

Abstract
Global use of erythropoietin (EPO) continues to increase as a proven agent for the treatment of anemia. Yet, EPO is no longer believed to have exclusive biological activity in the hematopoietic system and is now considered applicable for a variety of disorders such as diabetes, Alzheimer’s disease, and cardiovascular disease. Treatment with EPO is considered to be robust and can prevent metabolic compromise, neuronal and vascular degeneration, and inflammatory cell activation. On the converse side, observations that EPO administration is not without risk have fueled controversy. Here we present recent advances that have elucidated a number of novel cellular pathways governed by EPO to open new therapeutic avenues for this agent and avert its potential deleterious effects.
Keywords: angiogenesis, cardiac, diabetes, erythropoietin, neurodegeneration
1. Historical Background for Erythropoietin
Initially termed “hemopoietine”, erythropoietin (EPO) became evident as a factor that could stimulate new red blood cell development through the pioneering studies of Carnot and Deflandre in 1906 [1]. This team of investigators demonstrated that plasma removed from rabbits following a bleeding stimulus that was later injected into control untreated rabbits would lead to the development of immature red blood cells, or reticulocytosis. A number of other investigators followed these studies that demonstrated similar findings that plasma from animals that were bled would yield a significant reticulocytosis. Interestingly, more elegant experiments later demonstrated that a rise in hemoglobin levels with reticulocytosis occurred in parabiotic rats when only one partner was exposed to hypoxia, illustrating that depressed oxygen tensions could stimulate EPO production. Eventually, human EPO protein was purified that paved the way for the cloning of the EPO gene and the development of recombinant EPO for clinical use [2, 3].
2. Structural and Molecular Determinants of Erythropoietin Activity
EPO is a 30.4 kDa glycoprotein with approximately half of its molecular weight derived from carbohydrates that can vary among species [3]. EPO contains four glycosylated chains including three N-linked and one O-linked acidic oligosaccharide side chains. The glycosylated chains are important for the biological activity of EPO and can protect EPO from oxygen radical degradation. The presence of the carbohydrates also are important in the control of the metabolism of EPO, since EPO molecules with high sialic acid content can be easily cleared by the body through specific binding in the liver. In addition, the biological activity of EPO also relies upon two disulfide bonds formed between cysteines at positions 7 and 160 and at positions 29 and 33 [2].
The production and secretion of the mature EPO also relies upon the integrity of the N- and O-linked chains. The EPO gene is located on chromosome 7, exists as a single copy in a 5.4 kb region of the genomic DNA, and encodes a polypeptide chain containing 193 amino acids. During the production and secretion of EPO, a 166 amino acid peptide is initially generated following the cleavage of a 27 amino acid hydrophobic secretory leader at the amino-terminal. In addition, a carboxy-terminal arginine in position 166 is removed both in the mature human and recombinant human EPO (rhEPO) resulting in a circulatory mature protein of 165 amino acids [2, 3].
3. Cellular Expression and Signaling for Erythropoietin and its Receptor
EPO is considered to be ubiquitous in the body, since this trophic factor can be detected in the breath of healthy individuals [4, 5]. In addition, it has been suggested that EPO may provide developmental cognitive support in humans with the recent observation that elevated EPO concentrations during infant maturation have been correlated with increased Mental Development Index scores [6]. The primary organs of EPO production and secretion are the kidney, liver, brain, and uterus (Table 1). EPO production and secretion occurs foremost in the kidney [7]. Secondary sites of EPO production and secretion involve the liver and the uterus. Hepatocytes, hepatoma cells, and Kupffer cells of the liver can produce EPO and, in turn, EPO may provide a protective environment for these cells [8]. In regards to the uterine production of EPO, it is believed that the hypoproliferative neonatal anemia that invariably occurs in the early weeks after birth may partly result from the loss of EPO production and secretion by the placenta.
Table 1.
Tissue Sites of Erythropoietin (EPO) and its Biological Functions
| EPO Production and Expression Sites | Biological Function |
|---|---|
|
| |
| Kidney | Erythropoiesis |
| Liver | Protection and Erythropoiesis |
| Kupfer cells | |
| Hepatocytes | |
| Uterus | Proliferation |
| Brain | |
| Neurons | Neuroprotection |
| Astrocytes | Neuroprotection |
| Microvascular endothelial cells | Protection and proliferation |
| Peripheral Endothelial cells | Angiogenesis and migration |
| Muscle cells | Proliferation |
| Skeletal | |
| Smooth | |
| Cardiac | |
Further work has revealed several other organs as secretory tissues for EPO that include peripheral endothelial cells (ECs), muscle, and insulin-producing cells [2]. However, it is the discovery of EPO and its receptor in the nervous and vascular systems that has resulted in a heightened level of interest and enthusiasm for the potential clinical applications of EPO, such as in Alzheimer’s disease and cardiac insufficiency [9, 10]. In the nervous system, the major sites of EPO production and secretion are in the hippocampus, internal capsule, cortex, midbrain, cerebral ECs, and astrocytes [2, 3]. Additional sites in the vascular system also continue to surface as secretory tissues for EPO that include peripheral ECs, enterocytes, and skeletal, smooth, and cardiac muscle [3, 7].
EPO must bind to a target cell surface receptor to bring into play its biological function. Once the EPO gene was cloned, work was initiated to identify a receptor for EPO. EPO regulates bone marrow erythroid cell proliferation, differentiation, and survival through its binding to an erythroid progenitor cell surface EPO receptor (EPOR). The EPOR also is expressed in numerous non-erythroid blood lines that include neurons, microglia, astrocytes, and in cerebral ECs as well as on myelin sheaths of radicular nerves in human peripheral nerves [2, 3, 7], suggesting both a developmental and potential protective role for EPO not only in the central nervous system, but also in disease entities that involve the peripheral nervous system.
During the development of an organism, production of EPO and the expression of its receptor are altered. Elevated expression of the EPOR occurs in early embryonic neuronal tissues at levels similar to that observed in the adult spleen and bone marrow. However, the level of endogenous EPOR expression is significantly reduced following the maturation of the brain. During gestation, EPO production is increased, but later becomes suppressed following birth to be regulated by the tissue oxygen supply. A deficiency in tissue oxygen results in the production of EPO and an increase in the expression of the EPOR not only in peripheral organs [2, 3, 7], but also in the brain [11] that may be responsible for hypoxic tolerance in some species [12]. EPO secretion in the brain appears to be more sustained than in peripheral organs such as the kidney, suggesting that EPO production may originate in the brain and possibly cross the blood-brain barrier to reach the blood and peripheral organs [2, 3]. Furthermore, both primary neurons and neuronal cell lines have been found to retain the capacity to express EPO in an oxygen-dependent manner [4].
Although EPO is recognized as a critical modulator of erythropoiesis, a low concentration of red blood cells alone does not directly stimulate EPO production, but requires the presence of a diminished oxygen tension. Once a hypoxic stimulus is received, EPO is subsequently released into the peripheral blood circulation and upon arrival in the bone marrow, EPO binds to its receptor expressed on the surface of erythroid progenitor cells and leads to erythropoiesis. This results in an elevation in the number of mature erythrocytes and the improvement of oxygen supply. EPO also functions to stimulate colony-forming erythroid cells to induce these cells to proliferate, mature into erythrocytes, and possibly assist with reticulocyte release to the blood [13].
