

Abstract
The serotonin transporter (SERT) is a member of the Na+/Cl−-dependent neurotransmitter transporter family and constitutes the target of several clinically important antidepressants. Here, replacement of serine-545 in the recombinant rat SERT by alanine was found to alter the cation dependence of serotonin uptake. Substrate transport was now driven as efficiently by LiCl as by NaCl without significant changes in serotonin affinity. Binding of the antidepressant [3H]imipramine occurred with 1/5th the affinity, whereas [3H]citalopram binding was unchanged. These results indicate that serine-545 is a crucial determinant of both the cation dependence of serotonin transport by SERT and the imipramine binding properties of SERT.
Keywords: neurotransmitter, uptake, antidepressant binding, mutagenesis
Neurotransmitter transporters mediate the re-uptake of released neurotransmitters from the synaptic and nonsynaptic environment and play a key role in regulating synaptic transmission. Functional studies and sequence comparison of the transporters for different amino acid and monoamine transmitters have allowed their classification into two distinct families (1–3). The Na+/Cl−-dependent transporter family includes the biogenic amine, glycine, and γ-aminobutyric acid transporters, which share 12 putative transmembrane domains (TMDs) and a large extracellular loop between TMD3 and TMD4 that contains several potential N-glycosylation sites. The glutamate transporter family, in contrast, has a different transmembrane organization and needs K+ for substrate transport. All transmitter uptake systems display a stringent Na+ requirement for substrate translocation.
The serotonin transporter (SERT) has received particular attention because it represents the target of clinically important antidepressants as well as drugs of abuse and has been implicated in neurological and psychiatric disorders (4). Pharmacological studies indicate the existence of distinct binding sites for tricyclic antidepressants and selective serotonin (5-hydroxytryptamine, 5-HT) re-uptake inhibitors on the SERT protein, and complex interactions between these sites as well as the substrate translocation site have been reported (5–11). The cloning of SERT from different species (12–16) has fostered attempts to define structure–function relationships in this protein. Notably, analysis of chimeric human and rat SERT has revealed domains important for the selective binding of tricyclic antidepressants (10). However, little is known about the amino acid side chains implicated in substrate and Na+ binding as well as 5-HT transport.
Serine residues are known to be crucial for the binding of catecholamines to adrenergic receptors (17) and for [3H]dopamine uptake mediated by the dopamine transporter (18). Here, we substituted alanine for three serine residues located in TMD11 of the rat SERT. The effects of these substitutions on 5-HT transport and antidepressant binding were then analyzed in transfected cells. Our results show that a serine residue at position 545 is important for the cation dependence of 5-HT transport. Additionally, this mutation affects the velocity of substrate translocation as well as binding of imipramine, but not citalopram.
MATERIALS AND METHODS
Chemicals.
[3H]Imipramine (21 Ci/mmol; 1 Ci = 37 GBq) and [3H]5-HT (13.9 Ci/mmol) were obtained from Amersham, and [3H]citalopram (86.5 Ci/mmol) was from New England Nuclear. Unlabeled imipramine was purchased from ICN, and 5-HT from Sigma. Citalopram was a kind gift of J. Hyttel (H. Lundbeck, Copenhagen-Valby, Denmark).
Site-Directed Mutagenesis.
The XbaI/HpaI fragment of a pCis construct containing the coding sequence of rat SERT (14) was subcloned into the eukaryotic expression vector pRC/CMV (Invitrogen), to yield clone pRC-SERT. For the construction of mutants S545A, S555A, and S559A, single-stranded phagemid DNA derived from pRC-SERT was used as template, and point mutations were generated with synthetic oligonucleotides using an in vitro mutagenesis kit (Muta-gene; Bio-Rad) as described (19, 20). All mutations were verified by dideoxynucleotide sequencing.
Cell Culture.
Human embryonic kidney cells (HEK-293 cells; ATCC CRL 1573) were grown and transfected in 10-cm tissue culture dishes, or 24-well dishes, for membrane preparation and transport assay as described (11).
Preparation of Membranes.
Confluent HEK-293 cells were harvested and homogenized on ice in a Potter homogenizer. After nuclei and debris had been removed by centrifugation (11, 21), membranes were collected by sedimentation at 20,000 × g for 20 min at 4°C, resuspended in phosphate-buffered saline containing 5% (wt/vol) glycerol, frozen in liquid nitrogen, and stored at −70°C.
[3H]Antidepressant Binding and [3H]5-HT Uptake.
As reported previously (11, 21), binding and transport experiments were performed with the indicated concentration of tritiated ligands using TB1 buffer (120 mM NaCl/2 mM KCl/1 mM CaCl2/1 mM MgCl2/10 mM Hepes, pH 7.5) or TB2, TB3, and TB4 buffers, in which NaCl was replaced by 120 mM choline, 120 mM LiCl, or 120 mM N-methyl-d-glucamine (NMDG), respectively. For uptake measurements, HEK-cells were plated into 24-well dishes (diameter = 2 cm), and the culture medium was replaced by 0.2 ml of transport buffer TB1 containing 250 nM [3H]5-HT (unless otherwise stated). After 3 min at room temperature, the medium was rapidly removed, and the cells were washed twice with TB1 before extraction with 0.4 ml of 10% (wt/vol) sodium dodecyl sulfate. Radioactivity was determined by scintillation counting in a Beckman LS60001IC scintillation counter. To obtain comparable transport parameters with SERT wild-type (wt) and mutant proteins, HEK-293 cells were grown to 70% confluency in 24-well plates and transfected with 1 μg per well of the respective cDNA as described (11), and transport rates were determined in parallel at the same day.
