

Abstract
A key unanswered question in smooth muscle biology is whether phosphorylation of the myosin regulatory light chain (RLC) is sufficient for regulation of contraction, or if thin-filament-based regulatory systems also contribute to this process. To address this issue, the endogenous RLC was extracted from single smooth muscle cells and replaced with either a thiophosphorylated RLC or a mutant RLC (T18A/S19A) that cannot be phosphorylated by myosin light chain kinase. The actin-binding protein calponin was also extracted. Following photolysis of caged ATP, cells without calponin that contained a nonphosphorylatable RLC shortened at 30% of the velocity and produced 65% of the isometric force of cells reconstituted with the thiophosphorylated RLC. The contraction of cells reconstituted with nonphosphorylatable RLC was, however, specifically suppressed in cells that contained calponin. These results indicate that calponin is required to maintain cells in a relaxed state, and that in the absence of this inhibition, dephosphorylated cross-bridges can slowly cycle and generate force. These findings thus provide a possible framework for understanding the development of latch contraction, a widely studied but poorly understood feature of smooth muscle.
Extensive biochemical and physiological studies indicate that phosphorylation of Ser-19 of the myosin regulatory light chain (RLC) is both necessary and sufficient for the initiation of smooth muscle cell contraction (1–5). But it does not necessarily follow that the relaxed state in smooth muscle is merely due to the dephosphorylated state of the RLC. The notion that RLC phosphorylation is solely responsible for regulating myosin attachment and cycling has become increasingly untenable as numerous studies have shown that force generation is not a unique function of RLC phosphorylation (5). Following the onset of force development many smooth muscles enter a “latch” state, where high force continues to be produced but RLC phosphorylation and shortening velocity decline (6). These observations represent a long standing challenge to simple ideas about the role of RLC phosphorylation in regulating smooth muscle contraction. Modifications to elementary schemes for cross-bridge cycling in smooth muscle and its regulation by RLC phosphorylation have been introduced to explain the inconstant relationship between force and RLC phosphorylation (5, 7, 8). Many of these schemes require that dephosphorylated cross-bridges can produce force, a feature of smooth muscle that has yet to be directly demonstrated. If dephosphorylated cross-bridges can cycle, then there must be some other regulatory mechanism that determines when dephosphorylated cross-bridges contribute to the force produced by the cell and when they do not. A second regulatory system originating on the actin filament has been postulated to play a role in the regulation of smooth muscle contraction (1, 4, 9). Calponin is one such actin-binding protein that is almost exclusively expressed in smooth muscle (10). Calponin has been suggested as a possible regulator of smooth muscle contraction because it inhibits actomyosin ATPase and slows or blocks actin movement in motility assays (11, 12), most likely by inhibiting a kinetic step (13, 14). The relevance of these data to regulation of contraction in the smooth muscle cell are uncertain, however, since these observations were made on isolated proteins in solution under unphysiological conditions and without the constraints of a fixed filament lattice. Here we performed experiments in smooth muscle cells to assess the properties of dephosphorylated myosin and to determine the role of the thin filament protein calponin in regulation of smooth muscle.
MATERIALS AND METHODS
Preparation, Chemical Skinning, and Extraction of Single Smooth Muscle Cells.
Single smooth muscle cells were isolated by enzymatic treatment of the muscular layer of stomachs from the toad Bufo marinus (15). Previous mechanical studies (16) have shown that smooth muscle cells from the stomach of Bufo marinus exhibit an apparent slowing of cross-bridge cycling during force maintenance similar to other smooth muscles exhibiting latch contraction (6). The cells were skinned with saponin (17) and then kept in a solution containing 5 mM EGTA, 1 mM magnesium methanosulfate, 20 mM pipes, 75 mM potassium methanosulfate, and 5 mM DTT at pH 6.5 (rigor solution) for 20 min. Calponin and the RLC were extracted from the cells by treatment with 1 mM trifluoperazine (TFP) in 5 mM EDTA, 5 mM trans-1,2-diamino-cyclohexane-N,N,N′,N′-tetraacetic acid (CDTA), 10 mM KH2PO4, 150 mM KCl, 10 mM imidazole, 0.005% Triton X-100, and 5 mM DTT at pH 6.5 at 4°C for 20 min. The cells were then washed several times with the rigor solution to remove TFP and then reconstituted with light chain by incubation with 0.3 mg/ml RLC in the rigor solution at 4°C overnight. Unbound RLC was removed and the cells were washed with the rigor solution. RLCs were expressed and purified as described (18). For some experiments, skinned cells reconstituted with RLC were incubated in rigor solution containing 500 μg/ml of calponin at 4°C for 45 min or 200 μg/ml caldesmon for 3 hr at 4°C. Unbound calponin or caldesmon was removed by washing the cells with the rigor solution. Calponin was purified from the muscularis layer from stomachs of Bufo marinus (19) and caldesmon was purified from chicken gizzard (20). In some experiments, native RLC was exchanged with exogenous RLC (300 μg/ml) by incubating cells in an exchange solution (10 mM EDTA/10 mM CDTA/10 mM imidazole/10 mM KCl/5 mM DTT/0.1 mg leupeptin/ml/0.005% Triton X-100, at pH 6.5) at 4°C for 30 min, and then for 2 hr at 40°C. Skinned cells serving as controls were incubated with 300 μg/ml bovine serum albumin under the same conditions. After the exchange procedure, unbound RLC was removed by washing the cells with rigor solution.