In most tissues including the brain, hypoxia-dependent expression of EPO and EPOR are controlled by hypoxia-inducible factor 1 (HIF-1). HIF-1 is essential for the production and secretion of EPO in response to hypoxia. At the transcriptional level, the hypoxia- dependent gene transcription of EPO and EPOR directly results from the activation of the HIF-1 pathway under hypoxic conditions. Gene transcription of EPO is mediated by the transcription enhancer located in the 3′-flanking region of the EPO gene that specifically binds to HIF-1 [2, 3].
HIF-1 is a basic helix-loop-helix heterodimeric transcription factor containing two subunits, HIF-1α and HIF-1β. HIF-1β is a constitutively expressed 91–94 kDa subunit that was characterized previously as aryl hydrocarbon receptor nuclear translocator (ARNT). HIF-1α is a 120 kDa oxygen-labile subunit that undergoes rapid degradation via the ubiquitin-proteasome pathway under normoxic conditions. Upon hypoxic exposure, degradation of HIF-1α is impaired by blocking its association with von Hippel-Lindau protein that targets HIF-1α for proteasome destruction. HIF-1α subsequently translocates to the nucleus and heterodimerizes with HIF-1β to form a stable HIF-1 complex. The HIF complex binds to the conserved sequence (5′RCGTG3′) near the 5′ end of the hypoxia-responsive enhancer of the EPO gene to up-regulate EPO gene transcription. It is important to note that each of the HIF family members HIF-1α, HIF-1β, and HIF-3α play important roles in regulating the expression of EPO and the EPOR to foster protection against hypoxic cell injury [2, 3].
Hypoxia is not the only factor responsible for the expression of EPO and the EPOR. The production and secretion of EPO in female reproductive organs is estrogen-dependent. Administration of 17β-estradiol, which controls the cyclic development of the uterine endometrium, can lead to a rapid and transient increase in EPO mRNA in the uterus. Hypoxic induced EPO mRNA expression in uterine tissue occurs only in the presence of 17β-estradiol. Oviduct and ovary production of EPO is also 17β-estradiol dependent. Interestingly, a variety of stimulants may lead to increased EPO expression through activation of HIF, such as hypoglycemia, raised intracellular calcium, or intense neuronal depolarizations generated by mitochondrial reactive oxygen species. Anemic stress, insulin release, and several cytokines, including insulin-like growth factor, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), also can lead to increased expression of EPO and the EPOR [2, 3].
4. Erythropoietin during Cellular Metabolism, Function, and Injury
4.1 Diabetes and EPO
Diabetes mellitus (DM) is found in at least 16 million individuals in the United States and more than 165 million individuals worldwide [5]. Furthermore, by the year 2030, it is predicted that more than 360 million individuals will be afflicted with DM and its debilitating conditions [4]. Type 2 DM represents at least 80 percent of all diabetics and is dramatically increasing in incidence as a result of changes in human behavior and increased body mass index. Type 1 insulin-dependent diabetes mellitus accounts for only 5–10 percent of all diabetics. Both type 1 and type 2 DM represent important health concerns whether they begin early or later in life, since each can result in long-term complications throughout the body. In regards to the vascular and nervous systems, patients with DM can develop severe neurological and vascular disease that can lead to an increased risk for cognitive decline [4, 5]. Disease of the nervous system can become the most debilitating complications for DM and affect sensitive cognitive regions of the brain, such as the hippocampus that modulates memory function, resulting in significant functional impairment and dementia. In a prospective population based study of 6,370 elderly individuals, patients with DM had an approximate double risk for the development of dementia [4, 5]. DM also has been found to increase the risk for vascular dementia in elderly subjects [4, 5].
In clinical studies involving patients with DM, plasma EPO is often low in diabetic individuals whether or not anemia exists [4, 5]. Furthermore, the failure of these individuals to produce EPO in response to a declining hemoglobin level suggests an impaired EPO response in diabetic patients [4, 5]. Yet, increased EPO secretion during diabetic pregnancies may represent the body’s attempt at endogenous protection against the complications of DM [14]. EPO administration has been shown both in diabetics as well as non-diabetics with severe, resistant congestive heart failure to decrease fatigue, increase left ventricular ejection fraction, and significantly decrease the number of hospitalization days [15] (Table 2). In vitro studies with vascular cells exposed to elevated glucose also have elucidated a strong cytoprotective effect of EPO. Administration of EPO can significantly improve EC survival in a 1.0 ng/ml range [16]. Interestingly, EPO administration in patients also can significantly increase plasma levels of EPO well above this range of 1.0 ng/ml, suggesting that the effects of EPO observed during in vitro studies may parallel the cellular processes altered by EPO in patients with DM [6]. Furthermore, EPO can block apoptotic DNA degradation in ECs during elevated glucose similar to other models of oxidative stress in cardiac and vascular cell models [16–19].
Table 2.
Erythropoietin (EPO) Cellular Response in Disease
| Diseases modalities | Cellular response to EPO | References |
|---|---|---|
| Diabetes | ||
| Clinical studies | Complications decreased, cardiac function improved | 15 |
| In vitro elevated high glucose | EC survival increased, apoptotic DNA fragmentation Decreased | 16 |
| Neurodegeneration and inflammatory cell activation | ||
| Hypoxia and Free radical exposure | Neuronal and cerebrovascular EC survival increased, apoptotic DNA fragmentation and PS exposure decreased | 21, 22, 23 |
| Alzheimer’s disease | Aβ toxicity reduced, translocation of NF-κB, neuronal apoptotic injury reduced | 27 |
| Parkinson’s disease | Dopaminergic graft survival, striatal regeneration, and functional recovery enhanced | 28 |
| Neurotrauma | Lesion size decreased, caspase 3 activity and apoptotic DNA fragmentation decreased, neuronal function preserved | 30, 31, 33 |
| Epilepsy | Duration of epilepsy decreased, hippocampal neuron survival following status epileptics increased | 34, 35 |
| Inflammatory disease | Cytokine gene expression decreased, inflammatory function of cytokines inhibited | 2, 3 |
| Microglia | Microglial activation inhibited, microglial integrity preserved | 17, 22, 23, 25 |
| Cardiovascular and renal systems | ||
| Ischemic cardiac disease | Cardiac function improved, cardiomyocyte apoptosis inhibited, cardiac remodeling increased | 18, 19, 44, 45 |
| Renal protection | Podocyte injury induced reduced, actin cytoskeletal reorganization corrected | 43 |
| Angiogenesis | EC apoptosis inhibited, new capillary formation, endothelial progenitor cell mobilization increased | 11, 46 |
Abbreviations: EC: endothelial cell; NF-κB: nuclear factor-κB; NMDA: N-methyl-D-aspartate; PS: phosphatidylserine
4.2 Neurodegeneration, Inflammatory Cell Activation, and EPO
As a robust cytoprotectant, EPO can enhance the survival of cells in the nervous system during toxic insults [2, 3, 20]. In neuronal cells of the brain or retina, EPO can prevent injury from hypoxic ischemia [11, 21–25], excitotoxicity [2, 3], free radical exposure [23, 26], amyloid toxicity [27], and dopaminergic cell injury [28] (Table 2). More specifically, administration of EPO also represents a viable option for the prevention of retinal cell injury during glaucoma [29]. Systemic application of EPO also can improve functional outcome and reduce cell loss during spinal cord injury [30, 31], cerebral edema, [32], cortical trauma [33], and epileptic activity [34, 35].
