

Abstract
CLC chloride channels form a large and conserved gene family unrelated to other channel proteins. Knowledge of the transmembrane topology of these channels is important for understanding the effects of mutations found in human myotonia and inherited hypercalciuric kidney stone diseases and for the interpretation of structure–function studies. We now systematically study the topology of human ClC-1, a prototype CLC channel that is defective in human myotonia. Using a combination of in vitro glycosylation scanning and protease protection assays, we show that both N and C termini face the cytoplasm and demonstrate the presence of 10 (or less likely 12) transmembrane spans. Difficult regions were additionally tested by inserting cysteines and probing the effect of cysteine-modifying reagents on ClC-1 currents. The results show that D3 crosses the membrane and D4 does not, and that L549 between D11 and D12 is accessible from the outside. Further, since the modification of cysteines introduced between D11 and D12 and at the extracellular end of D3 strongly affect ClC-1 currents, these regions are suggested to be important for ion permeation.
Voltage-gated chloride channels of the CLC family are highly conserved during evolution and are expressed in organisms ranging from bacteria (1) and yeast (2) to plants (3) and animals (4). Their physiological functions in higher organisms include the regulation of cell volume, control of electrical excitability, and transepithelial transport.
The CLC proteins were identified by expression cloning of the Torpedo chloride channel ClC-0 (5). At present, nine mammalian members are known (for review, see ref. 6). ClC-1 is nearly specific for skeletal muscle (4). It ensures its electrical stability, which is evident from ClC-1 mutations leading to myotonia congenita, a disease characterized by defective muscle relaxation. Many different ClC-1 mutations were found in human myotonia and in animal models (7–11). The importance of CLC channels is underscored by another, recently identified disease. Inactivating mutations of ClC-5 cause a syndrome that is characterized by low molecular weight proteinuria, hypercalciuria, and kidney stones (12). ClC-2 may play a role in cell volume control (13) and in setting the intracellular chloride concentration, which in turn is important for synaptic transmission in certain neurons (14). However, the function of most CLC proteins is still unclear.
CLC proteins are structurally unrelated to other ion channel classes. Topological models are still mainly based on hydropathy analysis, suggesting the presence of about 12–13 transmembrane domains, originally termed D1 to D13 (5). This model has already had to be revised to accommodate new experimental findings (15, 16).
CLC channels function as multimers of identical (11) or homologous subunits (17). Dominant negative mutations suggested that ClC-1 channels have more than three subunits (11). However, ClC-0 is a homodimeric channel with one pore per subunit (16, 18, 19); this may also apply for ClC-1 (20).
In structure–function studies, mutations have been introduced into ClC-0 (18, 21), ClC-1 (9, 11), ClC-2 (13, 22), and ClC-5 (12). These mutations showed that several protein regions are important for gating and permeation. Unfortunately, the interpretation of these data is limited by the lack of structural information.
We put the topology of CLC channels on a firm experimental basis. We primarily used the glycosylation scanning procedure (23–25), which we complemented with protease protection assays (26, 27) and by introducing cysteines that were then probed with extracellular cysteine-modifying reagents for effects on currents (28–30). In addition to clarifying the transmembrane topology, the latter technique identified novel ClC-1 residues, which are important for channel function.
EXPERIMENTAL PROCEDURES
Construction of Glycosylation Mutants 1T2 to 12T13.
We started with pTLB⋅H1NQ, a mutant ClC-1 cDNA in the expression vector pTLN (17). The ClC-1 glycosylation site (N430) was mutated to glutamine and an epitope (LYPYDVPDYVSG) was inserted behind G972; the latter is of no importance for the present work. For C-terminally truncated pTLB⋅H1NQΔC, the sequence TGATAAGAATTC containing two stop codons and an EcoRI site was introduced after L629. Glycosylation transplants, encompassing a part (F413 to G444, referred to as “T,” or G409 to V456, indicated by “T*”) of the D8/D9 loop of ClC-1 including the native glycosylation site, were inserted into hydrophilic loops by recombinant PCR (31) as detailed in Fig. 1. All PCR-derived fragments were sequenced. Full-length glycosylation constructs for functional tests were gained from the truncated forms by swapping the respective stretches from pTLB⋅H1NQΔC into pTLB⋅H1NQ.
Figure 1.

Topological model of human ClC-1 illustrating the constructs used in this work. Hydrophobic domains are labeled D1 to D13 according to the original nomenclature for ClC-0 (5) based on Kyte–Doolittle hydropathy analysis. For amino acid sequence, see ref. 4. WT-glycosylation is represented by a branched line in loop D8/D9. An unused glycosylation site after D13 is indicated by an asterisk. Insertion sites of glycosylation transplants are indicated by triangles (solid = glycosylation positive), fusion sites with prolactin by ball-and-stick (solid = proteinase protection positive) after the amino acids shown in open circles. Amino acids in black have been replaced by cysteines and are sensitive to cysteine specific reagents.
Construction of ClC-1/Prolactin Fusion Proteins.