Fluorescence Imaging of Reconstituted RLCs.
For determination of the distribution of reincorporated RLC, extracted cells were reconstituted with a rhodamine-labeled RLC and immunolabeled using fluorescent-labeled monoclonal antibody LMM.1 against myosin heavy chain (21). High-resolution three-dimensional images were generated by processing a series of optical sections using a constrained deconvolution algorithm (22).
Measurement of Unloaded Shortening of Single Smooth Muscle Cells.
Cells were incubated in a chamber on an inverted Zeiss IM35 microscope. Brightfield images of the cells were captured with a sampling rate of 1 per sec using a CCD camera (MTI, CCD72) and stored on an optical disc (Panasonic). Digitization of the images and measurement of cell length were performed as described (23). The shortening velocity of each cell was determined by measuring the cell’s length in images taken before and at different times after the initiation of contraction. The initial rate of shortening was constant until the cell was about one-half of its initial length (Li). Shortening slowed after this cell length, probably due to compression of structures that resist shortening. Shortening velocities were determined 3 sec after activation and are given as Li/sec. Photolysis of 2 mM caged ATP (Dojindo Laboratories, Kumamoto, Japan) by illumination with a xenon lamp was used to initiate shortening from rigor. The shortening velocity using 1 or 5 mM caged ATP to initiate shortening was not significantly different from that using 2 mM caged ATP. Skinned cells were maximally activated by thiophosphorylation of the RLC on myosin by depleting the cells of MgATP (1 mM glucose and hexokinase in rigor solution for 30 min), followed by incubation with 1 mM ATPγS and 5 μM calmodulin in the rigor solution containing 30 μM free Ca2+ for 90 min at room temperature. Shortening was initiated by photogeneration of ATP after washing away the solution used to thiophosphorylate the RLC.
Measurement of Isometric Force in Single Smooth Muscle Cells.
For measurement of isometric force development the cells were mounted between a force tranducer and a length driver, and stretched to a passive tension that was about 5% of active force as described previously (24), except that in most experiments cells were glued at each end to stainless steel pins (tip diameter 10 μm) using Great Stuff (Insta-Foam Products, Joliet, IL). After attachment, the glue was allowed to cure for 20 min before contraction was initiated by photolytic release of ATP from 2 mM caged ATP. The photolysis was performed using a xenon arc lamp with a flash duration of less than 1 msec, controlled by a Strobex power pack (Model 236, Chadwick–Helmuth, El Monte, CA). Force was measured using a Cambridge force transducer (model 406, Cambridge Technology, Cambridge, MA).
RESULTS AND DISCUSSION
Characteristics of TFP-Extracted Smooth Muscle Cells.
To explore the contractile properties of cells containing dephosphorylated cross-bridges, we extracted the endogenous RLC from skinned cells using TFP and replaced it with a mutant RLC (T18A/S19A) that cannot be phosphorylated. In this way, we hoped to avoid uncertainties regarding the state of phosphorylation of endogenous RLC resulting from the presence of myosin light chain kinase in these cells. Calponin was also extracted by this procedure, which allowed us to explore its possible involvement in regulating actomyosin interaction in the organized contractile system. The TFP extraction procedure removes more than 90% of the RLC on myosin and essentially all the calponin in skinned smooth muscle cells (Fig. 1 Upper). While caldesmon was clearly present after TFP extraction (Fig. 1 Upper), a small and variable amount of it was lost in some preparations.
Figure 1.