Specific uptake or binding was defined as the difference between the data obtained with transfected and mock-transfected cells (11) from the same set of cultures. Each experiment was repeated at least twice. All data represent the mean of triplicate determinations for binding experiments and of quadruplicate determinations for uptake studies. Standard deviations were routinely <7% and are indicated where larger than the symbols used. Data were analyzed by a nonlinear regression analysis program (xact; Atari), which fitted sigmoidal uptake curves to the following equations: (i) saturation experiments: V = Vmax/[1 + (Km/S)nH], B = Bmax/[1 + (Kd/L)nH]; and (ii) competition experiments: V/Vmax = IC50nH/(InH + IC50nH), where V represents transport rate; Vmax, maximal transport rate; B, bound ligand; Bmax, maximal number of binding sites; L, ligand concentration; S, substrate concentration; I, inhibitor concentration; IC50, inhibitor concentration for half-maximal transport inhibition; and nH, the Hill coefficient. In case of competitive inhibition, Ki values were calculated from the respective IC50 values according to Cheng and Prusoff (22).
RESULTS
Effects of Serine Substitutions in SERT on 5-HT Uptake.
To determine whether the three serine residues (S545, S555, and S559) located in TMD11 have a role in the uptake of 5-HT by and antidepressant binding to the rat SERT, we replaced the codons for these residues in the SERT1 cDNA with alanine triplets and expressed the resulting mutants in HEK-293 cells for binding and uptake studies. Initial experiments indicated that the transport of [3H]5-HT was saturable with all three mutants; the Km, Hill coefficient, and Vmax values determined for [3H]5-HT transport by mutants S555A (Km = 1.13 ± 0.27 μM; nH = 1.4 ± 0.1; Vmax = 19.2 ± 4.5 pmol/min per well; n = 2) and S559A (Km = 1.15 ± 0.25 μM; nH = 1.45 ± 0.05; Vmax = 18.7 ± 4.3 pmol/min per well; n = 2) were similar to those observed with the wt SERT (Km = 1.0 ± 0.2 μM; nH = 1.47 ± 0.03; Vmax = 19 ± 4.4 pmol/min per well; see Table 1). In contrast, mutant S545A showed a reduced transport affinity with a Km value of 2.92 ± 0.47 μM and a loss of positive cooperativity as revealed by a Hill coefficient of nH = 1.1 ± 0.06. We therefore analyzed the properties of this mutant in more detail.
Table 1.
Comparison of [3H]5-HT transport parameters of wt and S545A mutant SERTs
| Medium |
Km,
μM
|
nH
|
Vmax,
%
|
|||
|---|---|---|---|---|---|---|
| wt | S545A | wt | S545A | wt | S545A | |
| NaCl | 1.00 ± 0.2 | 2.9 ± 0.47 | 1.47 ± 0.03 | 1.1 ± 0.06 | 100 | 185 ± 17 |
| LiCl | 1.90 ± 0.56 | 9.1 ± 0.92 | 1.06 ± 0.07 | 1.0 ± 0.04 | 23 ± 3 | 182 ± 3 |
All experiments were performed at least three times in quadruplicate. Vmax values are reported as percent of wt SERT in NaCl medium, 19 ± 4.4 pmol/min per well.
Characterization of [3H]5-HT Uptake by the S545A Mutant.
Kinetic studies showed that the initial rates of [3H]5-HT uptake were linear within the first 3 min for both S545A and wt SERTs (Fig. 1A). However, after 30 min cells transfected with the S545A cDNA had accumulated significantly higher amounts of [3H]5-HT than those expressing wt transporters (Fig. 1A). To extend this observation, saturation experiments with increasing concentrations of 5-HT were performed (Fig. 1B). This revealed that S545A SERT had a consistently (n = 3) higher Vmax (37 ± 11 pmol/min per well) than wt SERT (19 ± 4.4 pmol/min per well) and all other mutant SERTs tested simultaneously (see above). This is unlikely to reflect a better efficiency of S545A mutant transporter expression, since the levels of cell surface staining with a SERT-specific antibody (23) did not differ from those of cells expressing the wt and the S555A and S559A mutant proteins (data not shown). Accordingly, replacement of serine 545 by alanine appears to induce faster substrate translocation concomitant with a slight reduction of the apparent Km value. Interestingly, the binding affinity of 5-HT was not affected, as determined by its ability to displace [3H]citalopram (not shown); the respective Ki values were similar for the mutant and wt proteins (S545A, Ki = 1.1 ± 0.06 μM; wt, Ki = 1.24 ± 0.03 μM).
Figure 1.