Full-length prolactin cDNA was obtained by reverse transcription-PCR from bovine pituitary gland poly(A)+ RNA using Pfu DNA polymerase (Stratagene) and was cloned into pTLN to yield pTLN⋅Prl.
Constructions of NPrl, 1Prl, … 8Prl, 10Prl, 12Prl, 13Prl: Serial truncations of human ClC-1 cDNA, starting at the initiator ATG and extending to positions before or behind potential transmembrane spans, were fused to the prolactin cDNA C-terminal of Y74 (referred to as “prolactin epitope”). Fusions were generated by PCR-amplification of ClC-1 fragments and prolactin by using one BamHI-site containing primer, respectively, so that ligation introduced two additional residues (G and S) at the fusion site. In constructs ND3Prl, ND4Prl, and ND6Prl domains D3 (M193 to F235), D4 (K231 to Y262), or D6 (T293 to D340), respectively, are fused between the N terminus of ClC-1 (ending at G115) and the prolactin epitope on the C-terminal side. In SPD4Prl, the N terminus of ClC-1 of construct ND4Prl was replaced by the signal peptide of pre-prolactin, starting with the initiator methionine and ending with D61. N34T*5Prl consists of the ClC-1 N terminus fused to the sequence encoding D3 through D5 (M193 to R304), including the glycosylation transplant between D4 and D5 as in construct 4T*5. The C terminus was then fused to the prolactin epitope.
In Vitro Transcription.
Capped cRNA was transcribed by SP6 RNA polymerase from 0.5 μg of plasmid after linearization with MluI using the mMessageMachine cRNA synthesis kit (Ambion).
In Vitro Translation.
Glycosylation constructs were translated using rabbit reticulocyte lysate (Promega) in the presence or absence of canine pancreatic microsomes (Promega) and in the presence of [35S]methionine. For SDS/PAGE, 2–5 μl of translation mixture was diluted in 5–10 μl of sample buffer and separated on 8% gels. After drying, gels were examined with a bioimage-analyzer (Fuji BAS1500). Translation for proteinase protection experiments was done with microsomes but without radioactivity.
Protease Protection Assay and Western Blot Analysis.
Translation mixture was brought to 10 mM CaCl2 and chilled on ice. Aliquots of 10 μl were incubated with proteinase K (30 μg/ml, Boehringer Mannheim) in the presence or absence of 1% Triton X-100. Controls remained without proteinase and detergent. Proteolysis proceeded on ice for 60 min, and was stopped by adding 5 mM phenylmethylsulfonyl fluoride. After 10 min on ice, 50 μl of preheated (95°C) sample buffer was added and the sample was boiled for 15 min to inactivate the protease. Proteins were separated on a 5–20% SDS/PAGE gradient, blotted onto nitrocellulose (Schleicher & Schuell), and probed with an anti-prolactin antiserum (ICN) using protein A-coupled horseradish peroxidase (Bio-Rad) and the Renaissance chemiluminescence kit (DuPont/NEN).
Electrophysiology.
cRNA (10–15 ng) in 50 nl was injected into Xenopus oocytes, which were kept in modified Barth’s solution (88 mM NaCl/2.4 mM NaHCO3/1.0 mM KCl/0.41 mM CaCl2/0.33 mM CaNO3/0.82 mM MgSO4/10 mM Hepes, pH 7.6) for 2 to 3 days and analyzed in ND96 saline (96 mM NaCl/2 mM KCl/1.8 mM CaCl2/1 mM MgCl2/5 mM Hepes, pH 7.4). Standard two electrode voltage clamp measurements were performed at room temperature using a Turbotec amplifier (Npi Instruments) and pCLAMP 5.5 software (Axon Instruments, Foster City, CA).
Probing Cysteine Residues with SH Reactive Compounds.
We substituted L223, P228 in D3, E232 and G233 between D3 and D4, and L549 in the D11/D12 block with cysteine. PCR mutagenesis, cRNA synthesis, and injection into Xenopus oocytes were performed as described. Currents were measured in ND96 with 20 mM Hepes (pH 7.4) (NDHep) and monitored for reproducibility. The bath was then changed for 3 min to 10 mM sodium (2-sulfonatoethyl)methanethiosulfonate (MTSES) in NDHep, during which currents reached steady state. Oocytes were then washed extensively with NDHep and recorded again. By comparing currents before and after MTSES application, effects due to noncovalent interactions were excluded.
RESULTS
Probing ClC-1 Topology by Glycosylation.
The highly conserved consensus site for N-linked glycosylation between D8 and D9 is used in ClC-0, ClC-1, and the ClC-K channels (15, 16). Thus, in contrast to the initial topology model (5), this region must face the extracellular space. To discover which other regions are extracellular, we inserted glycosylation consensus sites between various hydrophobic domains of ClC-1 (Fig. 1). To detect exclusively the effects of newly introduced sites, we based these constructs on a mutant (ClC-1N430Q) lacking the wild-type (WT) glycosylation site. This mutant is functional (15). Unfortunately, most hydrophilic loops of ClC-1 are shorter than 33 residues, which is a lower limit for reliable glycosylation (32, 33). Therefore, we inserted a copy of the D8–D9 loop that is glycosylated in native CLC proteins (15). A similar strategy was used successfully, e.g., in the analysis of Glut-1 glucose transporter topology (34).