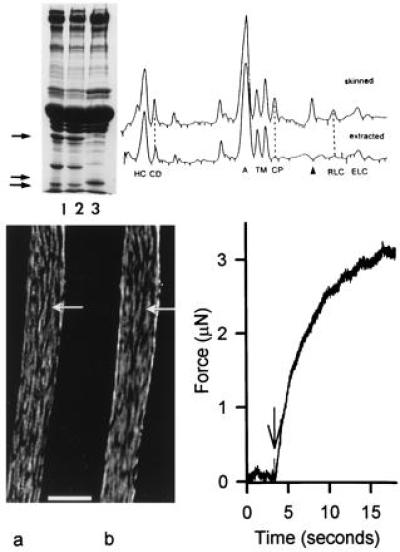
Properties of smooth muscle cells following TFP extraction and readdition of myosin RLCs. (Upper Left) SDS/10–15% PAGE gel of intact toad cells (lane 1), saponin skinned cells (lane 2), and cells treated with TFP and reconstituted with an expressed RLC (lane 3). The TFP treatment extracted more than 90% of the native RLC (lowermost arrow) and calponin (uppermost arrow) from the cells. The reconstituted expressed RLC has a slightly slower mobility than the native RLC due to a “tag” of 4 amino acids at the N terminus (lane 3, middle arrow); the gel lanes were from 3 different cell preparations and are representative of results obtained in over 30 such experiments. The relative amount of proteins present after extraction with TFP was determined by densitometric scanning of SDS/10–15% PAGE gels (Right) of skinned and extracted cells from the same preparation. The ratio of the peak areas (extracted/skinned cells) of known proteins are: myosin heavy chain (HC), 1.04; caldesmon (CD) 1.05; actin (A), 0.96; tropomyosin (TM, two bands), 0.78; calponin (CP), <0.07; RLC, <0.07. Two additional proteins with reduced content in extracted cells are the protein SM-22, which migrates between CP and the RLC, and filamin, which migrates slower than HC. The identity of the RLC, calponin, and caldesmon bands were determined from immunoblots. Immunoblots also indicated that there was no obvious extraction of essential light chains (ELC) with TFP treatment. Moreover, extracted cells reconstituted with RLC alone or with both RLC and ELC gave similar results. (Lower Left) Immunofluorescence image of a single-skinned smooth muscle cell reconstituted with a rhodamine-labeled RLC (a) and reacted with a fluorescent-labeled monoclonal antibody against myosin heavy chain (b). The pattern of myosin distribution in this extracted and reconstituted cell was similar to that in intact cells. (Bar = 5 μm.) (Lower Right) Force record of a cell reconstituted with thiophosphorylated RLC and activated from rigor by photolysis of 2 mM caged ATP. Following illumination with a 100 W Xenon arc lamp (arrow) the muscle develops force after a short delay.
The TFP extraction procedure per se is not deleterious to the cell. When extracted cells are reconstituted with RLC, the amount of RLC is similar both to the content before extraction as well as to that in the intact cell (Fig. 1 Left Upper, lane 3). Immunofluorescence experiments show that the exogenously added RLC colocalizes with myosin in cells where RLC has been extracted. The distribution of fluorescently labeled RLC (Fig. 1 Left Lower, lane a) is indistinguishable from that seen when a monoclonal antibody against myosin heavy chain is used to visualize myosin distribution in the same cell (Fig. 1 Left Lower, lane b). Measurements of isometric force in single cells reveal that after the extraction of endogenous RLC and reconstitution with thiophosphorylated RLCs, the contractile system functions normally. In cells reconstituted with thiophosphorylated RLC, the rate of force development (rate: 0.3 s−1) and the maximum force level (Fig. 1, Right Lower) are similar to that observed for intact electrically stimulated cells (24). The mean peak shortening velocity of cells reconstituted with thiophosphorylated RLC (0.097 Li/sec) was virtually identical to (Fig. 2 and Table 1) values obtained in stimulated intact cells or in skinned cells in which the endogenous RLCs were thiophosphorylated in situ in response to Ca2+ (Table 1, ref. 23). Finally, the myosin light chain kinase (MLCK) activation pathway appeared to be unaltered by the extraction/reconstitution procedure because TFP extracted cells reconstituted with wild-type (WT) RLC could be strongly activated after Ca2+⋅calmodulin-dependent thiophosphorylation.
Figure 2.

Shortening velocity of TFP-extracted smooth muscle cells following reconstitution with different myosin RLC. (A–C) Brightfield images of isolated smooth muscle cells before photolysis of caged ATP and 2, 4, and 6 sec after initiation of shortening. Cells have no RLC in A, but have been reconstituted with T18A/S19A RLC in B or with thiophosphorylated RLC in C. White arrows indicate the starting position of the ends of a typical cell under each condition. (D) Relative cell length as a function of time for cells deficient in calponin. The cells either had no RLC (▾) or were reconstituted with either T18A/S19A RLC (•) or thiophosphorylated RLC (▪). The mean velocity of RLC deficient cells was 0.014 ± 0.002 (n = 12 experiments). In each experiment, these basal velocities were determined and subtracted from the velocities of cells containing mutant RLCs (see text).
Table 1.
Mean relative shortening velocity of cells with varying levels of calponin
| Treatment to cells | Exogenous myosin RLC incorporated
|
|||
|---|---|---|---|---|
| Dephosphorylated WT | T18A/S19A | N smooth/ C skeletal | Thiophosphorylated WT | |
| RLC and calponin extracted with TFP* | 29 ± 1 (4) | 28 ± 2 (9) | 10 ± 3 (2) | 100 (12) |
| RLC exchanged at elevated temperature; calponin native levels† | 1 ± 2 (2) | 0 ± 1 (3) | — | 100 (3) |
| None‡ | — | — | — | 115 ± 19 (3) |
Mean relative maximal shortening velocity of single toad smooth muscle cells from which calponin and native RLC were extracted with TFP. Native RLC was replaced with either dephosphorylated WT RLC, thiophosphorylated WT RLC, a mutant RLC with Thr-18 and Ser-19 mutated to alanine (T18A/S19A), or a chimeric RLC composed of the N terminal from smooth muscle RLC and the C terminal from skeletal muscle RLC (N smooth/C skeletal). Neither dephosphorylated WT RLC, T18S/S19A RLC, or the N smooth/C skeletal support in vitro motility. Velocities are reported as mean ± SEM (10–28 cells per experiment) and are given in percent relative to the velocity of cells replaced with the thiophosphorylated WT RLC after subtraction of the basal activity of extracted cells without RLC. Mean velocity of cells reconstituted with the thiophosphorylated WT RLC without calponin was 0.097 ± 0.006 Li/sec (see Fig. 2 for details). Numbers in parentheses refer to the number of independent experiments.