Yet, of equal importance to the functional preservation of cells is the role of EPO during cellular inflammation. EPO can reduce cytokine gene expression in endothelial cells exposed to TNF-α [3], can decrease ulcer progression in cases of scleroderma [36], and can block primary microglial activation and proliferation during oxidative stress [22, 27]. Furthermore, EPO can inhibit microglial cell activation and proliferation to prevent phagocytosis of injured cells through pathways that involve cellular membrane phosphatidylserine (PS) exposure, protein kinase B [37], and the regulation of caspases [22, 26, 38]. EPO can directly inhibit several pro-inflammatory cytokines, such as IL-6, TNF-α, and monocyte chemoattractant protein 1 [2, 3], as well as reduce leukocyte inflammation [39]. In addition, EPO may foster the preservation of microglial cells for neuronal and vascular restructuring by preventing apoptotic injury in microglia [25].
4.3 Cardiovascular-Renal Protection, Angiogenesis, and EPO
Clinical studies have suggested an additional role for EPO in the cardiovascular system [2, 3] and in the renal system [10]. For example, patients with acute myocardial infarction have increased plasma EPO levels within seven days of the cardiac insult, suggesting a possible protective response from the body [40]. In addition, EPO administration in patients with anemia and congestive heart failure can improve exercise tolerance and renal function [10]. Randomized control studies with EPO administration in patients with congestive heart failure or diabetes combined with congestive heart failure also demonstrate an improved cardiac output and a decrease in medical resource utilization [3, 15]. Closely integrated with cardiac performance, pulmonary function also is believed to be enhanced during EPO administration, especially in the setting of ischemic reperfusion injury of the lung [41]. Serum levels of EPO also may function as a biomarker for cardiovascular injury [42]. At the cellular level, EPO plays a critical role in the vascular and renal systems. EPO can maintain erythrocyte [13] and podocyte [43] integrity, regulate the survival of ECs [21, 26], and function as a powerful endogenous protectant during cardiac injury [44].
It is important to note that as a large molecule, EPO may maintain the establishment of EC communication and function that could become crucial in a number of scenarios, such as repair of the blood-brain barrier during injury [16, 17]. In addition, by assuring EC integrity, EPO prevents ischemic cardiac demise by reducing myocardial injury and cardiomyocyte apoptosis [18, 19, 45], modulating cardiac remodeling [19], and improving cardiac function [18, 45] (Table 2). Overall, EPO can protect against myocardial cell apoptosis and decrease infarct size, resulting in improved left ventricular contractility. In isolated rat heart preparations following ischemia/reperfusion experiments, beneficial effects of treatment with EPO have been shown to significantly improve post-ischemic recovery of left ventricular pressure [2, 3].
Some of the results from experimental studies with EPO have correlated well with a number of positive clinical observations for EPO in cardiac patients. Early clinical studies in patients with anemia or on chronic hemodialysis have indicated that administration of EPO can increase left ventricular ejection fraction, stroke volume, and cardiac output, suggesting improved cardiac function secondary to the correction of anemia [2, 3, 15]. Other clinical randomized control studies in patients with mild anemia and severe or resistant congestive heart failure have demonstrated that EPO in combination with intravenous iron can lead to increased left ventricular ejection fraction and a reduction in hospitalization days by almost eighty percent. In addition to the correction of anemia, EPO can promote microvascular growth in the heart, suggesting that functional cardiac recovery with EPO may ensue also from the generation of new blood vessels [45].
Interestingly, EPO independently leads to angiogenesis for new cell growth such as capillary formation from pre-existing vessels into an avascular area [11, 46]. EPO has both a mitogenic and chemotactic effect that can lead to matrix metalloproteinase-2 production, cell proliferation, and new vessel formation [2, 3]. EPO also can stimulate postnatal neovascularization by increasing endothelial progenitor cell mobilization from the bone marrow. Angiogenesis has been observed in rat aortic rings four days following incubation with EPO in reconstituted basement membrane matrix and in ECs derived from human adult myocardial tissue treated by EPO [2, 3].
5. Erythropoietin and the Oversight of Novel Intrinsic Cellular Pathways
5.1 Oxidative Stress, Apoptosis, and EPO
EPO modulates a variety of signal transduction pathways for cytoprotection that can involve protein kinase B, signal transducer and activator of transcription pathways, forkhead transcription factors, caspases, and nuclear factor κB (Figure 1). Intimately linked to these cell longevity pathways with EPO are the injury mechanisms associated with oxidative stress and apoptosis. Oxidative stress represents a significant mechanism for the destruction of cells that can involve apoptotic cell injury. Apoptotic induced oxidative stress in conjunction with processes of mitochondrial dysfunction can contribute to a variety of disease states such as diabetes, ischemia, general cognitive loss, Alzheimer’s disease, and trauma [47–49]. As an early event in the dynamics of cellular apoptosis, membrane PS externalization can become a signal for the phagocytosis of cells. Cells expressing externalized PS may be removed by microglia [50]. In addition, membrane PS externalization on platelets has been associated with clot formation in the vascular cell system. In contrast to the early externalization of membrane PS residues, the cleavage of genomic DNA into fragments is considered to be a delayed event that occurs late during apoptosis [51, 52]. Several enzymes responsible for DNA degradation have been differentiated and include the acidic, cation independent endonuclease (DNase II), cyclophilins, and the 97 kDa magnesium - dependent endonuclease [47]. Three separate endonuclease activities are present in neurons that include a constitutive acidic cation-independent endonuclease, a constitutive calcium/magnesium-dependent endonuclease, and an inducible magnesium dependent endonuclease. EPO offers a unique opportunity to prevent the exposure of membrane PS residues, inhibit the committed stages of genomic DNA destruction, and the subsequent apoptotic cascades that may involve caspase activation [21–23, 26, 31].
Figure 1. Cytoprotection by erythropoietin (EPO) requires multiple signal transduction pathways.
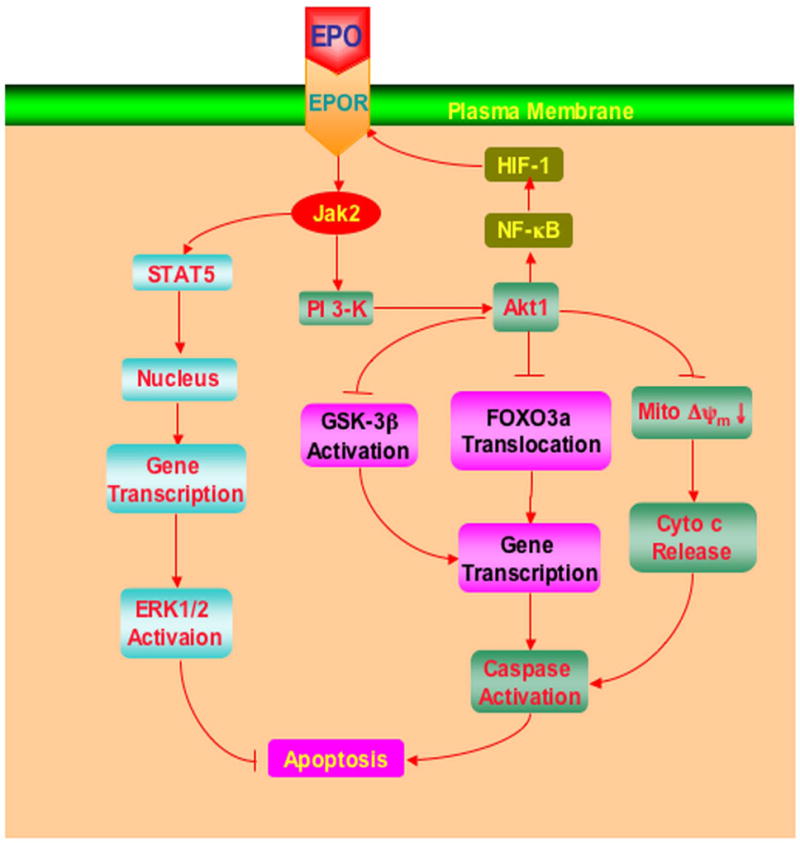
EPO and the EPO receptor (EPOR) can increase cell survival, promote progenitor cell development, and control inflammatory cell activation through pathways that involve the Janus-tyrosine kinase 2 (Jak2) protein, protein kinase B (Akt), and signal transducer and activator of transcription (STAT) proteins. Subsequent downstream signaling governs extracellular signal-related kinases (ERKs), the forkhead family member FOXO3a, glycogen synthase kinase-3β (GSK-3β), and nuclear factor-κB (NF-κB). Intimately linked to the ability of EPO to maintain cellular integrity and prevent inflammatory activation that ultimately can lead to cellular apoptosis are the maintenance of mitochondrial membrane potential (ΔΨm), the release of cytochrome c, (Cyto-c), and the prevention of caspase activation.