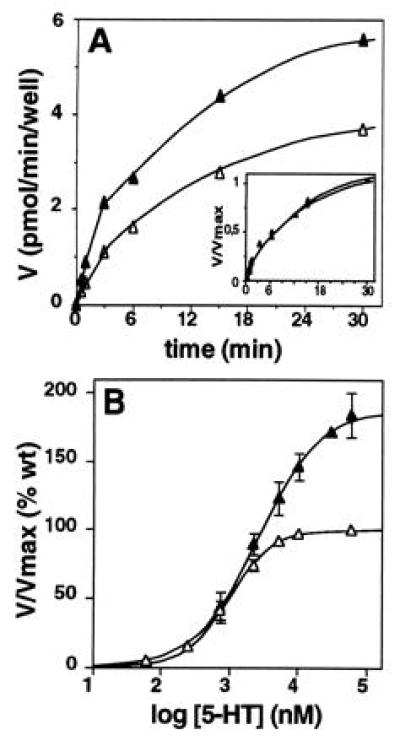
[3H]5-HT uptake into in HEK-293 cells transfected with the mutant S545A (▴) and wt SERT (▵) cDNAs. (A) Time course of [3H]5-HT (250 nM) accumulation. Note that the S545A mutant exhibited a higher Vmax (5.5 pmol/min per well) of uptake than the wt SERT (3.7 pmol/min per well). Inset shows that both mutant and wt have the same kinetic properties, with initial velocities being linear between 0 and 3 min. The data correspond to a representative experiment performed two times with similar results. (B) Concentration dependence of [3H]5-HT transport. The apparent affinity of [3H]5-HT uptake was reduced for S545A SERT (Km = 2.6 ± 0.5 μM) as compared with wt (Km = 1.0 ± 0.2 μM), whereas its Vmax was 1.85 ± 0.17 (n = 3) fold higher than with the wt transporter (S545A, Vmax = 37 ± 11 pmol/min per well; wt, Vmax = 19 ± 4.4 pmol/min per well). The data represent the mean ± SEM of three experiments; saturation characteristics were determined by the addition of different concentrations of unlabeled 5-HT to 250 nM [3H]5-HT, and uptake was measured for 3 min.
Cation Dependence of [3H]5-HT Transport.
Since the uptake of 5-HT by recombinant SERT is sodium dependent (13, 24), we analyzed the cation dependence of 5-HT transport mediated by the S545A mutant transporter. As shown in Fig. 2A, a similar Vmax value was reached with S545A upon replacement of NaCl by LiCl [Vmax(NaCl) = 25.2 ± 8.3 pmol/min per well; Vmax(LiCl) = 23.0 ± 7.3 pmol/min per well]. This corresponds to 98.5% ± 1.5% of the transport seen in NaCl, a value that is very different from the strong reduction in Vmax (≈80%) observed with the wt protein under the same conditions (Table 1). As indicated by a shift of the substrate concentration curve to the right, the affinity of 5-HT transport was reduced in the presence of LiCl [Km(NaCl) = 3.2 ± 0.7 μM; Km(LiCl) = 9.1 ± 0.9 μM]. A similar 2- to 3-fold increase of the Km value upon Na+ replacement by Li+ has also been observed for the wt SERT (P.S., unpublished results). In contrast, when NaCl was replaced by choline or NMDG, no transport was observed (1.3% ± 0.01% and 3.5% ± 2.3% of the uptake in NaCl medium, respectively) (Fig. 2B). These results indicate that the S545A SERT mutant has lost its selectivity for Na+, as Li+ can fully substitute for Na+ in providing the driving force for substrate translocation.
Figure 2.
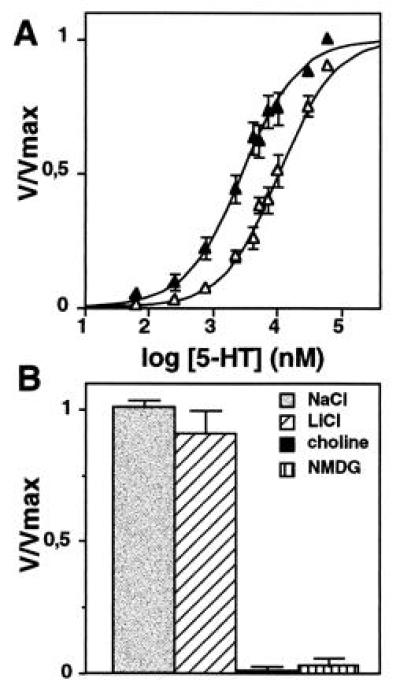
Cation dependence of [3H]5-HT uptake mediated by the S545A mutant. (A) Concentration dependence of [3H]5-HT transport in the presence (▴) or absence (▵) of Na+. Transfected cells were prewashed twice with buffer containing 120 mM NaCl or 120 mM LiCl, and uptake of 250 nM [3H]5-HT in the presence of increasing concentrations of unlabeled 5-HT was followed over 3 min. In LiCl medium, the Km value of [3H]5-HT transport was increased as compared with NaCl medium [Km(NaCl) = 3.2 ± 0.7 μM; Km(LiCl) = 9.1 ± 0.92 μM]. In contrast, Vmax values remained similar in both conditions: Vmax(NaCl) = 25.2 ± 8.3 pmol/min per well versus Vmax(LiCl) = 23 ± 7.3 pmol/min per well. The data represent the mean ± SEM of four experiments. (B) [3H]5-HT transport by S545A SERT is abolished upon replacement of Na+ or Li+ by choline or NMDG (120 mM). In these experiments, cells were prewashed with the appropriate buffer, and transport of 5-HT (30 μM) was followed over 20 min. Calculated Vmax values were as follows: NaCl, 23 ± 0.3 pmol/min per well; LiCl, 20.5 ± 2 pmol/min per well; choline, 0.3 ± 0.2 pmol/min per well; and NMDG, 0.8 ± 0.5 pmol/min per well. The data represent the mean ± SEM of two experiments.