The mean relative shortening velocity of cells from which calponin was not extracted but the native RLC was exchanged with mutant RLCs. Calponin content was 50–80% of control levels. Basal velocity was 0.012 ± 0.004 Li/sec and 0.010 ± 0.003 Li/sec (n = 3 and 4, respectively) for cells incubated in the exchange buffer at 4°C and in untreated skinned cells, respectively. Velocities are reported as mean ± SEM (10–26 cells per experiment) and are given in percent relative to the velocity of cells replaced with the thiophosphorylated WT RLC after subtraction of the basal activity of control cells incubated with 300 μg/ml albumin (0.022 ± 0.002 Li/sec, n = 3). Mean velocity of cells with the thiophosphorylated WT was 0.089 ± 0.012 Li/sec.
Shortening velocity of untreated skinned cells that were maximally activated by thiophosphorylation of the native RLC. Velocities are given in percent relative to the velocity of cells from the same preparation with thiophosphorylated WT RLCs, but without calponin.
Isotonic Contraction of TFP-Extracted Cells Reconstituted with Different RLCs.
While cells shortened rapidly in response to photorelease of ATP when thiophosphorylated RLC was introduced into the RLC extracted cells, they shortened at very slow velocities (0.014 ± 0.002 Li/sec, n = 12) when no RLCs were added back to the cells (Fig. 2A). This extremely slow shortening of the extracted cells is not surprising since RLC deficient myosin in vitro shows a slow rate of actin movement (25). This very low velocity does not appear to be due to the action of an unregulated MLCK on the small number of endogenous light chains retained after the TFP extraction, since extracted cells shortened when activated by ITP, a substrate for myosin, but not for MLCK (26).
Interestingly, the shortening velocity of cells reconstituted with an unphosphorylatable RLC (T18A/S19A) or dephosphorylated WT RLC was approximately 30% of the velocity of cells with the thiophosphorylated WT RLC (Table 1, Fig. 2). The relatively high shortening velocity of cells reconstituted with T18A/S19A RLC was not affected when MLCK was inhibited by wortmannin or the MLCK autoinhibitory peptide SM-1 (3, 27). Thus, it is unlikely that these cells were activated by phosphorylation of the small amount of native RLC remaining after TFP extraction. Shortening velocity with the nonphosphorylatable RLCs was considerably greater than in cells reconstituted with a control nonfunctional chimeric RLC containing the N-terminal half of the smooth RLC and the C-terminal half of the skeletal muscle myosin RLC (18) (N smooth/C skeletal, Table 1, P < 0.01, Student’s t test for unpaired data). These results suggest that unphosphorylated myosin can support significant rates of cell shortening, albeit at rates considerably less than in cells containing thiophosphorylated RLC.
Effect of Calponin on Isotonic Contraction.
How can the apparent ability of dephosphorylated RLC to support significant rates of shortening in these extracted cells be reconciled with the fact that intact smooth muscle cells do not shorten until the RLC is phosphorylated? A clear difference between the TFP-extracted cells and intact cells is the absence of calponin in the extracted cells. To investigate if calponin could restore more complete phosphorylation-dependent regulation to these cells, calponin was readded to extracted cells reconstituted with the thiophosphorylated or T18A/S19A RLC. The amount of bound calponin in these cells was somewhat higher than the amount present in intact or skinned cells (Fig. 3 Upper Left). The excess of calponin in these cells (as much as two times native levels) is presumably either bound to other proteins or is further saturating the actin filaments. Under these conditions, the velocity of cells with thiophosphorylated RLC remained high when calponin was added back, whereas the shortening of cells with the T18A/S19A RLC was completely inhibited (Fig. 3 Upper Right).
Figure 3.
