

Abstract
Cancer is a multi-step, multi-genetic event. Whether oncogenic mutations cooperate with one another to transform cells and how is not well understood. The Friend murine retroviral erythroleukemia model involves mitogenic activation of the erythropoietin receptor (EpoR) by the virus env gene (F-gp55), aberrant over-expression of the transcription factor PU.1, and inactivating mutations in p53. In this report we demonstrate that concurrent expression of F-gp55 and PU.1 in erythroid target cells, in vivo, cooperate to accelerate erythroleukemia induction. Early in the disease, prior to the detection of clonal leukemic cells, activation of the EpoR by F-gp55, but not erythropoietin, resulted in transcriptional upregulation of PU.1 through a trans regulatory mechanism. This could occur in the absence of an integrated provirus within the PU.1 gene locus. The regulation of PU.1 transcription in established erythroleukemia cell lines differed depending upon the level of PU.1 protein present. Our results suggest that the action of F-gp55 contributes to both early and late stages of Friend erythroleukemia and that persistence of F-gp55 expression may be required not only to initiate erythroleukemia but to also maintain erythroleukemia following Friend virus infection.
Keywords: erythroleukemia, oncogene cooperativity, PU.1 transcription, Friend virus
Introduction
The transition of a normal cell to a neoplastic one requires the accumulation of multiple genetic alterations (Weinberg, 1989; Fearon and Vogelstein, 1990). Despite the large number of gene mutations identified in various human cancers and the study of mouse models mimicking these mutations, the functional interdependency of these mutations in the development of cancer has not been extensively assessed. Leukemia is a cancer of blood precursor cells. Leukemic transformation involves alterations in the regulation of cell proliferation and self-renewal, survival, and terminal differentiation. Clues to genes involved in the pathogenesis of leukemia have come from studies of retroviral insertional mutagenesis in mice and the identification of fusion genes at sites of chromosomal translocations in human leukemic cells. Some alter growth signaling pathways (e.g., the t(9;22) product Bcr-Abl in chronic myeloid leukemia), however, many result in aberrant expression of transcription factors that are predicted to alter the normal hematopoietic differentiation program. The mouse model of Friend retrovirus-induced erythroleukemia has long been an excellent system for determining how viral and host cellular genes contribute to leukemic transformation (Ben-David and Bernstein, 1991; Hoatlin and Kabat, 1995; Ruscetti, 1999). As several genetic changes have been identified in Friend erythroleukemia this system offers the potential to determine whether the functional properties of multiple oncogenes cooperate to initiate and maintain leukemic transformation.
Friend virus is a retroviral complex composed of a replication-defective spleen focus-forming virus (SFFV) and a replication-competent Friend murine leukemia virus (F-MuLV). Acute erythroleukemia is due to the replication-defective SFFV component (Wolff and Ruscetti, 1985). The early, or first, stage of Friend erythroleukemia is manifest by a polyclonal increase in the number of erythroid progenitor cells that leads to an elevation in the number of circulating terminally differentiated red blood cells (i.e., elevated blood packed cell volume or hematocrit). Within these infected progenitor cells the SFFV envelope protein F-gp55 physically associates with the erythropoietin receptor (EpoR) at the cell surface, activating EpoR signaling pathways independent of erythropoietin (Epo) (Li et al., 1987; Li et al., 1990). These infected cells have limited self-renewal capacity, however, as they retain the capacity for terminal differentiation. Malignant, erythroleukemic clones emerge approximately 5 weeks following infection (Mager et al., 1981). The emergence of these malignant clones marks the second stage of Friend disease. These cells are blocked in their ability to differentiate into mature red blood cells, and are capable of transferring leukemia into recipient transplanted mice. In addition, they exhibit long-term growth factor-independent proliferation in culture, ex vivo. The second stage in Friend virus-induced erythroleukemia involves deregulated expression of the transcription factor PU.1, an ets gene family member, due to integration of a SFFV provirus within the PU 1 gene locus (Moreau-Gachelin et al., 1988; Moreau-Gachelin et al., 1989; Paul et al., 1989; Paul et al., 1991). Following expansion of PU.1 over-expressing clones, inactivating mutations in the suppressor oncogene p53 also occur (Mowat et al., 1985), however, p53 mutations appear to be late events and may not be required for leukemic transformation, in vivo (Howard et al., 1993; Tran Quang et al., 1997; Barnache et al., 1998; Kelley et al., 1998).