5.2 Jak2, STATS, and EPO
Cellular signal transduction with EPO requires the activation of the EPOR which specifically binds to and activates Janus-tyrosine kinase 2 (Jak2) through phosphorylation. Jak2 is a member of a family of Janus-type protein-tyrosine kinases including Jak1, Jak2, Jak3, and Tyk2 that are characterized by a kinase domain in the carboxyl portion, a kinase-like domain, and a large amino-terminal domain. The amino-terminal domain of Jak2 is responsible for the binding of Jak2 with the β-subunit of the EPOR at a region proximal to the membrane that contains the Box 1 sequence. The signal transducer and activator of transcription (STAT) proteins are direct substrates of Janus kinases. Activation of Janus kinases results in tyrosine phosphorylation and dimerization of STATs. Once activated, STATs translocate to the nucleus and bind to specific DNA sequences in the promoter regions of responsive genes to lead to gene transcription. Closely linked to these transcription pathways are the mitogen-activated protein kinases that include the extracellular signal-related kinases (ERKs), the c-Jun-amino terminal kinases, and p38 MAP kinase that can oversee erythroid proliferation and differentiation [3, 47]. Yet, in regards to cytoprotection, EPO has been shown to not only activate STAT 3 [17, 44], STAT 5 [13, 17], and ERK 1/2 [17], but also to employ these pathways for cell development and cell protection. For example, EPO significantly activates STAT3, STAT5, and ERK 1/2 in primary cerebral vascular cells, suggesting that EPO may require these cellular pathways to confer EC cytoprotection during oxidative stress [17].
5.3 PI 3-K, Akt, FOXO3a, and EPO
The ability of EPO to enhance cell survival during injury also directly relies upon the phosphatidylinositol 3-kinase (PI 3-K) pathway through protein kinase B (Akt1). Phosphorylation of Akt leads to its activation and protects against genomic DNA degradation and membrane PS exposure [47]. Up-regulation of Akt activity during injury paradigms, such as vascular and cardiomyocyte ischemia [2, 3], hypoxia [21, 53], β-amyloid toxicity [27], and oxidative stress [37, 51, 52] is vital for EPO protection, since inhibition of Akt activity blocks cellular protection and anti-inflammatory mechanisms by EPO [22, 23, 26]. EPO employs the PI 3-K/Akt pathway to prevent cellular apoptosis through several pathways that involve transcription factor regulation [17], maintenance of mitochondrial membrane potential (ΔΨm), prevention of cytochrome c release, and blockade of caspase activity [21, 22, 26].
Interestingly, a number of novel pathways that may mediate the ability of EPO to prevent cellular apoptosis are intimately tied to Akt. For example, Akt is a central regulatory element for the mammalian forkhead transcription factor family that oversees processes ranging from cell longevity to cell apoptosis [17, 54]. Of the forkhead transcription factors, FOXO3a is one member that has emerged as a versatile target for a number of disorders [55]. Akt controls the “pro-apoptotic” forkhead transcription factor FOXO3a by inhibiting the nuclear translocation of FOXO3a that would normally activate the transcription of apoptotic nuclear genes. As a result, control of FOXO3a is considered to be a viable therapeutic target for agents such as metabotropic glutamate receptors [56] and NAD+ precursors [25, 54, 57] to increase cell survival. In addition, FOXO3a interfaces with several pathways that regulate cellular lifespan [58] and function to control neoplastic growth [59]. In a similar manner, EPO controls the phosphorylation and degradation of FOXO3a to retain it in the cytoplasm through binding to 14-3-3 protein and foster vascular cell protection during oxidative stress [17] (Figure 2).
Figure 2. Erythropoietin (EPO) sequesters FOXO3a in the cytoplasm during oxygen-glucose deprivation (OGD).
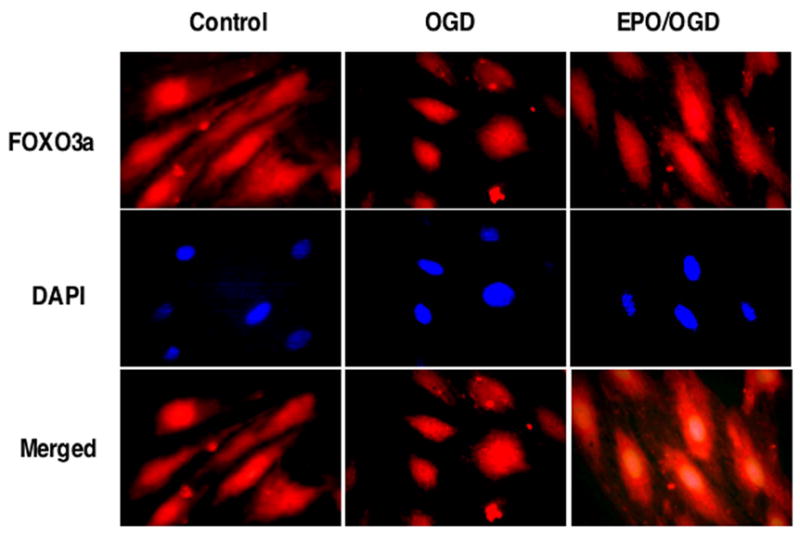
Administration of EPO (10 ng/ml) with an 8 hour period of OGD, OGD alone, or untreated cells (Control) was followed at 6 hours with immunofluorescent staining for FOXO3a (Texas-red) in endothelial cells (ECs). Nuclei of ECs were counterstained with DAPI. In merged images, cells with combined EPO and OGD show EC nuclei with minimal FOXO3a staining (blue/white) and show EC cytoplasm with significant FOXO3a staining (red) in contrast to cells with OGD alone with significant FOXO3a staining in both the cytoplasm and the nuclei of ECs, demonstrating the ability of EPO to sequester FOXO3a in the cytoplasm.