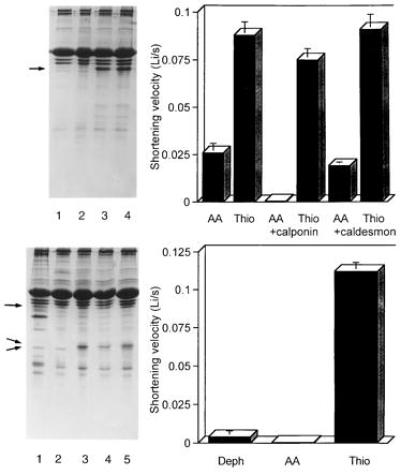
Effects of calponin on the shortening velocity of TFP-extracted skinned smooth muscle cells. (Upper Left) SDS/gel showing readdition of calponin (lanes 3 and 4) to TFP-treated cells from which calponin has been extracted (lanes 1 and 2). The T18A/S19A RLC (lanes 1 and 3) or thiophosphorylated RLC (lanes 2 and 4) was also added to the cells. Less homogenate was loaded than in Fig. 1 to ensure that the calponin band (arrow) could be clearly resolved and was not obscured by the actin band. (Upper Right) Effects of added calponin or caldesmon on the shortening velocity of cells with thiophosphorylated WT (thio) or T18A/S19A RLC (AA). Calponin reduced the velocity of cells containing T18A/S19A from 0.026 ± 0.005 Li/sec to essentially zero, since cell length was unaltered 3 sec after the illumination and after 1 min the cell length was 0.93 ± 0.02 of the initial length (n = 11–15 cells). Only one bar is shown for the controls since the velocities of the controls were similar for the experiments where calponin versus caldesmon was added. (Lower Left) SDS/gel showing skinned cells (lane 1), control cells incubated with BSA (lane 2), cells incubated with T18A/S19A RLC (lane 3), dephosphorylated RLC (lane 4), or thiophosphorylated RLC (lane 5). The amount of exogenous RLC exchanged into the myosin was about 90% (middle arrow); the lowermost arrow denotes the position of the endogenous RLC. The amount of calponin (uppermost arrow) in the cells following LC exchange was 70% that of control skinned cells as determined by densitometry. (Lower Right) Mean shortening velocity of cells in the presence of native levels of calponin after exchange of endogenous RLC with dephosphorylated RLC, T18A/S19A RLC, or thiophosphorylated RLC. See Table 1 for a summary of several such experiments.
Additional studies were carried out to determine if the variable decrease in caldesmon content or the loss of calmodulin that accompanied extraction of calponin contributed to the shortening capacity of unphosphorylatable cross-bridges. Addition of caldesmon to supernormal cellular levels (i.e., to more than 10 times that present in skinned cells) did not significantly affect the shortening velocity of cells containing either T18A/S19A or thiophosphorylated RLC (Fig. 3 Upper Right). Addition of calmodulin also did not affect the contraction of cells reconstituted with T18A/S19A or thiophosphorylated RLC. Two other proteins, most likely SM-22 and filamin, are partially extracted during TFP treatment. It is quite unlikely that the loss of SM-22 or filamin accounts for the ability of nonphosphorylated RLC to support shortening or force generation. This follows from prior results showing that smooth muscle cells that have lost SM-22, but not calponin, cannot contract unless the RLC is phosphorylated (17), and that filamin has no affect on actin-activated myosin ATPase under physiological conditions (28). As typified by the experiments in Fig. 3 and Table 1, the ability of myosin-containing nonphosphorylated RLC to support shortening is specifically inhibited following reconstitution with calponin. These findings suggest that in the intact cell calponin suppresses cross-bridge cycling by unphosphorylated myosin.
Since nonspecific binding of excess calponin to the thin filaments of skinned smooth muscle can inhibit shortening velocity and force (29, 30), we sought to assess the regulatory properties of T18A/S19A RLC and dephosphorylated WT RLC without perturbing the endogenous calponin. This was achieved by exchanging RLCs at elevated temperature in the absence of divalent cations, similar to procedures used for skinned striated muscle or in solution with purified myosin (31). The exchange procedure caused more than 90% replacement of the native RLC with only a small effect on the calponin content of the cells (Fig. 3 Lower Left). The contractile properties of cells exchanged with thiophosphorylated RLC are similar to those of cells from which the RLC was extracted and then reconstituted with thiophosphorylated RLC (Fig. 3 Lower Right, Table 1). In contrast to the cells lacking calponin, the shortening velocity of cells with native amounts of calponin and the T18A/S19A RLC was now low (Fig. 3 Lower Right, Table 1). In one experiment where cells were kept in the “exchange solution” for much longer periods than used for the experiment in Fig. 3, all calponin was lost and those cells contracted when unphosphorylatable RLC was exchanged for the native RLC. The results of these exchange experiments indicate that when calponin is present in physiological concentrations it inhibits slow cycling of unphosphorylated cross-bridges, whereas its effects on thiophosphorylated myosin are minor.
Isometric Contractile Properties of Cells: Effects of Calponin and RLCs.