In normal proerythroblasts progenitor cells a low level of PU.1 protein is present (Hromas et al., 1993). As these cells mature PU.1 production is shut off, suggesting that persistence of PU.1 protein is not compatible with continued erythroid differentiation (Hromas et al., 1993). In contrast the fully transformed erythroleukemia clones isolated from Friend virus-infected mice contain high levels of PU.1 protein (Schuetze et al., 1992). Accumulating evidence suggests that the level of PU.1 protein present in Friend erythroleukemia cells is an important factor contributing to the transition from the preleukemic first stage to the leukemic second stage of Friend disease (Schuetze et al., 1993; Moreau-Gachelin et al., 1996). In addition, persistent expression of high levels of PU.1 may also be required to maintain the leukemic phenotype. During DMSO-induced erythroid differentiation of Friend erythroleukemia cell lines one of the early events is a marked decline in the level of PU.1 protein expression (Schuetze et al., 1992). However, when Friend erythroleukemia cells contain an exogenous plasmid that constitutively expresses PU.1, DMSO-induced differentiation is blocked (Yamada et al., 1997).
Recently it was shown in cultured avian erythroblasts that activation of the EpoR by receptor mutations or through association with F-gp55, but not Epo, was required for over expression of PU.1 to block erythroid differentiation (Tran Quang et al., 1997; Pereira et al., 2000). Due to the nature of these studies leukemogenicity could not be assessed, and how these pathologic modes of EpoR activation cooperate with PU.1 was not determined. Herein we show in mice that concurrent expression of the F-gp55 and PU.1 cooperate to accelerate induction of both the early and late stages of Friend erythroleukemia. We found that activation of the EpoR by F-gp55, but not Epo, resulted in transcriptional upregulation of PU.1 and this occurred in the absence of an integrated SFFV provirus within the PU.1 gene locus, demonstrating that SFFV can regulate PU.1 promoter activity through a trans regulatory mechanism.
Results
Concurrent expression of F-gp55 and PU.1 in murine erythroid progenitor cells, in vivo, accelerated both stages of Friend erythroleukemia
To determine if SFFV activated EpoR signals cooperate with PU.1 to induce erythroleukemia in mice we asked whether co-expression of both oncogenes in the same target cell, in vivo, would alter the kinetics of leukemia induction. Recombinant SFFV retroviruses that expressed both F-gp55 and PU.1, F-gp55 alone, or PU.1 alone were generated (Figure 1a). To distinguish retrovirally derived PU.1 protein from endogenous PU.1 protein a myc-epitope was introduced at the N-terminus of retrovirally derived PU.1. All three retroviruses were of equivalent titer, and transduced the expected gene products following infection of tissue culture cells (data not shown).
Figure 1.
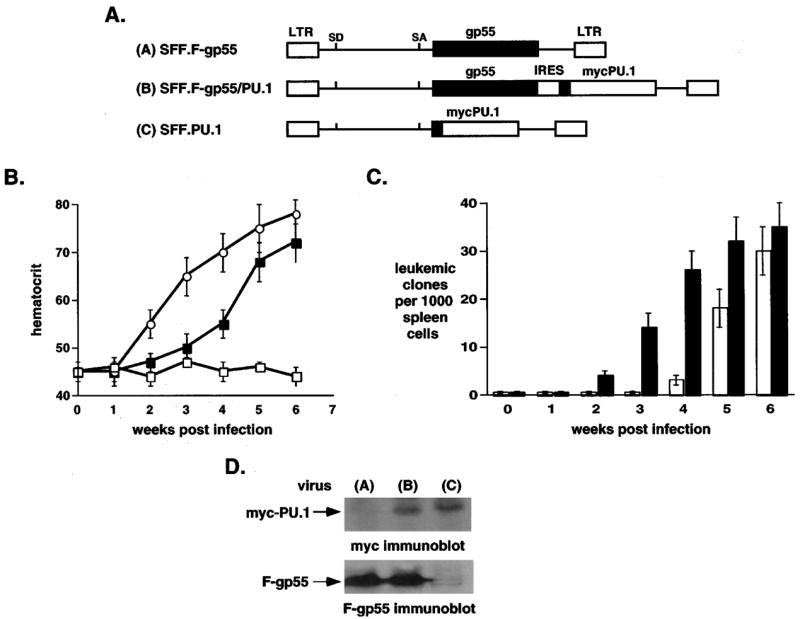