Core glycosylation increases the molecular weight by about 2 kDa. With ClC-1 (≈110 kDa) the relative increase in mass is just ≈2% if a single site is glycosylated. The resulting shift is difficult to detect by SDS/PAGE. To increase the relative mass difference, we used a truncated construct which ends about 50 amino acids downstream of the last transmembrane domain. As membrane integration is a sequential process starting at the N terminus, this should not influence upstream topology.
Fig. 2 shows the size analysis of WT and mutant proteins synthesized in vitro in the presence and absence of microsomes. The shortened WT protein, WTΔC, and the equivalent glycosylation knockout, NQΔC, yielded a ≈74-kDa band when translated without microsomes. With microsomes, an additional band of ≈77 kDa reflecting the glycosylated protein was seen with WTΔC but not with NQΔC. The shift to higher masses was incomplete, indicating that part of the protein is still synthesized at free ribosomes in the cytosol also with microsomes. This could not be improved by changing the cRNA/microsome ratio. The ≈42-kDa band in Fig. 2 is due to a fraction of truncated cRNA, which results from a premature transcription stop of the SP6 RNA polymerase (C. Lorenz and T.J.J., unpublished observations). This band was larger in constructs 1T2ΔC, 2T3ΔC, and 3T*4ΔC because it contained the glycosylation transplant.
Figure 2.
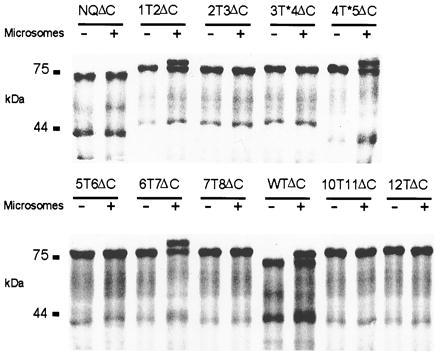
Autoradiograph of in vitro translation products with glycosylation sites engineered between hydrophobic domains. RNA was translated in reticulocyte lysate in absence (−) or presence (+) of microsomal membranes. All constructs are shortened at the C terminus as indicated by ΔC. WTΔC contains the native glycosylation site. In NQΔC the glycosylation site is removed by mutating N430 to Q. “T” in other constructs indicates the standard glycosylation transplant, “T*” the longer variant, and the flanking numbers the respective hydrophobic domains. Numbers at left indicate the molecular mass in kDa.
Glycosylation occurred between D1/D2, D6/D7, and D8/D9 (equivalent to WT glycosylation). In contrast, the standard glycosylation transplant could not be glycosylated when inserted in the short hydrophilic stretches flanking D4 (data not shown). Since these are the shortest interdomain segments, we introduced a larger transplant. The segment behind D4 could now be glycosylated (construct 4T*5ΔC), while the one before D4 (construct 3T*4ΔC) remained unglycosylated.
Chloride Channel Function of Glycosylation Constructs.
To exclude that these transplants had perturbed channel structure, we expressed full-length cRNAs containing these inserts in Xenopus oocytes and measured whole cell currents. It is highly unlikely that mutants with a changed transmembrane topology still yield functional chloride channels. In contrast, a loss of currents does not prove an altered topology, as the insertions may have abolished channel function by other means.
In general, currents from glycosylation constructs were smaller than those expressed from identical amounts of WT cRNA (Fig. 3). Western blot analysis of Xenopus oocyte crude membrane preparations showed that the reduction in currents was paralleled by lower protein levels (data not shown). Thus, insertion constructs are translated less efficiently or degraded more efficiently, or both. Currents of 3T*4, 5T6, and 12T13 were not significantly different from water-injected controls. By contrast, the remaining six constructs elicited currents of 5–30% as compared with WT and NQ mutants. Thus, these insertions did not alter the transmembrane topology. Importantly, this includes all mutants that were glycosylated. Currents of constructs 1T2 and 6T7 showed qualitative changes, indicating that insertions at these positions changed channel properties, but did not abolish its function. With 1T2 the voltage dependence was shifted to more positive values, and outward currents slowly activated. Mutant 6T7 has almost completely lost the inactivation behavior at hyperpolarizing potentials and its rectification is strongly changed.
Figure 3.

Two-electrode voltage clamp experiments of Xenopus oocytes expressing full-length glycosylation constructs. Mean conductance at −40 mV of WT ClC-1 and its mutants in comparison to water injected control oocytes (mean ± SEM, n = 4 to 9). (Inset) Pulse protocol and typical current traces for WT and constructs 1T2, 4T*5, and 6T7.
Proteinase Protection Assay.