The conclusions from the studies described thus far are based on measurements of shortening velocity, and while telling us that cross-bridges containing unphosphorylated RLC can cycle in the absence of calponin, they do not tell us how such cross-bridges might contribute to force production. Experiments were therefore carried out to measure the isometric force of single cells containing different RLCs and thin-filament regulatory proteins. Single cells were stretched slightly, thereby producing a small amount of rigor force prior to photogeneration of ATP (Fig. 4, arrows). When MLCK was inhibited with wortmannin in cells that were skinned but not extracted, isometric force dropped to zero upon photorelease of ATP and did not subsequently increase in the cell shown in Fig. 4A or in two other similar cells. When extracted cells were challenged with ATP, force initially decreased and then slowly began to increase slightly (Fig. 4B). The small amount of steady-state force produced by the extracted cells is probably due to the cycling of a small number of RLC containing cross-bridges that remained after extraction, since in one preparation from which all the native RLC was removed no force was produced following photorelease of ATP. Cells without calponin but reconstituted with T18A/S19A RLC produced, on average, about 65% of the force of cells reconstituted with thiophosphorylated RLC (Fig. 4 C and D, Table 1). The response of cells reconstituted with T18A/S19A was not affected when MLCK was inhibited with wortmannin indicating that the cells are not cooperatively activated by phosphorylation of the small amount of native RLC remaining in some of the cells. As typified by the records in Fig. 4 C and D, isometric force increased about 4-fold more slowly in cells containing nonphosphorylatable RLC compared with phosphorylated RLC. Cells reconstituted with both calponin and T18A/S19A RLC produced almost no force (n = 4) (Fig. 4E), similar to skinned cells in the presence of wortmannin where calponin was not removed and endogenous RLCs were not phosphorylated (Fig. 4A). Reconstitution of calponin in extracted cells had no significant effect on force produced by cells containing thiophosphorylated RLC; such cells produced 124 ± 21% (n = 5) of the force produced by cells from the same preparation reconstituted with thiophosphorylated RLC alone. Addition of caldesmon to supernormal cellular levels had no significant effect on the relatively high force production characteristic of T18A/S19A reconstituted cells. Three cells exposed to caldesmon at 200 μg/ml and reconstituted with T18A/S19A produced on average 72 ± 16% of the force of three other cells reconstituted with thiophosphorylated WT RLC, comparable to cells reconstituted with T18A/S19A in the absence of added caldesmon or calponin. These results indicate that cross-bridges containing unphosphorylated RLC can produce significant force and that this interaction is inhibited by calponin either directly, or indirectly as part of some regulatory complex.
Figure 4.
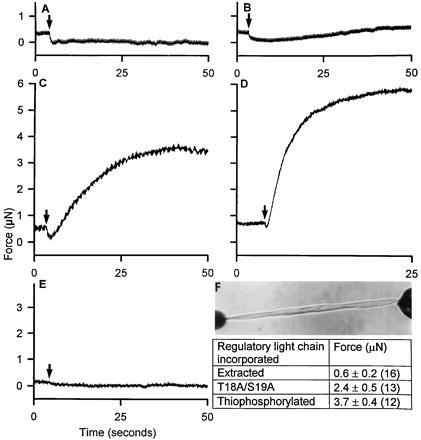
Isometric force generation by single-skinned smooth muscle cells containing different myosin RLCs with and without calponin. (F) Shows a cell, reconstituted with the thiophosphorylated WT RLC, mounted for measurement of isometric force. The force records are from a skinned cell activated in the presence of 100 μM wortmannin (A), from a TFP-extracted cell deficient in RLC and calponin (B), from TFP-extracted cells containing T18A/S19A RLC (C) or the thiophosphorylated WT RLC (D), and from a TFP-extracted cell incubated in 100 μg/ml calponin containing T18A/S19A (E). Numbers in parentheses refer to the number of cells.
Interpretation of Results.
While the ability of nonphosphorylated cross-bridges to produce force has long been predicted from physiological measurements on intact smooth muscle (2, 6, 7), the current results seem at first difficult to reconcile with many (32–34), but not all (35, 36), biochemical experiments on purified smooth muscle myosin that have concluded that dephosphorylated myosin does not cycle. Recent mutational studies (37) on expressed smooth muscle subfragments suggest a resolution of this apparent paradox. It is becoming increasingly clear that for myosin to attain a completely inhibited state solely by dephosphorylation of the RLC, a number of interactions involving multiple domains of the molecule must occur. If any of these interactions are prohibited, then dephosphorylated myosin can show a range of activities, up to 50% of that seen with phosphorylated myosin (37). Within the cell, it is not difficult to envision that either the structure of the native filament itself or accessory proteins that bind near the head/rod junction (e.g., telokin) could restrict the necessary intramolecular interactions in myosin. This could cause partial activation of dephosphorylated myosin, thus allowing dephosphorylated cross-bridges to produce force and shorten at significant rates. Similarly, effects on intramolecular interactions of smooth muscle myosin may well explain why in the presence of high concentrations of MgCl2 actin-activated ATPase is 40% of the rate following RLC phosphorylation (35) and why under strong reducing conditions the rate of sliding of actin filaments in in vitro motility assays may be 20% of that with myosin where RLC is phosphorylated (36). While differences in intramolecular interactions may well explain why purified myosin behaves differently from that in the cell, another possibility is that because the intracellular actin concentration is much higher than can be achieved in vitro actin-myosin interactions involving low actin affinity states may be uniquely expressed in the cell. The existence of such low affinity states has been suggested (38) to explain differences in smooth muscle myosin activity in solution versus in vitro motility assays. While the explanation for the observed differences will have to await further study, the current results clearly indicate that within the cell dephosphorylated smooth muscle myosin can support both shortening and significant force production.
The observation that calponin inhibits the ability of nonphosphorylated myosin to support shortening and force production is consistent with previous biochemical observations indicating that calponin inhibits actomyosin ATPase and slows or blocks sliding of actin filaments on myosin in motility assays (1, 11, 12). While one recent immunolocalization study failed to find calponin on thin filaments in close proximity to myosin (39), at least two others have shown that calponin is found in regions of smooth muscle cells thought to contain molecules characteristic of the contractile domain of the smooth muscle cells (19, 40). We have recently found, using high-resolution fluorescence energy transfer imaging, that calponin (and actin and tropomyosin) containing fibrils are closely apposed to regions containing myosin (U.M., J.C., and F.S.F., unpublished observations). Hence, calponin is indeed present in regions of the smooth muscle cells used in this study, where it could act to regulate the interaction of dephosphorylated myosin and actin.
