fertilizations, in situ hybridization and RT-PCR can be found at http://www.hhmi.ucla.edu/derobertis/index.html.
Protein purification, immunoprecipitation and crosslinking
Flag-tagged secreted proteins were obtained by transient tranfection of human 293T cells or from Drosophila S2 cells stably expressing CTGF (Fig. S2). Full-length Flag-CTGF from S2 cells was affinitypurified with anti-Flag M2 affinity gel column and eluted with Flag peptide according to manufacturer’s directions (Sigma, St Louis, MO). Culture medium obtained from Drosophila S2 cells transfected with empty vector was purified in the same way and used as negative control in all experiments. Protein concentrations were determined using bovine serum albumen (BSA) standards stained with Coommassie blue in SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Immunoprecipitation analyses and chemical crosslinking with disuccinimidyl suberate (DSS) were performed as described previously 4,13.
TGF-β-receptor binding
[125I]-TGF-β1 (specific activity 3000 Ci/mmol, NEN) was crosslinked to recombinant TGF-β-receptor II-Fc (TβRII-Fc) (R&D systems, Minneapolis, MN). Different concentrations of [125I]-TGF-β1 were pre-incubated for 1 h in absence or presence of 10 nM of purified CTGF. 1 nM of TβRII-Fc was added to the reactions for 30 min and the samples were crosslinked with DSS for 30 min4. Binding of [125I]-TGF-β1 to cell surface receptors of NRK9F cultured cells was performed as described previously17. Briefly, 100 pM [125I]-TGF-β1 was pre-incubated with or without 6 nM of purified CTGF for 1 h. Proteins were added to semi-confluent normal rat kidney (NRK) cells pre-equilibrated in binding buffer (128 mM NaCl, 5 nM KCl, 5 mM MgSO4, 1.2 mM CaCl2, 50 mM HEPES, 2 mg ml-1 BSA, pH 7.5) and incubated for 4 h at 4 °C on an oscillating platform at 120 rpm. After extensive washes with ice-cold binding buffer, DSS was added in binding buffer without BSA for 15 min at 4 °C. Cells were extracted in detachment buffer (0.25 M sucrose, 10 mM Tris, 1 mM EDTA, pH 7.4, 0.3 mM PMSF), sedimented by centrifugation and the pellets solubilized with solubilization buffer (125 mM NaCl, 10 mM Tris, 1 mM EDTA, pH 7.0, 1% Triton X-100). Cell debris were removed by centrifugation, and samples were subjected to SDS-PAGE.
Surface plasmon resonance (SPR) analysis
Real-time binding experiments were performed on the Biacore 2000 apparatus (Biacore AB, Uppsala, Sweden) as described previously12. Carrier-free recombinant human BMP4 and TGF-β1 from human platelets (R&D Systems) were immobilized on the sensor-chip surface (CM5, certified grade, Biacore AB). HBS-EP buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.005% v/v surfactant P20) was used as running buffer.
Cell culture assays and immunocytochemistry
Alkaline phosphatase (AP) induction assay13 was performed in the multipotent mesodermal cell line 10T1/2 (ATCC) using the fluorescent substrate 4-methyl umbelliferyl-phosphate (Sigma). AP values were normalized using the amount of protein in each tissue-culture well (Bio-Rad Dc protein kit). Mink lung epithelial cells (Mv1Lu - CCL 64, ATCC) were cultured in alpha-minimal essential media (MEM; Gibco, Grand Island, NY) supplemented with 10% foetal bovine serum. Cells plated in 96-well plates were transiently co-transfected with 0.1 μg of p3TP-lux (ref. 18) and 0.015 μg of pBSKSβ-gal expression plasmid using FuGENE (Roche, Indianapolis, IN) as a transfection reagent. Before growth factors were added, cells were washed with phosphate buffer saline and pre-incubated for 2 h with alpha-MEM containing 2% NuSerum (BD Biosciences, San Jose, CA). Growth factors diluted in alpha-MEM containing 2% NuSerum were pre-incubated at room temperature for 1 h before they were added to cells. After 18 h cells were lysed in Reporter Lysis Buffer (Promega, Madison, WI), and enzyme activity measured using the Luciferase Assay System and the β-Galactosidase Enzyme Assay System (Promega). Enzyme activities were measured in a microplate luminometer and enzyme-linked immunosorbent assay (ELISA) plate reader, respectively. Smad phosphorylation was detected on western blots using 1:6000 dilution of anti-phosphoSmad1 (specific for Smad 1, 5 and 8, gift of C. H. Heldin), 1:500 dilution of anti-phosphoSmad2 (Upstate Biotechnology, Waltham, MA), and 1:400 actin antibodies (Sigma) for protein loading controls. Purified growth factors were premixed for 1 h in serum-free medium (DMEM, F12, Iscove’s, 1:1:1) at room temperature, and added to 80% confluent 293T or Mv1Lu cells for 2 h. Immunocytochemistry was performed with an endothelial-specific anti-CD31 (PECAM-1) monoclonal antibody22 (MEC 13.3, Pharmingen, San Diego, CA) diluted 1:400, and an anti-vWF polyclonal antibody (Dako Co., Burlingame, CA) diluted 1:200. Staining was visualized after incubation with secondaryantibodies conjugated to fluorescein isothiocyanate (FITC) or Cy3 (Jackson Immunoresearch Laboratories) using fluorescent microscopy. Coverslips were mounted in Vectashield mounting media containing DAPI (Vector, Carpinteria, CA). Negative controls were performed by omitting the primary antibody during the immunostaining reaction.