Glycosylation of a given site proves that it is extracellular. However, absence of glycosylation is no proof that it is intracellular, because the access of the glycosyltransferase could be impaired by sterical hindrance. Proteinase protection, however, can probe for an intracellular localization. An epitope is fused to the protein at different positions. If the epitope ends up in the lumen of the endoplasmic reticulum, it can be degraded by proteinase K only if the integrity of the membrane is destroyed by detergents (35). Conversely, complete degradation in the absence of detergent indicates a cytosolic location.
We constructed a series of truncated ClC-1 proteins in which a prolactin epitope was fused behind putative transmembrane domains (Fig. 1). Portions of prolactin lacking its endogenous signal sequence are topogenically neutral and respond faithfully to upstream signal-anchor and stop-transfer sequences (23, 36, 37). These constructs were translated in vitro in the presence of microsomes and treated with proteinase K in the absence or presence of Triton X-100. Fragments containing the prolactin epitope were detected by Western blot analysis.
As expected, the hydrophilic N terminus of ClC-1 confers no protection on the epitope in construct NPrl (Fig. 4), whereas D1 (construct 1Prl) acts as a signal-anchor and translocates the epitope into the endoplasmic reticulum. The protected fragment is ≈20 kDa in size, which results from 17.7 kDa of the prolactin domain and 2.3 kDa of the transmembrane domain D1. Protection of prolactin-containing fragments of similar sizes are found with constructs 6Prl and 8Prl, consistent with the extracellular localization indicated by our glycosylation analysis. 3Prl only shows a weak signal at ≈20 kDa, suggesting that the translocation of the epitope occurred less efficiently. Addition of detergent always led to complete digestion. No protection could be observed with 2Prl, 4Prl, 5Prl, 7Prl, 10Prl, and 13Prl.
Figure 4.

Proteinase protection assay. Increasing portions of ClC-1 starting at the N terminus (N) were fused C-terminally to a prolactin reporter-epitope (Prl). Numbers indicate the last hydrophobic domain of ClC-1 that is included in the fusion protein. In vitro translation was performed in the presence of pancreatic microsomes. Products were treated with proteinase K with (+) or without (−) Triton X-100. Controls were treated equivalently, but proteinase K and detergent were omitted (−/−).
The lack of protease protection with 4Prl apparently contradicts the extracellular localization of the D4–D5 linker inferred from glycosylation of construct 4T*5ΔC (Fig. 2). The glycosylation assay is more trustworthy because no transmembrane domains were deleted in that construct and because the full-length protein 4T*5 yielded typical chloride currents. This suggests that the transfer of D4 across the membrane may be more complicated than that of other domains.
We therefore tested the capability of D3 or D4 to act individually as signal-anchor sequences (Fig. 5). We fused the ClC-1 N terminus to D3 and D4 and then to the prolactin epitope (constructs ND3Prl and ND4Prl). Prolactin was protected against digestion in control constructs ND1Prl and ND6Prl, but neither D3 nor D4 alone were able to translocate the epitope. We also tested whether D4 might act as a stop-transfer sequence by replacing the ClC-1 N terminus in ND4Prl by the prolactin signal sequence to initiate translocation (construct SPD4Prl). When translated with microsomes, the product was shortened by the signal peptidase. The truncated peptide was completely resistant to proteinase K. This indicates that D4 cannot redirect the C terminus into the cytoplasm.
Figure 5.

Testing of individual domains for their topogenic activity. Modular fusions of individual domains with the ClC-1 N terminus and the prolactin reporter epitope. In construct SPD4Prl the N terminus is formed by the signal sequence of pre-prolactin (SP). (Upper) Immunoblot of in vitro translation products treated as described above. (Lower) Schematic drawings of topologies compatible with the experimental results.
Thus, D3 and D4 cannot individually translocate the prolactin epitope. However, construct 3Prl (Fig. 4) indicates that D3 can translocate the epitope (albeit inefficiently) if preceded by D1 and D2, and glycosylation shows that the region before D5 is extracellular. In addition, constructs 2Prl and 5Prl suggest that the stretches preceding D3 and following D5, respectively, are cytoplasmic. To test whether domains D3, D4, and D5 may insert into the membrane only if translated together, we designed construct N34T*5Prl. Following the ClC-1 N terminus, the construct contains the complete D3–D5 region (including the extended glycosylation transplant tested in construct 4T*5ΔC) fused to the reporter epitope. Translating this construct in the presence of microsomes indeed resulted in glycosylation (as indicated by its larger size), whereas the prolactin epitope was digestable by proteinase K (Fig. 5). Thus, the stretch after D3/D4 is only translocated if it is followed by D5. Glycosylation provides strong evidence that the stretch after D4 is extracellular, while the partial protease protection of construct 3Prl indicates that this also applies for the segment preceding D4. The latter evidence, however, is somewhat weaker because it was obtained with a severely truncated protein.
Cysteine Substitution Mutants.