A model for the regulation of smooth muscle contraction in the intact cell emerges from these studies. In the resting state, slow cycling of dephosphorylated cross-bridges is inhibited by calponin bound to the thin filament. During activation with Ca2+, the attachment of strongly bound phosphorylated cross-bridges could directly or via tropomyosin override calponin inhibition and turn on the actin filaments, thus allowing attachment and cycling of unphosphorylated cross-bridges. In addition, or perhaps alternatively, the inhibitory effect of calponin could be reversed by Ca2+/calmodulin or Ca2+/caltropin or by phosphorylation of calponin (14, 41, 42). Activation of any of these pathways that cancel the inhibitory action of calponin could in principle allow dephosphorylated cross-bridges to manifest their ability to slowly cycle. Such slowly cycling dephosphorylated cross-bridges are essentially the “latch” bridges proposed by Murphy and colleagues (5, 6). In contrast, however, to the suggestion by Murphy and colleagues that latch bridges uniquely arise when attached phosphorylated myosin is dephosphorylated, these results indicate that slow cycling high force attachments between actin and myosin can occur without prior RLC phosphorylation. These results indicating the involvement of a second regulatory system, in conjunction with the primary regulation by light chain phosphorylation, could explain how dephosphorylated cross-bridges could be noncycling in the relaxed state, but slowly cycle when the cells are activated.
Acknowledgments
This article is dedicated to the memory of our friend and colleague, Fredric S. Fay, who died unexpectedly on March 18, 1997. We gratefully acknowledge Dr. M. Walsh for providing anti-calponin and purified calponin, Dr. G. Kargacin for advice on skinning procedures, Dr. M. Ikebe for providing caldesmon, and Drs. S. Lowey and M. Ikebe for helpful conversations throughout this work. This research was supported by grants from the Swedish Medical Research Council (U.M.), the Wenner–Gren Center Foundation (U.M.), the Swedish Society of Medicine (U.M.), the National Institutes of Health (K.M.T. and F.S.F.), the National Science Foundation (F.S.F.), and the American Heart Association (K.M.T.).
ABBREVIATIONS
- RLC
regulatory light chain
- TFP
trifluoperazine
- WT
wild type
- Li
initial length
- MLCK
myosin light chain kinase
References
- 1.Winder S J, Walsh M P. Cell Signaling. 1993;5:677–686. doi: 10.1016/0898-6568(93)90029-l. [DOI] [PubMed] [Google Scholar]
- 2.Somlyo A P, Somlyo A V. Nature (London) 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 3.Itoh T, Ikebe M, Kargacin T J, Hartshorne D J, Kemp B E, Fay F S. Nature (London) 1989;338:164–167. doi: 10.1038/338164a0. [DOI] [PubMed] [Google Scholar]
- 4.Stull J T, Gallagher P J, Herring B P, Kamm K E. Hypertension. 1991;17:723–732. doi: 10.1161/01.hyp.17.6.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hai C M, Murphy R A. Annu Rev Physiol. 1989;51:285–298. doi: 10.1146/annurev.ph.51.030189.001441. [DOI] [PubMed] [Google Scholar]
- 6.Dillon P F, Aksoy M O, Driska S P, Murphy R A. Science. 1981;211:495–497. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- 7.Somlyo A V, Goldman Y E, Fujimori T, Bond M, Trentham D R, Somlyo A P. J Gen Physiol. 1988;91:165–192. doi: 10.1085/jgp.91.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vyas T B, Mooers S R, Barayan N J, Witherell J C, Siegman M J, Butler T M. Am J Physiol. 1992;263:C210–C219. doi: 10.1152/ajpcell.1992.263.1.C210. [DOI] [PubMed] [Google Scholar]
- 9.Marston S B, Trevett R, Walters M. Biochem J. 1980;185:355–365. doi: 10.1042/bj1850355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi K, Hiwada K, Kokubu T. Biochem Biophys Res Commun. 1986;141:20–26. doi: 10.1016/s0006-291x(86)80328-x. [DOI] [PubMed] [Google Scholar]
- 11.Shirinsky V P, Biryukuv K G, Hettasch J M, Sellers J S. J Biol Chem. 1992;267:15886–15892. [PubMed] [Google Scholar]
- 12.Haeberle J R. J Biol Chem. 1994;269:12424–12431. [PubMed] [Google Scholar]