5.4 Wnt, GSK-3β, NF-κB, and EPO
Wnt proteins, derived from the Drosophila Wingless (Wg) and the mouse Int-1 genes, have been shown to play a role in both cell development and cell demise with recent recognition that the Wnt pathway also is dependent upon Akt signaling [16, 47, 60, 61]. Current experimental work suggests that some Wnt family members may offer glucose tolerance and increased insulin sensitivity [4], suggesting a potential protective cellular mechanism for EPO to improve clinical cardiac function in diabetic patients [15] and decrease complications in woman with diabetic pregnancies [14]. New in vitro studies demonstrate that the Wnt1 protein is necessary and sufficient to impart cellular protection during elevated glucose exposure [16]. Administration of exogenous Wnt1 protein can significantly prevent apoptotic EC injury during elevated glucose exposure. Interestingly, EPO maintains the expression of Wnt1 during elevated glucose exposure and prevents loss of Wnt1 expression that would occur in the absence of EPO during elevated glucose. More importantly, blockade of Wnt1 with a Wnt1Ab can neutralize the protective capacity of EPO, illustrating that Wnt1 is a critical component in the cytoprotection of EPO during elevated glucose exposure [16]. Furthermore, EPO also blocks glycogen synthase kinase-3β (GSK-3β), a downstream pathway of Wnt, that can influence cell survival and inflammation. As a result, EPO and GSK-3β are both considered to be therapeutic targets for a number of disorders [38, 47, 62, 63].
Expression and cytoprotection of EPO also is dependent, in part, upon Akt and the activation of nuclear factor-κB (NF-κB). NF-κB itself can be cytoprotective and lead to the induction of several anti-apoptotic genes, such as inhibitors of apoptotic protein that can specifically inhibit caspases 3, 7, and 9. NF-κB also plays a key role in the expression of EPO during HIF-1 induction. Akt can significantly increase NF-κB and HIF-1 activation resulting in the enhancement of EPO expression. Although in instances that involve some EPO receptor-positive tumors or specific ischemia-reperfusion cardiac injury models, EPO under most conditions uses NF-κB to prevent apoptosis through the enhanced expression and translocation of NF-κB to the nucleus to elicit anti-apoptotic gene activation [25, 27, 64].
6. Future Directions for Clinical Efficacy, Safety, and Toxicity of Erythropoietin
In light of the multiple cytoprotective pathways that are governed by EPO, it may come as no surprise that EPO has been identified as a possible candidate for a number of disease entities that involve cardiac, nervous, and vascular system diseases. At present, there are at least 100 trials with the National Institutes of Health website (clinicaltrials.gov) that are either recruiting or in preparation to examine the clinical effects of EPO in patients with a variety of disorders that include anemia, cancer, cardiac ischemia, or spinal cord trauma. Although some cardiac injury experimental models do not consistently demonstrate a benefit with EPO [65], initial studies in patients with anemia or on chronic hemodialysis have suggested a direct cardiac benefit from EPO administration [2, 3]. Subsequent work has demonstrated that EPO administration can improve exercise tolerance and renal function in patients with anemia and congestive heart failure [10] and that this may be tied to improved pulmonary function [41]. Of significant interest is a recent randomized, concealed, multicenter trail of 1460 patients who received 40,000 U of epoetin alfa up to a 3 week maximum following intensive care unit admission and demonstrated a reduced mortality in patients with trauma [66].
Unfortunately, agents such as EPO may not be tolerated by all individuals, especially those with co-morbid conditions such as congestive heart failure, hypertension, and neoplasms. Some studies suggest that EPO may contribute to vascular stenosis with intima hyperplasia [67]. Furthermore, adverse effects during treatment with EPO are not uncommon, such as an increased incidence of thrombotic vascular effects [66]. In addition, the use of EPO in patients with hypertension must proceed with caution, since both acute and long-term administration of EPO can significantly elevate mean arterial pressure [68].
The potential progression of cancer has been another significant concern raised with EPO administration [69, 70]. Not only has both EPO and its receptor been demonstrated in tumor specimens, but under some conditions EPO expression has been suggested to block tumor cell apoptosis through Akt [3, 70], enhance tumor progression, increase metastatic disease, and negate the effects of radiotherapy by assisting with tumor angiogenesis [71]. It should be noted though that the potential risk of EPO administration to either initiate tumor growth or lead to tumor progression is not entirely understood. In regards to the possible tumor promoting ability of EPO [72], a number of competing factors must be considered that include the possible benefits of EPO administration in patients with cancer that involve the synergistic effects of EPO with chemotherapeutic modalities [73] and the treatment of cancer-related anemia. The deployment of further large scale prospective trials that can more clearly examine the attributes and contraindications for EPO, especially in patients with neoplastic disease, are required.
However, in addition to the concerns outlined in patients with cancer, other important considerations for EPO exist. Irrespective of the problems associated with EPO abuse and gene doping [74], EPO has been correlated with the alteration of red cell membrane properties leading to a cognitive decrement in rodent animal models [2, 3]. In addition, development of potentially detrimental side-effects during EPO therapy, such as for cerebral ischemia with increased metabolic rate and blood viscosity [75], could severely limit or halt the use of EPO for neurovascular diseases. As a result, alternate strategies have been suggested. New investigations are studying the role of targeted bioavailability for EPO such as in bone marrow stromal cells genetically engineered to secrete EPO [76] and controlled release of EPO from encapsulated cells [77, 78]. Enhancement of EPO entry into the central nervous system continues to attract significant interest as well as does the use of novel intranasal routes for EPO administration [79]. Other avenues of study are directed to the development of derivations of EPO to reduce erythropoietic activity and the potential associated vascular complications [30]. Yet, these lines of investigation are not without limitations, since chemical derivatives of EPO can become absent of clinical efficacy [2, 3] as well as possibly loose the ability to promote sustainable cytoprotective effects, such as neurogenesis [80] and angiogenesis [11, 46].
As both basic experimental studies and clinical trials continue to outline the advantageous effects of EPO, raves for this unique cytoprotective agent will continue to unfold at a surprisingly rapid pace. EPO is now well accepted as an agent not only necessary for the induction of erythropoiesis, but also required for cellular maintenance, survival, and the control of inflammatory pathways. As a therapeutic entity, EPO appears to have applicability for a broad range of disorders that may extend from chronic organ failure to fragile cognitive loss. Yet, it is the further elucidation of the primary cellular pathways that are governed by EPO that should guide us in designing clinical applications for this agent and assist us in eliminating, or at the very least, reducing the risks of EPO that may be closely intertwined with its substantial benefits for patient care.
Acknowledgments
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, Nelson Foundation Award, NIH NIEHS (P30 ES06639), and NIH NINDS/NIA.
Biography
 Kenneth Maiese
Kenneth Maiese
Kenneth Maiese is a physician-scientist whose interests focus on the basic and clinical mechanisms that control cellular plasticity and longevity as well as inflammatory mechanisms in the nervous and vascular systems. He is presently the Director of the Division of Cellular and Molecular Cerebral Ischemia and is Professor in Neurology, Anatomy & Cell Biology, Molecular Medicine, and the Institute of Environmental Health Sciences at Wayne State University School of Medicine. Dr. Maiese graduated from the University of Pennsylvania Suma cum Laude with Distinction and received his medical degree as a Teagle and Grupe Foundation Scholar from Weill Medical College of Cornell University. He obtained his internship and residency at The New York Hospital-Cornell Medical Center, subsequently completed his clinical and basic science postdoctoral training at Cornell and the National Institute of Aging, then joined the faculty of Weill Medical College of Cornell University. His investigations are designed to translate basic science into successful therapeutic treatments for conditions such as metabolic disorders, cardiovascular disease, diabetes, stroke, and Alzheimer’s disease. To highlight some of his accomplishments, Dr. Maiese has been cited early in his career with several young scientist awards and his work has received the distinction by the National Institutes of Health as being “High Impact Research and Potential Public Health Benefit” with continuous funding from numerous sources that include the American Diabetes Association, the American Heart Association, the Bugher Foundation, a Johnson and Johnson Focused Giving Award, and the National Institutes of Health. Dr. Maiese also has been fortunate to receive recognition with outstanding teaching awards and election to America’s Top Physicians and The Best of U.S. Physicians. He chairs national grant committees and is a chartered panel member or consultant for several national and international foundations as well as multiple study sections and special emphasis panels for the National Institutes of Health. He serves as the Editor-in-Chief for two international journals as well as an Associate Editor or a member of the editorial board for several journals, executive committees, technology transfer panels, and scientific advisory councils. Given the broad applications of his work, Dr. Maiese is frequently honored as the chairperson and/or the plenary speaker for a number of international symposiums in a range of disciplines that include cell biology, neuroscience, vascular biology, cardiac disease, molecular oncology, drug discovery, and renal physiology.