We reprobed the D3/D4 region in the context of the full-length channel. As enzymatic modification of residues may be hindered by steric constraints, we replaced single residues by cysteines and monitored their accessibility to comparatively small cysteine-specific reagents using electrophysiology. Methanethiosulfonate derivatives have been used successfully to probe the pore-forming residues of several channel classes (29, 38–40), including the cystic fibrosis transmembrane conductance regulator chloride channel (41, 42). These reagents covalently add protruding charged moieties (-SCH2CH2SO3− in the case of MTSES) to cysteines, which may interfere with ion permeation if they are next to the pore. If these membrane impermeable reagents affect currents, this proves that the respective residues are accessible from the exterior. A negative result, however, does not yield any information, as the residue may have been modified without any functional effect. In contrast to methods detecting modified cysteines biochemically (43), our functional assay does not require a cysteine-less ClC-1 mutant (that may be nonfunctional) as a starting construct, provided that WT ClC-1 currents are unaffected by MTSES. This is indeed the case (Fig. 6).
Figure 6.

Effects of MTSES on Xenopus oocytes expressing ClC-1 WT or cysteine mutants indicated in Fig. 1. (Upper) Examples of current traces. (Lower) Relative changes in conductance at −40 mV 3 min after application of 10 mM MTSES (mean ± SEM, n = 5 to 9) and subsequent washout.
We replaced L223 and P228 in D3 as well as E232 and G233 between D3 and D4 (see Fig. 1 for positions), since we knew that mutations at similar positions in ClC-0 affect pore properties (M. Pusch and T.J.J., unpublished data). These substitutions did not abolish currents when expressed in Xenopus oocytes. While currents from mutant P228C were like WT, L223C and E232C showed a weaker inactivation at hyperpolarizing potentials. The currents for mutant G233C were smaller and slightly outwardly rectifying. Currents from mutants L223C, P228C, E232C, and G233C, but not from WT ClC-1, were drastically reduced by incubating the oocytes for 3 min in 10 mM of MTSES (Fig. 6). This shows that the end of D3 and amino acids before D4 are accessible to small charged molecules from the exterior and confirm the protease protection experiment with construct 3Prl. In mutant E232C a negatively charged amino acid is replaced by a neutral one. Although a modification with MTSES reintroduces the negative charge, WT inactivation behavior was not restored.
We used the same technique to test whether residues in the middle of the broad hydrophobic region D11/D12, which enters the cytoplasm at both ends (constructs 10Prl and 12Prl; Fig. 4), may be accessible from the exterior. When we mutated leucine 549, which is located next to a highly conserved glutamate residue, to cysteine, channel activity was retained and like WT. Extracellular application of MTSES drastically inhibited these currents (Fig. 6). Thus, D11/D12 crosses the membrane twice.
Several other introduced cysteines in the vicinity of those described above did not confer MTSES-sensitivity (data not shown).
DISCUSSION
Several techniques are available to probe the transmembrane topology of multispan transmembrane proteins, including probing with epitope-specific antibodies, gene fusions with reporter enzymes, glycosylation scanning, protease protection assays, and cysteine scanning (for review, see ref. 44). All of these techniques have their specific problems, and sometimes different techniques have suggested different topologies. Here we chose three complementary techniques to increase the reliability of the results.
In the first set of experiments we used an in vitro glycosylation scanning procedure. Since N-glycosylation exclusively takes place in the endoplasmic reticulum, carbohydrate transfer to an artificially introduced acceptor site indicates its extracellular position. Absence of glycosylation, however, does not prove an intracellular localization. We therefore reprobed ClC-1 topology with the protease protection assay. It indicates a cytoplasmic localization of a reporter epitope by its lability to added protease, while those in the lumen are protected. Additionally, we checked two particular areas by modifying artificially introduced cysteines with extracellularly applied membrane-impermeable reagents. Importantly, all crucial constructs from the first and third set of experiments yielded functional chloride channels, demonstrating that their topology was unchanged.
The first CLC topology models were exclusively based on a hydropathy analysis according to Kyte and Doolittle (45) that indicated 13 hydrophobic stretches long enough to potentially span the lipid bilayer. These were termed D1 to D13 (ref. 5; see Fig. 1). The broad hydrophobic region preceding the hydrophilic C terminus was arbitrarily divided into domains D9 through D12. Hydropathy analysis also identified a C-terminal region (D13) of weak hydrophobicity. In contrast to the D12–D13 interdomain stretch, D13 is highly conserved.
CLC proteins have no cleavable signal peptide. This is compatible with (but does not prove) a cytoplasmic N terminus. The orientation of signal-anchor sequences is strongly influenced by the charge distribution in flanking sequences. Positive charges are placed preferentially in the cytosol (“positive-inside rule”; refs. 46 and 47). With ClC-1, the first transmembrane span D1 was directly preceded by five positive and two negative charges, but there were only two positive residues directly behind D1. This suggests an intracellular N terminus. This was confirmed by our experiments, which demonstrate that D1 is a signal-anchor sequence that assumes type II orientation (46). The second hydrophobic domain, D2, was a stop-transfer sequence whose orientation was again compatible with the charge distribution at its ends.