- 13.Horiuchi K Y, Chacko S. Biochem Biophys Res Commun. 1991;176:1487–1493. doi: 10.1016/0006-291x(91)90455-g. [DOI] [PubMed] [Google Scholar]
- 14.Miki M, Walsh M P, Hartshorne D J. Biochem Biophys Res Commun. 1992;187:867–871. doi: 10.1016/0006-291x(92)91277-w. [DOI] [PubMed] [Google Scholar]
- 15.Fay F S, Hoffman R, LeClaire S, Merriam P. Methods Enzymol. 1982;85:284–292. doi: 10.1016/0076-6879(82)85027-1. [DOI] [PubMed] [Google Scholar]
- 16.Warshaw D M, Rees D D, Fay F S. J Gen Physiol. 1988;91:761–779. doi: 10.1085/jgp.91.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kargacin G J, Fay F S. J Gen Physiol. 1987;90:49–73. doi: 10.1085/jgp.90.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trybus K M, Chatman T A. J Biol Chem. 1993;267:4412–4419. [PubMed] [Google Scholar]
- 19.Walsh M P, Carmichael J D, Kargacin G J. Am J Physiol. 1993;265:1371–1378. doi: 10.1152/ajpcell.1993.265.5.C1371. [DOI] [PubMed] [Google Scholar]
- 20.Katayama E, Horiuchi K Y, Chacko S L. Biochem Biophys Res Commun. 1989;160:1316–1322. doi: 10.1016/s0006-291x(89)80147-0. [DOI] [PubMed] [Google Scholar]
- 21.Trybus K M, Henry J. J Cell Biol. 1989;109:2879–2886. doi: 10.1083/jcb.109.6.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrington W A, Lynch R M, Moore E D W, Isenberg G, Fogarty K E, Fay F S. Science. 1995;268:1483–1487. doi: 10.1126/science.7770772. [DOI] [PubMed] [Google Scholar]
- 23.Kargacin G J, Ikebe M, Fay F S. Am J Physiol. 1990;259:C315–C324. doi: 10.1152/ajpcell.1990.259.2.C315. [DOI] [PubMed] [Google Scholar]
- 24.Warshaw D M, Fay F S. J Gen Physiol. 1983;82:157–199. doi: 10.1085/jgp.82.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trybus K M, Waller G S, Chatman T A. J Cell Biol. 1994;124:963–969. doi: 10.1083/jcb.124.6.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cassidy P, Kerrick W G L. Biochim Biophys Acta. 1982;705:63–69. doi: 10.1016/0167-4838(82)90336-3. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi S, Kakita S L, Takahashi I, Kawahara K, Tsukuda E, Sano T, Yamada K, Kase H, Matsuda Y, Hashimoto Y, Nonomura Y. J Biol Chem. 1992;267:2157–2163. [PubMed] [Google Scholar]
- 28.Dabrowska R, Goch A, Osinska H, Szpacenko A, Sosinski J. J Muscle Res Cell Motil. 1985;6:29–42. doi: 10.1007/BF00712309. [DOI] [PubMed] [Google Scholar]
- 29.Itoh T, Suzuki S, Suzuki A, Nakamura F, Naka M, Tanaka T P. Eur J Physiol. 1994;427:301–308. doi: 10.1007/BF00374538. [DOI] [PubMed] [Google Scholar]
- 30.Jaworowski Å, Anderson K I, Arner A, Engström M, Gimona M, Strasser P, Small J V. FEBS Lett. 1995;365:167–171. doi: 10.1016/0014-5793(95)00451-e. [DOI] [PubMed] [Google Scholar]
- 31.Moss R L. Circ Res. 1992;70:865–884. doi: 10.1161/01.res.70.5.865. [DOI] [PubMed] [Google Scholar]
- 32.Cross R A, Cross K E, Sobieszek A. EMBO J. 1986;5:2637–2641. doi: 10.1002/j.1460-2075.1986.tb04545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sellers J R. J Biol Chem. 1985;260:15815–15819. [PubMed] [Google Scholar]
- 34.Trybus K M. J Cell Biol. 1989;109:2887–2894. doi: 10.1083/jcb.109.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner P D, Vu N D. J Biol Chem. 1987;262:15556–15562. [PubMed] [Google Scholar]
- 36.Haeberle J R, Hemric M E. Biophys J. 1995;68:306S–310S. [PMC free article] [PubMed] [Google Scholar]
- 37.Trybus K M, Freyzon Y, Faust L Z, Sweeney H L. Proc Natl Acad Sci USA. 1997;94:48–52. doi: 10.1073/pnas.94.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bresnick A R, Wolff-Long V L, Bauman O, Pollard T D. Biochemistry. 1995;34:12576–12583. doi: 10.1021/bi00039a012. [DOI] [PubMed] [Google Scholar]
- 39.Mabuchi K, Li Y, Tao T, Wang C L A. J Muscle Res Cell Motil. 1996;17:243–260. doi: 10.1007/BF00124246. [DOI] [PubMed] [Google Scholar]
- 40.North A J, Gimona M, Cross R A, Small J V. J Cell Sci. 1994;104:437–444. doi: 10.1242/jcs.107.3.437. [DOI] [PubMed] [Google Scholar]
- 41.Wills F L, McCubbin W D, Kay C M. Biochemistry. 1994;33:5562–5569. doi: 10.1021/bi00184a027. [DOI] [PubMed] [Google Scholar]
- 42.Winder S J, Walsh M P. J Biol Chem. 1990;265:10148–10155. [PubMed] [Google Scholar]