 Zhao Zhong Chong
Zhao Zhong Chong
Zhao Zhong Chong is a Research Assistant Professor in the Division of Cellular and Molecular Cerebral Ischemia and the Department of Neurology whose research is directed upon the molecular mediators of cellular protection and inflammation. Dr. Chong received his undergraduate training from Binzhou Medical College, his Masters degree from Chongqing University of Medical Science, and his M.D. and Ph.D. degrees from Peking Union Medical College. Dr. Chong initially concentrated his work on neuroprotective mechanisms in the brain which has led to the development of novel agents to prevent thrombosis in both the heart and the brain. In the Division of Cellular and Molecular Cerebral Ischemia, Dr. Chong subsequently has characterized the role of specific kinases that are responsible for both the maintenance and destruction of DNA in both vascular and neuronal cells. His work has led to multiple publications and presentations at international meetings. Dr. Chong has broadened his translational research efforts with his focus upon unique cell receptor systems in the nervous and vascular systems, such as those that involve erythropoietin and the metabotropic glutamate receptor system, to examine the specific genetic mechanisms that may be developed to formulate therapy for degenerative disorders of the neuronal and vascular systems.
 Yan Chen Shang
Yan Chen Shang
Yan Chen Shang is a Research Assistant in the Division of Cellular and Molecular Cerebral Ischemia who has considerable experience in working with primary cells, cell lines, and animal models of neuronal and vascular diseases. Ms. Shang received her undergraduate training from Shangdong University. Her interests center upon inflammatory cells of the body and their integration and participation in metabolic and degenerative disorders, such as diabetes mellitus. Ms. Shang’s present work has further elucidated several components of cellular inflammation and the activation of phagocytic cells that can destroy viable cells in the brain.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carnot P, DeFlandre C. Sur l’activite hemopoietique de serum au cours de la regeneration du sang. C R Acad Sci (Paris) 1906;143:384–386. [Google Scholar]
- 2.Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004;25:577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293:90–95. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maiese K, Chong ZZ, Shang YC. Mechanisitic insights into diabetes mellitus and oxidative stress. Curr Med Chem. 2007;14:1689–1699. doi: 10.2174/092986707781058968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maiese K, Morhan SD, Chong ZZ. Oxidative stress biology and cell injury during type 1 and type 2 diabetes mellitus. Curr Neurovasc Res. 2007;4:63–71. doi: 10.2174/156720207779940653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bierer R, Peceny MC, Hartenberger CH, Ohls RK. Erythropoietin concentrations and neurodevelopmental outcome in preterm infants. Pediatrics. 2006;118:e635–640. doi: 10.1542/peds.2005-3186. [DOI] [PubMed] [Google Scholar]
- 7.Fliser D, Haller H. Erythropoietin and treatment of non-anemic conditions--cardiovascular protection. Semin Hematol. 2007;44:212–217. doi: 10.1053/j.seminhematol.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Schmeding M, Neumann UP, Boas-Knoop S, Spinelli A, Neuhaus P. Erythropoietin reduces ischemia-reperfusion injury in the rat liver. Eur Surg Res. 2007;39:189–197. doi: 10.1159/000101009. [DOI] [PubMed] [Google Scholar]
- 9.Assaraf MI, Diaz Z, Liberman A, Miller WH, Jr, Arvanitakis Z, Li Y, et al. Brain erythropoietin receptor expression in Alzheimer disease and mild cognitive impairment. J Neuropathol Exp Neurol. 2007;66:389–398. doi: 10.1097/nen.0b013e3180517b28. [DOI] [PubMed] [Google Scholar]
- 10.Palazzuoli A, Silverberg D, Iovine F, Capobianco S, Giannotti G, Calabro A, et al. Erythropoietin improves anemia exercise tolerance and renal function and reduces B-type natriuretic peptide and hospitalization in patients with heart failure and anemia. Am Heart J. 2006;152:1096 e1099–1015. doi: 10.1016/j.ahj.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Lu Z, Keogh CL, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043–1054. doi: 10.1038/sj.jcbfm.9600417. [DOI] [PubMed] [Google Scholar]
- 12.Ravid O, Shams I, Ben Califa N, Nevo E, Avivi A, Neumann D. An extracellular region of the erythropoietin receptor of the subterranean blind mole rat Spalax enhances receptor maturation. Proc Natl Acad Sci U S A. 2007;104:14360–14365. doi: 10.1073/pnas.0706777104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sathyanarayana P, Menon MP, Bogacheva O, Bogachev O, Niss K, Kapelle WS, et al. Erythropoietin modulation of podocalyxin and a proposed erythroblast niche. Blood. 2007;110:509–518. doi: 10.1182/blood-2006-11-056465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teramo K, Kari MA, Eronen M, Markkanen H, Hiilesmaa V. High amniotic fluid erythropoietin levels are associated with an increased frequency of fetal and neonatal morbidity in type 1 diabetic pregnancies. Diabetologia. 2004;47:1695–1703. doi: 10.1007/s00125-004-1515-3. [DOI] [PubMed] [Google Scholar]
- 15.Silverberg DS, Wexler D, Iaina A, Schwartz D. The interaction between heart failure and other heart diseases, renal failure, and anemia. Semin Nephrol. 2006;26:296–306. doi: 10.1016/j.semnephrol.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Chong ZZ, Shang YC, Maiese K. Vascular injury during elevated glucose can be mitigated by erythropoietin and Wnt signaling. Curr Neurovasc Res. 2007;4:194–204. doi: 10.2174/156720207781387150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chong ZZ, Maiese K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br J Pharmacol. 2007;150:839–850. doi: 10.1038/sj.bjp.0707161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao E, Boucher M, Chuprun JK, Zhou RH, Eckhart AD, Koch WJ. Darbepoetin alfa, a long-acting erythropoietin analog, offers novel and delayed cardioprotection for the ischemic heart. Am J Physiol Heart Circ Physiol. 2007;293:H60–68. doi: 10.1152/ajpheart.00227.2007. [DOI] [PubMed] [Google Scholar]
- 19.Toma C, Letts DP, Tanabe M, Gorcsan J, 3rd, Counihan PJ. Positive effect of darbepoetin on peri-infarction remodeling in a porcine model of myocardial ischemia-reperfusion. J Mol Cell Cardiol. 2007;43:130–136. doi: 10.1016/j.yjmcc.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Lykissas MG, Korompilias AV, Vekris MD, Mitsionis GI, Sakellariou E, Beris AE. The role of erythropoietin in central and peripheral nerve injury. Clin Neurol Neurosurg. 2007;109:639–644. doi: 10.1016/j.clineuro.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106:2973–2979. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- 22.Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003;138:1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res. 2003;71:659–669. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- 24.Keogh CL, Yu SP, Wei L. The effect of recombinant human erythropoietin on neurovasculature repair after focal ischemic stroke in neonatal rats. J Pharmacol Exp Ther. 2007;322:521–528. doi: 10.1124/jpet.107.121392. [DOI] [PubMed] [Google Scholar]