The D3/D5 stretch was the only region in which a protease protection experiment apparently contradicted the glycosylation assay. The latter results suggest that the D4–D5 linker is extracellular—and this is strong evidence because the full-length glycosylation construct is functional—while the prolactin fused at the end of D4 was not translocated into the endoplasmic reticulum. We could resolve this problem by showing that D3–D5 correctly insert into the membrane only when translated together. By contrast, D3 or D4 could not insert into the membrane individually. Indeed, neighboring domains act synergistically during membrane insertion in several other proteins (27, 37, 48–50). The results from both protease protection (construct 3Prl) and cysteine scanning suggest that D3 crosses the membrane and that its C-terminal part is accessible from the exterior. Thus, there is strong cumulative evidence that D3 does indeed span the lipid bilayer, but that D4 does not (Fig. 1). This is consistent with our previous suggestion (51), which was based on a comparative analysis of several CLC proteins. While D4 presents as a peak of moderate hydrophobicity in Kyte–Doolittle plots of ClC-0 and ClC-1 and is conserved between these proteins, there is nearly no sequence conservation to more distantly related CLC proteins like ClC-5 or ClC-7. The latter channels even lack an hydrophobic stretch in that segment.
Glycosylation and protease protection assays consistently show that domains D6 to D8 are transmembrane domains. Compatible with the “positive-inside” rule, this places the D7–D8 linker into the cytosol. Though poorly conserved by sequence, the linker is strongly positively charged in all known CLC proteins. The extracellular localization of the D8–D9 linker is consistent with its glycosylation in ClC-0 and ClC-K (15, 16), and most mammalian CLC proteins have glycosylation consensus sites in this loop.
The broad hydrophobic region D9 to D12 (Fig. 1) is found in all CLC proteins. It is interrupted by a hydrophilic stretch of variable length, as seen in hydrophobicity plots of ClC-0 and ClC-2 through ClC-5. Protease protection experiments indicate that this stretch is cytoplasmic. The hydrophobic regions before and after this stretch are broad enough to span the membrane at least twice. However, we felt that it would be inappropriate to probe these regions of uninterrupted hydrophobicity by glycosylation scanning or protease protection assays. From studies on ClC-0 we knew that mutations in a highly conserved region in the middle of D11/D12 affect gating and rectification (M. Pusch, U. Ludewig, and T.J.J., unpublished data). Currents mediated by the L549C mutant situated in that region were sensitive to modification with MTSES, showing that this residue is accessible from the exterior. Because the stretches before D11 and after D12 are cytoplasmic, D11/D12 must span the membrane twice, or at least dip deeply into the lipid bilayer. We have not yet performed a similar analysis in the D9–D10 stretch, as we do not have any clues from mutagenesis studies as to which amino acid to choose. As the D8–D9 linker is extracellular and the segment after D10 intracellular, this segment must cross the membrane either once or three times. This implies that CLC channels have 10 (or 12) transmembrane spans in total.
The large hydrophilic C terminus is poorly conserved within the gene family and varies in length. Both sides of the weakly hydrophobic, conserved region D13 are cytoplasmic. No glycosylation occurs at the engineered site after D12 in construct 12TΔC nor at the naturally occurring site after D13 [as shown for ClC-K1 and ClC-K2 (15)], and prolactin epitopes are not protected when fused either before or behind D13.
It is interesting to compare our experimental results with predictions from algorithms that use alignments of several members of a gene family as input. We used two such programs [tmap (52) and predictprotein (53–55)] with ClC-0 to ClC-7, ClC-Ka, Saccharomyces cerevisiae scClC, and Escherchia coli ecClC as inputs. Both programs correctly indicate that D4 does not cross the membrane, but only tmap correctly predicts that D3 does so. predictprotein is consistent with four transmembrane domains in the D9–D12 region, whereas TMAP lumps D9 and D10 together into one domain. Thus, only the predictions by TMAP are consistent with the experimental data.
The topology determined here is consistent with several electrophysiological findings. In ClC-0, mutation K519E next to the end of D12 causes an outward rectification consistent with a charge reversal close to the cytoplasmic end of the pore (9). The cytoplasmic localization of this residue was confirmed by cysteine modification experiments (19). In addition, indirect evidence obtained with ClC-2 (13) is compatible with the cytoplasmic localization of D13 shown here. ClC-2 gating depends on an N-terminal inactivation domain. It could be transplanted to the C terminus either before or behind D13 without loss of function. Recent experiments have identified the D7–D8 linker as a putative receptor for this inactivation domain (22). Thus, all four regions must be on the same (cytoplasmic) side of the membrane, which is fully consistent with the present work.