[3H]Imipramine and [3H]Citalopram Binding to the S545A Mutant.
To obtain further insight into the properties of the S545A transporter protein, we characterized the binding of the antidepressants imipramine and citalopram to this mutant. These ligands are known to bind to different sites, or conformations, of the rat SERT (11). Saturation experiments (Fig. 3) performed on identical membrane preparations of S545A SERT-expressing cells revealed twice as many [3H]imipramine binding sites as [3H]citalopram binding sites [Bmax(imipramine)/Bmax(citalopram) = 2.2 ± 0.3; n = 4). Scatchard analysis showed a linear binding isotherm for [3H]imipramine; however, the binding affinity was significantly reduced (Kd = 34.2 ± 4.3 nM; see Table 2). The Kd of [3H]citalopram binding was identical for the mutant and the wt transporters (Table 2), suggesting that citalopram binding was not affected in the mutant. These results indicate that substitution of alanine for serine-545 affects high-affinity imipramine, but not [3H]citalopram, binding.
Figure 3.
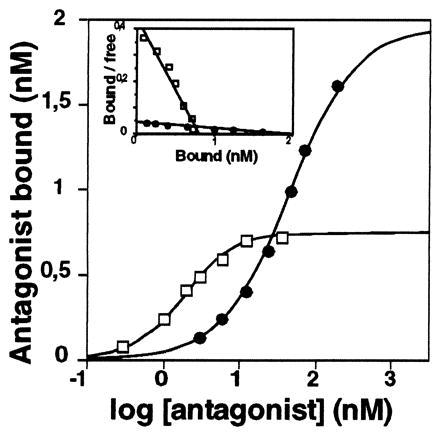
[3H]Imipramine (•) and [3H]citalopram (□) binding to S545A SERT membranes. This representative experiment shows that more [3H]imipramine than [3H]citalopram binds to the mutant transporter. The calculated binding parameters for this experiment were as follows: [3H]imipramine, Kd = 42 nM; nH = 1.06; and Bmax = 12.8 pmol/mg protein; [3H]citalopram, Kd = 1.75 nM; nH = 1.35; and Bmax = 4.9 pmol/mg protein; ratio Bmax(imipramine)/Bmax(citalopram) = 2.6. Inset shows a Scatchard representation of the data, indicating the evident difference in Bmax values. This experiment was performed four times; the following mean (±SEM) values were obtained: [3H]imipramine, Kd = 41 ± 2 nM; nH = 1.04 ± 0.01; and Bmax = 2.04 ± 0.57 nM; [3H]citalopram, Kd = 2.25 ± 0.2 nM; nH = 1.33 ± 0.02; and Bmax = 0.90 ± 0.18 nM; ratio Bmax(imipramine)/Bmax(citalopram) = 2.2 ± 0.3.
Table 2.
Comparison of antidepressant binding parameters of wt and S545A mutant SERTs
| Antidepressant | Medium |
Kd,
nM
|
nH
|
||
|---|---|---|---|---|---|
| wt | S545A | wt | S545A | ||
| Citalopram | NaCl | 2.15 ± 0.28 | 2.42 ± 0.24 | 1.42 ± 0.03 | 1.35 ± 0.04 |
| Citalopram | LiCl | 6.4 ± 3.1* | 4.4 ± 0.15 | — | 1.1 ± 0.05 |
| Imipramine | NaCl | 6.90 ± 0.9 | 34.2 ± 4.3 | 1.17 ± 0.01 | 1.05 ± 0.01 |
All experiments were performed at least three times in triplicate.
Value taken from ref. 11.
5-HT and Antidepressant Interactions in the S545A Mutant.
To elucidate whether the known interactions between the antidepressant and 5-HT binding sites were affected in the S545A SERT, we analyzed [3H]5-HT uptake and antidepressant binding in the presence of imipramine and citalopram. The results obtained are summarized and compared with those determined at wt SERT in Table 3. Both antidepressants showed a reduced potency in inhibiting [3H]5-HT uptake mediated by the S545A mutant transporter. The Ki values obtained were more than 20-fold higher than the respective Kd values determined by direct radioligand binding (Table 2). Moreover, citalopram displaced [3H]imipramine binding with a potency (Ki) similar to its binding affinity (Kd) (see Tables 2 and 3). However, imipramine antagonized [3H]citalopram binding with a 3-fold higher Ki than the Kd determined by direct radioligand binding (Tables 2 and 3). The interactions between imipramine and citalopram had been shown to be complex as revealed by both competitive and noncompetitive displacement profiles (11). In contrast, at S545A, imipramine purely competitively inhibited [3H]citalopram binding (Fig. 4A). Similarly, increasing amounts of citalopram (4 and 8 nM) only competitively antagonized [3H]imipramine binding (Fig. 4B).