- 25.Li F, Chong ZZ, Maiese K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr Neurovasc Res. 2006;3:187–201. doi: 10.2174/156720206778018758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J Cereb Blood Flow Metab. 2003;23:320–330. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- 27.Chong ZZ, Li F, Maiese K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr Neurovasc Res. 2005;2:387–399. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLeod M, Hong M, Mukhida K, Sadi D, Ulalia R, Mendez I. Erythropoietin and GDNF enhance ventral mesencephalic fiber outgrowth and capillary proliferation following neural transplantation in a rodent model of Parkinson’s disease. Eur J Neurosci. 2006;24:361–370. doi: 10.1111/j.1460-9568.2006.04919.x. [DOI] [PubMed] [Google Scholar]
- 29.Tsai JC, Song BJ, Wu L, Forbes M. Erythropoietin: a candidate neuroprotective agent in the treatment of glaucoma. J Glaucoma. 2007;16:567–571. doi: 10.1097/IJG.0b013e318156a556. [DOI] [PubMed] [Google Scholar]
- 30.King VR, Averill SA, Hewazy D, Priestley JV, Torup L, Michael-Titus AT. Erythropoietin and carbamylated erythropoietin are neuroprotective following spinal cord hemisection in the rat. Eur J Neurosci. 2007;26:90–100. doi: 10.1111/j.1460-9568.2007.05635.x. [DOI] [PubMed] [Google Scholar]
- 31.Okutan O, Solaroglu I, Beskonakli E, Taskin Y. Recombinant human erythropoietin decreases myeloperoxidase and caspase-3 activity and improves early functional results after spinal cord injury in rats. J Clin Neurosci. 2007;14:364–368. doi: 10.1016/j.jocn.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 32.Verdonck O, Lahrech H, Francony G, Carle O, Farion R, Van de Looij Y, et al. Erythropoietin protects from post-traumatic edema in the rat brain. J Cereb Blood Flow Metab. 2007;27:1369–1376. doi: 10.1038/sj.jcbfm.9600443. [DOI] [PubMed] [Google Scholar]
- 33.Cherian L, Goodman JC, Robertson C. Neuroprotection with erythropoietin administration following controlled cortical impact injury in rats. J Pharmacol Exp Ther. 2007;322:789–794. doi: 10.1124/jpet.107.119628. [DOI] [PubMed] [Google Scholar]
- 34.Mikati MA, Hokayem JA, Sabban ME. Effects of a single dose of erythropoietin on subsequent seizure susceptibility in rats exposed to acute hypoxia at p10. Epilepsia. 2007;48:175–181. doi: 10.1111/j.1528-1167.2006.00900.x. [DOI] [PubMed] [Google Scholar]
- 35.Nadam J, Navarro F, Sanchez P, Moulin C, Georges B, Laglaine A, et al. Neuroprotective effects of erythropoietin in the rat hippocampus after pilocarpine-induced status epilepticus. Neurobiol Dis. 2007;25:412–426. doi: 10.1016/j.nbd.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Ferri C, Giuggioli D, Sebastiani M, Colaci M. Treatment of severe scleroderma skin ulcers with recombinant human erythropoietin. Clin Exp Dermatol. 2007;32:287–290. doi: 10.1111/j.1365-2230.2007.02363.x. [DOI] [PubMed] [Google Scholar]
- 37.Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y, Shang Y, Sun S, Liang H, Liu R. Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3beta/caspase-3 mediated signaling pathway. Apoptosis. 2007;12:1365–1375. doi: 10.1007/s10495-007-0065-9. [DOI] [PubMed] [Google Scholar]
- 39.Contaldo C, Meier C, Elsherbiny A, Harder Y, Trentz O, Menger MD, et al. Human recombinant erythropoietin protects the striated muscle microcirculation of the dorsal skinfold from postischemic injury in mice. Am J Physiol Heart Circ Physiol. 2007;293:H274–283. doi: 10.1152/ajpheart.01031.2006. [DOI] [PubMed] [Google Scholar]
- 40.Ferrario M, Massa M, Rosti V, Campanelli R, Ferlini M, Marinoni B, et al. Early haemoglobin-independent increase of plasma erythropoietin levels in patients with acute myocardial infarction. Eur Heart J. 2007;28:1805–1813. doi: 10.1093/eurheartj/ehm065. [DOI] [PubMed] [Google Scholar]
- 41.Wu H, Ren B, Zhu J, Dong G, Xu B, Wang C, et al. Pretreatment with recombined human erythropoietin attenuates ischemia-reperfusion-induced lung injury in rats. Eur J Cardiothorac Surg. 2006;29:902–907. doi: 10.1016/j.ejcts.2006.02.036. [DOI] [PubMed] [Google Scholar]
- 42.Fu Q, Van Eyk JE. Proteomics and heart disease: identifying biomarkers of clinical utility. Expert Rev Proteomics. 2006;3:237–249. doi: 10.1586/14789450.3.2.237. [DOI] [PubMed] [Google Scholar]
- 43.Eto N, Wada T, Inagi R, Takano H, Shimizu A, Kato H, et al. Podocyte protection by darbepoetin: preservation of the cytoskeleton and nephrin expression. Kidney Int. 2007;72:455–463. doi: 10.1038/sj.ki.5002311. [DOI] [PubMed] [Google Scholar]
- 44.Asaumi Y, Kagaya Y, Takeda M, Yamaguchi N, Tada H, Ito K, et al. Protective role of endogenous erythropoietin system in nonhematopoietic cells against pressure overload-induced left ventricular dysfunction in mice. Circulation. 2007;115:2022–2032. doi: 10.1161/CIRCULATIONAHA.106.659037. [DOI] [PubMed] [Google Scholar]
- 45.Westenbrink BD, Lipsic E, van der Meer P, van der Harst P, Oeseburg H, Du Marchie Sarvaas GJ, et al. Erythropoietin improves cardiac function through endothelial progenitor cell and vascular endothelial growth factor mediated neovascularization. Eur Heart J. 2007;28:2018–2027. doi: 10.1093/eurheartj/ehm177. [DOI] [PubMed] [Google Scholar]
- 46.Reinders ME, Rabelink TJ, Briscoe DM. Angiogenesis and endothelial cell repair in renal disease and allograft rejection. J Am Soc Nephrol. 2006;17:932–942. doi: 10.1681/ASN.2005121250. [DOI] [PubMed] [Google Scholar]
- 47.Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Harris SE, Fox H, Wright AF, Hayward C, Starr JM, Whalley LJ, et al. A genetic association analysis of cognitive ability and cognitive ageing using 325 markers for 109 genes associated with oxidative stress or cognition. BMC Genet. 2007;8:43. doi: 10.1186/1471-2156-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okouchi M, Ekshyyan O, Maracine M, Aw TY. Neuronal apoptosis in neurodegeneration. Antioxid Redox Signal. 2007;9:1059–1096. doi: 10.1089/ars.2007.1511. [DOI] [PubMed] [Google Scholar]
- 50.Chong ZZ, Kang J, Li F, Maiese K. mGluRI Targets Microglial Activation and Selectively Prevents Neuronal Cell Engulfment Through Akt and Caspase Dependent Pathways. Curr Neurovasc Res. 2005;2:197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003;64:557–569. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- 52.Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003;74:37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Park TS, Gidday JM. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol Heart Circ Physiol. 2007;292:H2573–2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]