However, the topology determined here contradicts some other reports. It was reported (56) that a variant of ClC-K2 lacking D2 induced currents identical to WT ClC-K2, suggesting—in clear contrast to the present data—that D2 does not span the bilayer (16). A deletion of an upstream transmembrane domain will grossly change the topology of a multispan membrane protein, predicting a loss of function. Indeed, deleting D2 in ClC-0 and ClC-5 abolishes currents (6, 12), and deleting D2 in ClC-5 causes Dent’s disease (12). Thus, ClC-K currents [which could not be reproduced by others (15, 57)] should be reexamined. Additionally, a serum against a C-terminal fusion protein of a rabbit ClC-K protein was reported to inhibit currents in renal membrane vesicles when applied from the extracellular face (57). This is difficult to reconcile with the cytoplasmic location of the C terminus shown here.
In summary, we have shown experimentally that ClC-1 (and probably all CLC proteins) has 10 (or 12) transmembrane domains. Both its N and C termini reside in the cytoplasm. In contrast to a previous model based solely on hydropathy analysis according to Kyte and Doolittle (5), D4 does not cross the membrane, but is accessible at either end from the exterior.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, the Muscular Dystrophy Association, and the Fonds der Chemischen Industrie.
ABBREVIATIONS
- MTSES
sodium (2-sulfonatoethyl)methanethiosulfonate
- WT
wild type
References
- 1.Fujita N, Mori H, Yura T, Ishihama A. Nucleic Acids Res. 1994;22:1637–1639. doi: 10.1093/nar/22.9.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greene J R, Brown N H, DiDomenico B J, Kaplan J, Eide D J. Mol Gen Genet. 1993;241:542–553. doi: 10.1007/BF00279896. [DOI] [PubMed] [Google Scholar]
- 3.Hechenberger M, Schwappach B, Fischer W N, Frommer W B, Jentsch T J, Steinmeyer K. J Biol Chem. 1996;271:33632–33638. doi: 10.1074/jbc.271.52.33632. [DOI] [PubMed] [Google Scholar]
- 4.Steinmeyer K, Ortland C, Jentsch T J. Nature (London) 1991;354:301–304. doi: 10.1038/354301a0. [DOI] [PubMed] [Google Scholar]
- 5.Jentsch T J, Steinmeyer K, Schwarz G. Nature (London) 1990;348:510–514. doi: 10.1038/348510a0. [DOI] [PubMed] [Google Scholar]
- 6.Jentsch T J, Günther W, Pusch M, Schwappach B. J Physiol (London) 1995;482P:19S–26S. doi: 10.1113/jphysiol.1995.sp020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George A L, Crackower M A, Abdalla J A, Hudson A J, Ebers G C. Nat Genet. 1993;3:305–309. doi: 10.1038/ng0493-305. [DOI] [PubMed] [Google Scholar]
- 8.Koch M C, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann Horn F, Grzeschik K H, Jentsch T J. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- 9.Pusch M, Steinmeyer K, Koch M C, Jentsch T J. Neuron. 1995;15:1–20. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 10.Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch T J. Nature (London) 1991;354:304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- 11.Steinmeyer K, Lorenz C, Pusch M, Koch M C, Jentsch T J. EMBO J. 1994;13:737–743. doi: 10.1002/j.1460-2075.1994.tb06315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lloyd S E, Pearce S H, Fisher S E, Steinmeyer K, Schwappach B, Scheinman S J, Harding B, Bolino A, Devoto M, Goodyer P, Rigden S P, Wrong O, Jentsch T J, Craig I W, Thakker R V. Nature (London) 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 13.Gründer S, Thiemann A, Pusch M, Jentsch T J. Nature (London) 1992;360:759–762. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- 14.Staley K J, Smith R, Schaak J, Wilcox C, Jentsch T J. Neuron. 1996;17:543–551. doi: 10.1016/s0896-6273(00)80186-5. [DOI] [PubMed] [Google Scholar]
- 15.Kieferle S, Fong P, Bens M, Vandewalle A, Jentsch T J. Proc Natl Acad Sci USA. 1994;91:6943–6947. doi: 10.1073/pnas.91.15.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middleton R E, Pheasant D J, Miller C. Biochemistry. 1994;33:13189–13198. doi: 10.1021/bi00249a005. [DOI] [PubMed] [Google Scholar]
- 17.Lorenz C, Pusch M, Jentsch T J. Proc Natl Acad Sci USA. 1996;93:13362–13366. doi: 10.1073/pnas.93.23.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludewig U, Pusch M, Jentsch T J. Nature (London) 1996;383:340–343. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]
- 19.Middleton R E, Pheasant D J, Miller C. Nature (London) 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- 20.Fahlke C, Knittel T, Gurnett C A, Campbell K P, George A L. J Gen Physiol. 1997;109:93–104. doi: 10.1085/jgp.109.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pusch M, Ludewig U, Rehfeldt A, Jentsch T J. Nature (London) 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- 22.Jordt S E, Jentsch T J. EMBO J. 1997;16:1582–1592. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavez R A, Hall Z W. J Biol Chem. 1991;266:15532–15538. [PubMed] [Google Scholar]