Table 3.
IC50 and Ki values of wt and S545A mutant SERTs
| Competitor | Substrate | IC50,
nM
|
Ki, nM
|
||
|---|---|---|---|---|---|
| wt | S545A | wt | S545A | ||
| Citalopram | [3H]5-HT (250 nM) | 10.5 ± 2 | 68 ± 6 | 8.4 ± 1.6 | 62 ± 5 |
| [3H]Imipramine* | 3.7 ± 1.2 | 6.4 ± 1.1 | NC | 2.86 ± 0.49 | |
| [3H]Citalopram* | 7.6 ± 0.8 | ND | 3.04 ± 0.32 | ND | |
| Imipramine | [3H]5-HT (250 nM) | 592 ± 105 | 1,600 ± 208 | 474 ± 84 | 1,473 ± 191 |
| [3H]Imipramine (48 nM) | 27.2 ± 4.8 | ND | 12.1 ± 2.1 | ND | |
| [3H]Citalopram (4 nM) | 83.2 ± 11 | 300 ± 56 | NC | 107 ± 28 | |
All experiments were performed at least twice in quadruplicate (uptake) or triplicate (binding). NC, not calculated because of noncompetitive interaction between imipramine and citalopram; ND, not done.
Binding data taken from ref. 11.
Figure 4.
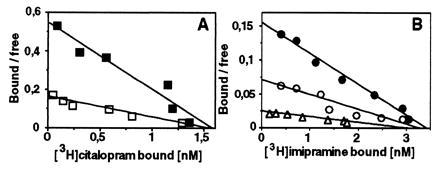
Scatchard plots of [3H]citalopram (A) and [3H]imipramine (B) binding to S545A membranes in the presence of unlabeled imipramine and citalopram, respectively. (A) Imipramine at 200 nM (□) is a competitive inhibitor of [3H]citalopram binding as revealed by increased apparent Kd values [control (▪) = 2.8 nM; imipramine = 9 nM] without significant changes in the Bmax (control = 14 pmol/mg protein; imipramine = 14.1 pmol/mg protein). This experiment was performed twice; mean (±SEM) values were as follows: control, Kd = 2.9 ± 0.1 nM; Bmax = 16 ± 2.5 pmol/mg protein; imipramine, Kd = 11 ± 2 nM; Bmax = 16 ± 2 pmol/mg protein. (B) This representative experiment showed that citalopram at concentrations of 4 nM (○) and 8 nM (▵) is a competitive inhibitor of [3H]imipramine binding as revealed by increased apparent Kd values [control (•) = 22 nM; citalopram (4 nM) = 48 nM and citalopram (8 nM) = 155 nM]; the corresponding Bmax values were as follows: control = 32.9 pmol/mg protein; citalopram (4 nM) = 32.8 pmol/mg protein; and citalopram (8 nM) = 32.7 pmol/mg protein. This experiment was repeated twice with similar results.
DISCUSSION
Following the premise that serine residues might be involved in 5-HT recognition by the SERT, we have mutated three serine residues located in TMD11 of the rat SERT (S545, S555, and S559). Whereas mutation of the two latter residues did not alter the transport characteristics of SERT, replacement of serine-545 by alanine resulted in significant changes of transport parameters. Consequently, the effects of this point mutation on 5-HT uptake and antidepressant binding parameters were analyzed in more detail. We report that serine at position 545 is involved in the Na+ dependence of 5-HT transport as well as in high-affinity binding of imipramine but not citalopram.
Mutant S545A Is a Fast Translocating Transporter.
The S545A mutant accumulates 5-HT with an increased velocity as compared with the wt protein, suggesting modified kinetics of 5-HT transport. The uptake process can be simply described by the following Michaelis–Menten equation:
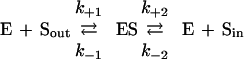 |
where E is the concentration of transporter and Sout and Sin are the concentrations of 5-HT outside and inside the cell, respectively; k+1, k−1 and k+2, k−2 are kinetic constants related to 5-HT binding (k+1, k−1) and 5-HT translocation (k+2, k−2). The constant k+2 includes the kinetic constants of the multiple steps associated with substrate translocation. Under our experimental conditions (fast washing of the cells after a 3-min period of substrate uptake), we can assume that the efflux of 5-HT (k−2) was negligible. Therefore, the maximal transport rate (Vmax) is equivalent to Vmax = k+2ES. Thus, the increased Vmax observed with the mutant S545A most probably results from an increased k+2. According to the Michaelis-Menten equation [Km = (k−1 + k+2)/k+1], an increase in k+2 should increase the apparent Km value of the substrate, given that the substrate binding affinity (k−1/k+1) does not change. Indeed, we found a 2- to 3-fold-increased Km value for 5-HT transport by the mutant S545A but no alteration in the binding of 5-HT as estimated by its unchanged potency in displacing [3H]citalopram from both S545A and wt SERT-containing membranes. This is consistent with substitution of serine-545 not impairing 5-HT binding, but creating a SERT with a faster substrate translocation rate.