- 54.Chong ZZ, Lin SH, Maiese K. The NAD + precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004;24:728–743. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- 55.Maiese K, Chong ZZ, Shang YC. “Sly as a FOXO”: New paths with Forkhead signaling in the brain. Curr Neurovasc Res. 2007;4:295–302. doi: 10.2174/156720207782446306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chong ZZ, Li F, Maiese K. Group I Metabotropic Receptor Neuroprotection Requires Akt and Its Substrates that Govern FOXO3a, Bim, and beta-Catenin During Oxidative Stress. Curr Neurovasc Res. 2006;3:107–117. doi: 10.2174/156720206776875830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li F, Chong ZZ, Maiese K. Cell Life Versus Cell Longevity: The Mysteries Surrounding the NAD(+) Precursor Nicotinamide. Curr Med Chem. 2006;13:883–895. doi: 10.2174/092986706776361058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Wang Z, Kong D, Murthy S, Dou QP, Sheng S, et al. Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3′-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J Biol Chem. 2007;282:21542–21550. doi: 10.1074/jbc.M701978200. [DOI] [PubMed] [Google Scholar]
- 60.Chong ZZ, Li F, Maiese K. Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell Signal. 2007;19:1150–1162. doi: 10.1016/j.cellsig.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Speese SD, Budnik V. Wnts: up-and-coming at the synapse. Trends Neurosci. 2007;30:268–275. doi: 10.1016/j.tins.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chong ZZ, Li F, Maiese K. The pro-survival pathways of mTOR and protein kinase B target glycogen synthase kinase-3beta and nuclear factor-kappaB to foster endogenous microglial cell protection. Int J Mol Med. 2007;19:263–272. [PMC free article] [PubMed] [Google Scholar]
- 63.Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev. 2007;31:920–931. doi: 10.1016/j.neubiorev.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spandou E, Tsouchnikas I, Karkavelas G, Dounousi E, Simeonidou C, Guiba-Tziampiri O, et al. Erythropoietin attenuates renal injury in experimental acute renal failure ischaemic/reperfusion model. Nephrol Dial Transplant. 2006;21:330–336. doi: 10.1093/ndt/gfi177. [DOI] [PubMed] [Google Scholar]
- 65.Olea FD, Vera Janavel G, De Lorenzi A, Cuniberti L, Yannarelli G, Cabeza Meckert P, et al. High-dose erythropoietin has no long-term protective effects in sheep with reperfused myocardial infarction. J Cardiovasc Pharmacol. 2006;47:736–741. doi: 10.1097/01.fjc.0000211766.59636.0d. [DOI] [PubMed] [Google Scholar]
- 66.Corwin HL, Gettinger A, Fabian TC, May A, Pearl RG, Heard S, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med. 2007;357:965–976. doi: 10.1056/NEJMoa071533. [DOI] [PubMed] [Google Scholar]
- 67.Reddy MK, Vasir JK, Hegde GV, Joshi SS, Labhasetwar V. Erythropoietin induces excessive neointima formation: a study in a rat carotid artery model of vascular injury. J Cardiovasc Pharmacol Ther. 2007;12:237–247. doi: 10.1177/1074248406297326. [DOI] [PubMed] [Google Scholar]
- 68.Kanbay M, Akcay A, Delibasi T, Uz B, Kaya A, Koca C, et al. Comparison of effects of darbepoetin alfa and epoetin alfa on serum endothelin level and blood pressure. Adv Ther. 2007;24:346–352. doi: 10.1007/BF02849903. [DOI] [PubMed] [Google Scholar]
- 69.Kokhaei P, Abdalla AO, Hansson L, Mikaelsson E, Kubbies M, Haselbeck A, et al. Expression of erythropoietin receptor and in vitro functional effects of epoetins in B-cell malignancies. Clin Cancer Res. 2007;13:3536–3544. doi: 10.1158/1078-0432.CCR-06-2828. [DOI] [PubMed] [Google Scholar]
- 70.Maiese K, Li F, Chong ZZ. Erythropoietin and cancer. JAMA. 2005;293:1858–1859. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ceelen W, Boterberg T, Smeets P, Van Damme N, Demetter P, Zwaenepoel O, et al. Recombinant human erythropoietin alpha modulates the effects of radiotherapy on colorectal cancer microvessels. Br J Cancer. 2007;96:692–700. doi: 10.1038/sj.bjc.6603568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rades D, Golke H, Schild SE, Kilic E. The Impact of Tumor Expression of Erythropoietin Receptors and Erythropoietin on Clinical Outcome of Esophageal Cancer Patients Treated with Chemoradiation. Int J Radiat Oncol Biol Phys. 2007 doi: 10.1016/j.ijrobp.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 73.Sigounas G, Sallah S, Sigounas VY. Erythropoietin modulates the anticancer activity of chemotherapeutic drugs in a murine lung cancer model. Cancer Lett. 2004;214:171–179. doi: 10.1016/j.canlet.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 74.Diamanti-Kandarakis E, Konstantinopoulos PA, Papailiou J, Kandarakis SA, Andreopoulos A, Sykiotis GP. Erythropoietin abuse and erythropoietin gene doping: detection strategies in the genomic era. Sports Med. 2005;35:831–840. doi: 10.2165/00007256-200535100-00001. [DOI] [PubMed] [Google Scholar]
- 75.Frietsch T, Maurer MH, Vogel J, Gassmann M, Kuschinsky W, Waschke KF. Reduced cerebral blood flow but elevated cerebral glucose metabolic rate in erythropoietin overexpressing transgenic mice with excessive erythrocytosis. J Cereb Blood Flow Metab. 2007;27:469–476. doi: 10.1038/sj.jcbfm.9600360. [DOI] [PubMed] [Google Scholar]
- 76.Eliopoulos N, Gagnon RF, Francois M, Galipeau J. Erythropoietin delivery by genetically engineered bone marrow stromal cells for correction of anemia in mice with chronic renal failure. J Am Soc Nephrol. 2006;17:1576–1584. doi: 10.1681/ASN.2005101035. [DOI] [PubMed] [Google Scholar]
- 77.Orive G, De Castro M, Ponce S, Hernandez RM, Gascon AR, Bosch M, et al. Long-term expression of erythropoietin from myoblasts immobilized in biocompatible and neovascularized microcapsules. Mol Ther. 2005;12:283–289. doi: 10.1016/j.ymthe.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 78.Ponce S, Orive G, Hernandez RM, Gascon AR, Canals JM, Munoz MT, et al. In vivo evaluation of EPO-secreting cells immobilized in different alginate-PLL microcapsules. J Control Release. 2006;116:28–34. doi: 10.1016/j.jconrel.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 79.Yu YP, Xu QQ, Zhang Q, Zhang WP, Zhang LH, Wei EQ. Intranasal recombinant human erythropoietin protects rats against focal cerebral ischemia. Neurosci Lett. 2005;387:5–10. doi: 10.1016/j.neulet.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 80.Gonzalez FF, McQuillen P, Mu D, Chang Y, Wendland M, Vexler Z, et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci. 2007;29:321–330. doi: 10.1159/000105473. [DOI] [PubMed] [Google Scholar]