- 24.Chang X B, Hou Y X, Jensen T J, Riordan J R. J Biol Chem. 1994;269:18572–18575. [PubMed] [Google Scholar]
- 25.Turk E, Kerner C J, Lostao M P, Wright E M. J Biol Chem. 1996;271:1925–1934. doi: 10.1074/jbc.271.4.1925. [DOI] [PubMed] [Google Scholar]
- 26.Chavez R A, Hall Z W. J Cell Biol. 1992;116:385–393. doi: 10.1083/jcb.116.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skach W R, Lingappa V R. Cancer Res. 1994;54:3202–3209. [PubMed] [Google Scholar]
- 28.Akabas M H, Kaufmann C, Cook T A, Archdeacon P. J Biol Chem. 1994;269:14865–14868. [PubMed] [Google Scholar]
- 29.Kürz L L, Zühlke R D, Zhang H J, Joho R H. Biophys J. 1995;68:900–905. doi: 10.1016/S0006-3495(95)80266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez-Garcia M T, Chiamvimonvat N, Marban E, Tomaselli G F. Proc Natl Acad Sci USA. 1996;93:300–304. doi: 10.1073/pnas.93.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higuchi R. In: PCR Protocols. Innis M A, Gelfand D H, Sinnsky J J, White T J, editors. San Diego: Academic; 1990. pp. 177–183. [Google Scholar]
- 32.Nilsson I M, von Heijne G. J Biol Chem. 1993;268:5798–5801. [PubMed] [Google Scholar]
- 33.Landolt-Marticorena C, Reithmeier R A. Biochem J. 1994;302:253–260. doi: 10.1042/bj3020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hresko R C, Kruse M, Strube M, Mueckler M. J Biol Chem. 1994;269:20482–20488. [PubMed] [Google Scholar]
- 35.Perara E, Lingappa V R. J Cell Biol. 1985;101:2292–2301. doi: 10.1083/jcb.101.6.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothman R E, Andrews D W, Calayag M C, Lingappa V R. J Biol Chem. 1988;263:10470–10480. [PubMed] [Google Scholar]
- 37.Skach W R, Lingappa V R. J Biol Chem. 1993;268:23552–23561. [PubMed] [Google Scholar]
- 38.Akabas M H, Stauffer D A, Xu M, Karlin A. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- 39.Kuner T, Wollmuth L P, Karlin A, Seeburg P H, Sakmann B. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 40.Pascual J M, Shieh C C, Kirsch G E, Brown A M. Neuron. 1995;14:1055–1063. doi: 10.1016/0896-6273(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 41.Akabas M H, Karlin A. Biochemistry. 1995;34:12496–12500. doi: 10.1021/bi00039a002. [DOI] [PubMed] [Google Scholar]
- 42.Cheung M, Akabas M H. Biophys J. 1996;70:2688–2695. doi: 10.1016/S0006-3495(96)79838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loo T W, Clarke D M. J Biol Chem. 1995;270:843–848. doi: 10.1074/jbc.270.2.843. [DOI] [PubMed] [Google Scholar]
- 44.Traxler B, Boyd D, Beckwith J. J Membr Biol. 1993;132:1–11. doi: 10.1007/BF00233047. [DOI] [PubMed] [Google Scholar]
- 45.Kyte J, Doolittle R F. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 46.von Heijne G, Gavel Y. Eur J Biochem. 1988;174:671–678. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- 47.Sipos L, von Heijne G. Eur J Biochem. 1993;213:1333–40. doi: 10.1111/j.1432-1033.1993.tb17885.x. [DOI] [PubMed] [Google Scholar]
- 48.Skach W R, Shi L B, Calayag M C, Frigeri A, Lingappa V R, Verkman A S. J Cell Biol. 1994;125:803–815. doi: 10.1083/jcb.125.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henn D K, Baumann A, Kaupp U B. Proc Natl Acad Sci USA. 1995;92:7425–7429. doi: 10.1073/pnas.92.16.7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin J, Addison R. J Biol Chem. 1995;270:6935–6941. doi: 10.1074/jbc.270.12.6935. [DOI] [PubMed] [Google Scholar]
- 51.Jentsch T J, Günther W, Pusch M, Schwappach B. J Physiol (London) 1995;482:19S–25S. doi: 10.1113/jphysiol.1995.sp020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Persson B, Argos P. J Mol Biol. 1994;237:182–192. doi: 10.1006/jmbi.1994.1220. [DOI] [PubMed] [Google Scholar]
- 53.Rost B, Casadio R, Fariselli P, Sander C. Protein Sci. 1995;4:521–533. doi: 10.1002/pro.5560040318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rost B. Methods Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- 55.Rost B, Fariselli B, Casadio R. Protein Sci. 1996;7:1704–1718. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adachi S, Uchida S, Ito H, Hata M, Hiroe M, Marumo F, Sasaki S. J Biol Chem. 1994;269:17677–17683. [PubMed] [Google Scholar]
- 57.Zimniak L, Winters C J, Reeves W B, Andreoli T E. Am J Physiol. 1996;270:F1066–F1072. doi: 10.1152/ajprenal.1996.270.6.F1066. [DOI] [PubMed] [Google Scholar]