Sodium-Independent Transport of 5-HT by the S545A Mutant.
Transport of 5-HT is a sodium-dependent process as determined in [3H]5-HT uptake experiments (13, 24) and electrophysiological recordings from Xenopus oocytes expressing recombinant wt SERT (25). In uptake experiments, replacement of external Na+ by Li+ resulted in a reduction of the maximal transport rate to ≈15–20% of the control value (13, 24). Sodium ions are thought to be cotransported with 5-HT molecules at a 1:1 stoichiometry and to provide the electrochemical gradient for substrate transport (24). It is proposed that binding of Na+ as well as of 5-HT to the extracellular side of the transporter triggers a conformational change, which allows the protein to initiate a complete transport cycle (26). Interestingly, SERT bears at least two sodium binding sites as shown by a sigmoidal curve of Na+ dependence of imipramine binding to platelet membranes and the cooperative binding of two Na+ ions associated with transient currents recorded in oocytes (8, 25). Electrophysiological analysis of the γ-aminobutyric acid transporter, GAT1, also supports the view that Na+ binding promotes a conformational change which facilitates substrate binding and thereby translocation (27, 28). Biochemical evidence for Na+-induced conformational changes has also been obtained for the rat glutamate transporter (29).
Our finding that 5-HT transport mediated by the S545A mutant is as efficient in Li+- as in Na+-containing media suggests that the mutant SERT is already in an activated conformation, which normally is assumed only upon Na+ binding. This conformation can immediately bind 5-HT and proceed into the transport cycle. Thus, Na+ ions seem to be essential for an initial step of the transport cycle, but not for 5-HT translocation into the cytoplasm per se. Such a Na+-induced “pre-activation step” may in addition reflect the positive cooperativity of substrate transport seen with the wt rat (this study) and human SERT (30), as well as with GAT1 (28). In contrast, no positive cooperativity was observed with the S545A mutant. This is consistent with this mutant existing already in a conformational state that normally is achieved only upon Na+ binding.
Serine-545:
A Putative Determinant of the External Gate? Electrophysiological studies of Na+/Cl−-dependent transporters support the concept that transporters share numerous similarities with ion channels, such as ligand-dependent gating processes, selectivity filters, and hydrophilic permeation pathways (25, 31). Consistent with this idea, binding of 5-HT and Na+ to the transporter may promote a conformational change leading to the opening of a gate, and thus initiate substrate translocation. We propose that serine-545 is a determinant of the Na+ binding domain of such a gate, which is constitutively open in mutant S545A. The 5-HT translocation step then can also be driven by other cations like Li+; the Na+ selectivity of the transport-associated permeation pathway thus appears not stringent. This implies that the transport of 5-HT by SERT is Na+ dependent, whereas its translocation across the membrane is not.
Previously proposed models of the transmembrane organization of the SERT and related transporter proteins place serine-545 into the center of the TMD11 (12, 32). Notably, this residue is followed by a highly conserved proline residue. We propose that this proline induces a kink in the transmembrane α-helix and thus makes S545 accessible to Na+.
Imipramine Binding to Mutant S545A.
SERT is the target of tricyclic antidepressants and selective 5-HT re-uptake inhibitors. We have previously identified two allosterically interacting antidepressant binding sites (ABS) on rat SERT heterologously expressed in HEK-293 cells (11). ABS1 binds imipramine with high affinity, whereas ABS2 constitutes a high-affinity citalopram binding site that recognizes imipramine only with lower affinity (>100 nM). We also provided evidence that ABS2 most probably corresponds to a large binding pocket that accomodates citalopram, imipramine, and 5-HT binding subsites (11). Moreover, it has been shown that the high-affinity imipramine binding site is Na+ dependent (11, 26, 33), whereas its low-affinity site is not (11, 33). The latter could be identified only by the ability of imipramine to displace [3H]citalopram (11) or a labeled cocaine analog (26) in LiCl-containing medium. Here, we demonstrated by direct ligand binding studies on the S545A mutant that [3H]imipramine indeed binds twice as many sites as [3H]citalopram, thus confirming the existence of two imipramine binding sites on the rat SERT. At the wt SERT, a large difference between the affinities of imipramine for ABS1 and ABS2 together with negative allosteric coupling of both sites prevent the identification of ABS2 by direct radioligand binding. The saturation and competition experiments reported here reveal that citalopram and 5-HT binding to ABS2 is not changed in the S545A mutant. Since imipramine competitively inhibited [3H]citalopram binding to S545A SERT (Ki = 107 nM; Table 3) with an affinity comparable to that preventing [3H]citalopram binding to the wt SERT in the absence of Na+ ions (Ki = 112 nM), we conclude that imipramine binding to ABS2 is not altered in the mutant protein. Thus, serine residue 545 is not directly involved in the binding of substrate and antagonists to subsites present within ABS2. The observed increase in the overall Kd value of imipramine binding to the S545A mutant therefore most likely results from a reduced affinity of ABS1, and thus reflects imipramine binding to both ABS1 and ABS2. We assume that the imipramine affinities of the two sites are only slightly different, as suggested by the linear Scatchard plot obtained in our [3H]imipramine binding experiments.