Supplementary Material
ACKNOWLEDGEMENTS
We thank K. M. Lyons and S. Ivkovic for unpublished information, K. Masuhara and C.H. Heldin for antibodies, J. Massague for reporter plasmid, Naoto Ueno for the protocol to immobilize BMP4 to Biacore chips, M. L. King and Z. Ying for entering the CTGF full-length sequence in GenBank, S.-Y. Li and A. Cuellar for technical assistance, and C. Coffinier, E. Delot, J. I. Kim, J. Larraín, K. M. Lyons, M. Oelgeschläger, E. Pera, O. Wessely for discussions and comments on the manuscript. J.G.A. was a Latin American PEW fellow. This work was supported by the National Institutes of Health (R37 HD21502-16). E.M.D.R. is an Investigator of the Howard Hughes Medical Institute. Correspondence and requests for material should be addressed to E.M.D.R.
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
References
- 1.Moussad EE, Brigstock DR. Mol. Genet. Metab. 2000;71:276–292. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 2.Abreu J, Coffinier C, Larraín J, Oelgeschläger M, De Robertis EM. Gene. 2002;287:39–47. doi: 10.1016/s0378-1119(01)00827-7. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Oganesian A, Keene DR, Sandell LJ. J. Cell Biol. 1999;144:1069–1080. doi: 10.1083/jcb.144.5.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larraín J, et al. Development. 2000;127:821–830. doi: 10.1242/dev.127.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakayama N, et al. Dev. Biol. 2001;232:372–387. doi: 10.1006/dbio.2001.0200. [DOI] [PubMed] [Google Scholar]
- 6.Sakuta H, et al. Science. 2001;293:111–115. doi: 10.1126/science.1058379. [DOI] [PubMed] [Google Scholar]
- 7.Hunt LT, Barker WC. Biochem. Biophys. Res. Commun. 1987;144:876–882. doi: 10.1016/s0006-291x(87)80046-3. [DOI] [PubMed] [Google Scholar]
- 8.Bork P. FEBS Lett. 1993;327:125–130. doi: 10.1016/0014-5793(93)80155-n. [DOI] [PubMed] [Google Scholar]
- 9.Smith WC, Harland RM. Cell. 1992;70:829–840. doi: 10.1016/0092-8674(92)90316-5. [DOI] [PubMed] [Google Scholar]
- 10.Sasai Y, et al. Cell. 1994;79:779–790. doi: 10.1016/0092-8674(94)90068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masuhara K, et al. Bone. 1995;16:91–96. doi: 10.1016/s8756-3282(94)00014-x. [DOI] [PubMed] [Google Scholar]
- 12.Iemura S, et al. Proc. Natl. Acad. Sci. USA. 1998;95:9337–9342. doi: 10.1073/pnas.95.16.9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccolo S, Sasai Y, Lu B, De Robertis EM. Cell. 1996;86:589–598. doi: 10.1016/s0092-8674(00)80132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katagiri T, et al. Biochem. Biophys. Res. Commun. 1990;172:295–299. doi: 10.1016/s0006-291x(05)80208-6. [DOI] [PubMed] [Google Scholar]
- 15.Persson V, et al. FEBS Lett. 1998;434:83–87. doi: 10.1016/s0014-5793(98)00954-5. [DOI] [PubMed] [Google Scholar]
- 16.Larraín J, et al. Development. 2001;128:4439–44347. doi: 10.1242/dev.128.22.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massagué J. Methods Enzymol. 1987;46:174–195. doi: 10.1016/s0076-6879(87)46020-5. [DOI] [PubMed] [Google Scholar]
- 18.Wrana JL, et al. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 19.Robson P, Stein P, Zhou B, Schultz RM, Baldoin HS. Dev. Biol. 2001;234:317–329. doi: 10.1006/dbio.2001.0274. [DOI] [PubMed] [Google Scholar]
- 20.Segarini PR, et al. J. Biol. Chem. 2001;276:40659–40667. doi: 10.1074/jbc.M105180200. [DOI] [PubMed] [Google Scholar]
- 21.Kireeva ML, et al. Exp. Cell Res. 1997;233:63–77. doi: 10.1006/excr.1997.3548. [DOI] [PubMed] [Google Scholar]
- 22.Roberts AB, et al. Proc. Natl. Acad. Sci. USA. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. J. Invest. Dermatol. 1996;107:404–411. doi: 10.1111/1523-1747.ep12363389. [DOI] [PubMed] [Google Scholar]
- 24.Grotendorst GR, Okochi H, Hayashi N. Cell Growth Differ. 1996;7:469–480. [PubMed] [Google Scholar]
- 25.Holmes A, et al. J. Biol. Chem. 2001;276:10594–10601. doi: 10.1074/jbc.M010149200. [DOI] [PubMed] [Google Scholar]
- 26.Kothapalli D, Frazier KS, Welply A, Segarini PR, Grotendorst GR. Cell Growth Differ. 1997;8:61–68. [PubMed] [Google Scholar]
- 27.Kim HS, et al. Proc. Natl Acad. Sci. USA. 1997;94:12981–12986. doi: 10.1073/pnas.94.24.12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams JC, Tucker RP. Dev. Dyn. 2000;218:280–299. doi: 10.1002/(SICI)1097-0177(200006)218:2<280::AID-DVDY4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Inoki I, et al. FASEB J. 2002;16:219–221. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- 30.Pearce JJ, Penny G, Rossant J. Dev. Biol. 1999;209:98–110. doi: 10.1006/dbio.1999.9240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.