Pharmacology and Electrophysiology of SERT: A Joint Model.
The conclusions emerging from this and previous studies (11) together with the evidence that SERT shares characteristics of ion channels (25) prompt us to propose a joint model for 5-HT transport as shown in Fig. 5. This model combines our pharmacological and mutagenesis data with the alternating channel access model of transport described by Mager and colleagues (25). Accordingly, completion of a transport cycle is a dynamic process, during which SERT undergoes the following distinct conformational alterations: (i) At rest (state 1), the external and internal gates are only slightly open to allow for a leakage current driven by Na+, Li+, or K+. (ii) Binding of Na+ and 5-HT to a gate domain promotes a conformational change (state 2) which prepares the transporter for translocation. Serine-545 is likely to constitute an important determinant of this gate binding domain. Imipramine may interact at this site with high affinity, since its high-affinity binding is Na+ dependent (11, 26, 33) and competitively inhibited by 5-HT with an affinity [IC50 = 252 nM (11)] higher than its Km for transport (1 μM; Table 1). (iii) In a subsequent step, the external gate opens (state 3), and 5-HT, Na+, or Li+ and Cl− bind to the substrate translocation domain. Substitution of alanine for serine-545 is postulated to create a SERT which is constitutively held in this state. The translocation binding domain contains also subsites for high-affinity citalopram and low-affinity imipramine binding. In mutant S545A, these antidepressant subsites are not altered; however, citalopram and imipramine have a reduced potency to antagonize uptake, suggesting that their inhibitory action may take place at a step upstream of the opening of the ligand-dependent gate. (iv) In the terminal step (state 4), the external gate is closed and the internal gate is opened. This allows the substrate to be released on the cytoplasmic side together with the cotransported ions. At this stage, a K+ ion is trapped in the transporter permeation pathway. Its release into the extracellular medium upon reassuming state 1 will terminate the transport cycle.
Figure 5.
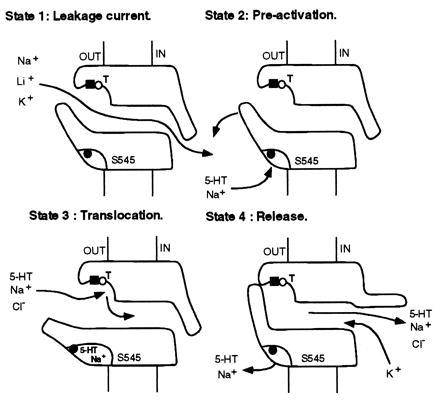
Model for the localization of 5-HT, antidepressant, and ion binding sites in relation to the 5-HT transport mechanism. State 1: At rest, transport gates are slightly opened, which allows for a leakage current driven by Na+, Li+, or K+ (25). State 2: Binding of 5-HT and Na+ to a gate site which may include serine-545 triggers a conformational change that pre-activates the transporter. This binding domain associated with the gate is proposed to contain the high-affinity imipramine binding site (•). State 3: The substrate and the cotransported ions bind to the translocation domain (T), which probably contains overlapping low-affinity imipramine (○) and citalopram (▪) binding sites. State 4: The external gate closes, the internal gate opens, and 5-HT, Na+, and Cl− are released to the cytoplasm. The mechanism of dissociation of 5-HT and Na+ from the gating domain remains speculative at this stage. Subsequently a K+ ion enters the permeation pathway to be released in to the extracellular medium. The depicted localization of both gates at the edges of the transporter is hypothetical and was chosen for simplicity.
Conclusion.
Our data show that serine residue 545 of rat SERT plays a key role in the sodium dependence of 5-HT transport and in high-affinity imipramine binding. They also demonstrate that Li+ can be efficiently cotransported with the substrate, suggesting that the permeation pathway is not selective for Na+. The control of uptake properties by a hydroxyl-containing side chain in TMD11 may also apply to related carriers, since serine-545 is conserved or isofunctionally replaced by threonine in several Na+/Cl−-dependent neurotransmitter transporters.
Acknowledgments
We thank Anja Niehuis for expert technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 269) and the Biomed and Health Research Program BMH1-CT93-1110. C.S. holds a European Molecular Biology Organization postdoctoral fellowship.
ABBREVIATIONS
- ABS
antidepressant binding site
- 5-HT
5-hydroxytryptamine (serotonin)
- NMDG
N-methyl-d-glucamine
- SERT
serotonin transporter
- TMD
transmembrane domain
- wt
wild-type
References
- 1.Amara S G, Arriza J L. Curr Opin Neurobiol. 1993;3:337–344. doi: 10.1016/0959-4388(93)90126-j. [DOI] [PubMed] [Google Scholar]
- 2.Schloss P, Püschel A W, Betz H. Curr Opin Cell Biol. 1994;6:595–599. doi: 10.1016/0955-0674(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 3.Worrall D M, Williams D C. Biochem J. 1994;297:425–436. doi: 10.1042/bj2970425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meltzer H Y, Lowry M T. In: Psychopharmacology, the Third Generation of Progress. 3rd Ed. Meltzer H Y, editor. New York: Raven; 1987. pp. 513–526. [Google Scholar]
- 5.Sette M, Briley M S, Langer S Z. J Neurochem. 1983;40:622–628. doi: 10.1111/j.1471-4159.1983.tb08026.x. [DOI] [PubMed] [Google Scholar]
- 6.Segonzac A, Raisman T, Tateishi T, Schoemaker H, Hicks P E, Langer S Z. J Neurochem. 1985;44:349–356. doi: 10.1111/j.1471-4159.1985.tb05423.x. [DOI] [PubMed] [Google Scholar]
- 7.Biessen E A L, Norder J A, Horn A S, Robillard G T. Biochem Pharmacol. 1988;37:3959–3966. doi: 10.1016/0006-2952(88)90080-9. [DOI] [PubMed] [Google Scholar]
- 8.Humphreys C J, Levin J, Rudnick G. Mol Pharmacol. 1988;33:657–663. [PubMed] [Google Scholar]
- 9.O’Riordan C, Phillips O M, Williams D C. J Neurochem. 1990;54:1275–1280. doi: 10.1111/j.1471-4159.1990.tb01959.x. [DOI] [PubMed] [Google Scholar]
- 10.Barker E L, Kimmel H L, Blakely R D. Mol Pharmacol. 1994;46:799–807. [PubMed] [Google Scholar]
- 11.Schloss P, Betz H. Biochemistry. 1995;34:12590–12595. doi: 10.1021/bi00039a014. [DOI] [PubMed] [Google Scholar]
- 12.Blakely R D, Berson H E, Fremeau R T, Jr, Caron M G, Peek M M, Prince H K, Bradley C C. Nature (London) 1991;354:66–70. doi: 10.1038/354066a0. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman B J, Mezey E, Brownstein M J. Science. 1991;254:579–580. doi: 10.1126/science.1948036. [DOI] [PubMed] [Google Scholar]
- 14.Mayser W, Betz H, Schloss P. FEBS Lett. 1991;295:203–206. doi: 10.1016/0014-5793(91)81418-8. [DOI] [PubMed] [Google Scholar]
- 15.Ramamoorthy S, Bauman A L, Moore K L, Han H, Yang Feng T, Chang A S, Ganapathy V, Blakely R D. Proc Natl Acad Sci USA. 1993;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corey J L, Quick M W, Davidson N, Lester H A, Guastella J. Proc Natl Acad Sci USA. 1994;91:1188–1192. doi: 10.1073/pnas.91.3.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strader C D, Fong T M, Tota M R, Underwood D. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 18.Kitayama S, Shimada S, Xu H, Markham L, Donovan D M, Uhl G. Proc Natl Acad Sci USA. 1992;89:7782–7785. doi: 10.1073/pnas.89.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langosch D, Herbold A, Schmieden V, Borman J, Kirsch J. FEBS Lett. 1993;336:540–544. doi: 10.1016/0014-5793(93)80872-r. [DOI] [PubMed] [Google Scholar]
- 20.Borman J, Rundström N, Betz H, Langosch D. EMBO J. 1993;12:3729–3737. doi: 10.1002/j.1460-2075.1993.tb06050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schloss P, Mayser W, Betz H. Biochem Biophys Res Commun. 1994;198:637–645. doi: 10.1006/bbrc.1994.1093. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Y C, Prusoff W H. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 23.Sur C, Betz H, Schloss P. Neuroscience. 1996;73:217–231. doi: 10.1016/0306-4522(96)00030-9. [DOI] [PubMed] [Google Scholar]
- 24.Gu H, Wall S C, Rudnick G. J Biol Chem. 1994;269:7124–7130. [PubMed] [Google Scholar]
- 25.Mager S, Min C, Henry D J, Chavkin C, Hoffman B J, Davidson N, Lester H A. Neuron. 1994;12:845–859. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 26.Humphreys C J, Wall S C, Rudnick G. Biochemistry. 1994;33:9118–9125. doi: 10.1021/bi00197a014. [DOI] [PubMed] [Google Scholar]
- 27.Cammack J N, Rakhilin S V, Schwartz E A. Neuron. 1994;13:949–960. doi: 10.1016/0896-6273(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 28.Mager S, Kleinberger-Doron N, Keshet G I, Davidson N, Kanner B I, Lester H A. J Neurosci. 1996;16:5405–5414. doi: 10.1523/JNEUROSCI.16-17-05405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grunewald M, Kanner B I. J Biol Chem. 1995;270:17017–17024. doi: 10.1074/jbc.270.28.17017. [DOI] [PubMed] [Google Scholar]
- 30.Quian Y, Galli A, Ramamoorthy S, Risso S, DeFelice L J, Blakely R D. J Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonders M S, Amara S G. Curr Opin Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- 32.Schloss P, Mayser W, Betz H. FEBS Lett. 1992;307:76–80. doi: 10.1016/0014-5793(92)80905-v. [DOI] [PubMed] [Google Scholar]
- 33.Wood M D. Neuropharmacology. 1987;26:1081–1085. doi: 10.1016/0028-3908(87)90251-6. [DOI] [PubMed] [Google Scholar]