Co-expression of PU.1 and F-gp55 in murine erythroid progenitor cells accelerated both stages of Friend erythroleukemia, in vivo. (a) Schematic representation of retroviral constructs. A myc epitope tag (black box) was placed at the N-terminus of retroviral PU.1. (b) Weekly blood hematocrit levels of retrovirally-infected mice. SFF.F-gp55 virus, filled squares; SFF.F-gp55/PU.1 virus, open circles; SFF.PU.1 virus, open squares. For each virus five mice were infected. Results represent the mean and the standard deviation from the mean. (c) Tumorigenic assays. Mice were infected with SFF.F-gp55 virus (white columns), or SFF.F-gp55/PU.1 virus (black columns). Each week following infection splenic cells were isolated and 1000 cells plated in methylcellulose media without any added cytokines. Ten days following initiation of culture erythroid colonies were identified and scored. Three mice were analysed at each time point. Results represent the mean and the standard deviation from the mean. (d) Immunoblot analysis of splenic extracts from mice infected with viruses. Splenic extracts were prepared from mice 6 weeks following infection with virus: SFF.F-gp55, lane 1, SFF.F-gp55/PU.1, lane 2, and SFF.PU.1, lane 3. Anti-myc immunoblot, upper panel, and anti-F-gp55 immunoblot, lower panel
Mice were infected and weekly hematocrit determined (Figure 1b). Mice infected with F-gp55/PU.1 virus developed earlier onset of erythrocytosis compared to mice infected with F-gp55 virus (Figure 1b: 2 weeks versus 3 weeks), while mice infected with PU.1 retrovirus did not exhibit any alteration in red blood cell numbers. This result indicated that concurrent transduction of F-gp55 and PU.1 into erythroid target cells accelerated the first stage of Friend disease.
To determine whether the transition to the second stage of Friend disease (i.e., leukemic transformation) was altered, sets of mice were sacrificed each week following infection and splenic cells placed in methylcellulose cultures lacking any added Epo (Figure 1c). Compared to F-gp55 infected mice Epo-independent leukemic cells developed, ex vivo, at earlier times following infection with F-gp55/PU.1 virus (Figure 1c: 5 weeks versus 3 weeks, respectively). Mice infected with the PU.1 retrovirus did not develop splenomegaly nor did splenic cells give rise to any Epo-independent colonies (data not shown). To confirm that the appropriate gene products were expressed in infected mice, immunoblots of protein extracts from the spleens of retrovirally-infected mice were performed (Figure 1d). This analysis demonstrated that F-gp55 and myc-PU.1 were expressed in the appropriate infected spleens (Figure 1d). Collectively, these results indicated that concurrent transduction of F-gp55 and PU.1 into erythroid target cells accelerated the development of the second stage of Friend disease.
Activation of the EpoR by F-gp55, but not Epo, resulted in increased PU.1 transcription, independent of SFFV proviral integration
To determine whether signals arising from a F-gp55 activated EpoR, versus Epo activated EpoR, affected PU.1 activity we first developed cell lines in which the only difference was the mode of EpoR activation. We chose the IL3-dependent myeloid cell line 32D. 32D cells express endogenous PU.1 protein, do not express GATA-1 (which can interact with PU.1 and inhibit its transcriptional activity (Zhang et al., 1999; Rekhtman et al., 1999)), and do not express the EpoR (Beckman et al., 1999). The derivation and characterization of the 32D.EpoR cells, which proliferate in response to added Epo, have been previously described (Beckman et al., 1999). Infection of 32D.EpoR cells with SFF-gp55 retrovirus led to the establishment of Epo-independent cells: 32D.EpoR/F-gp55 cells. Immunoblot analysis of protein extracts from these cells demonstrated that F-gp55 was expressed (Figure 2a).
Figure 2.
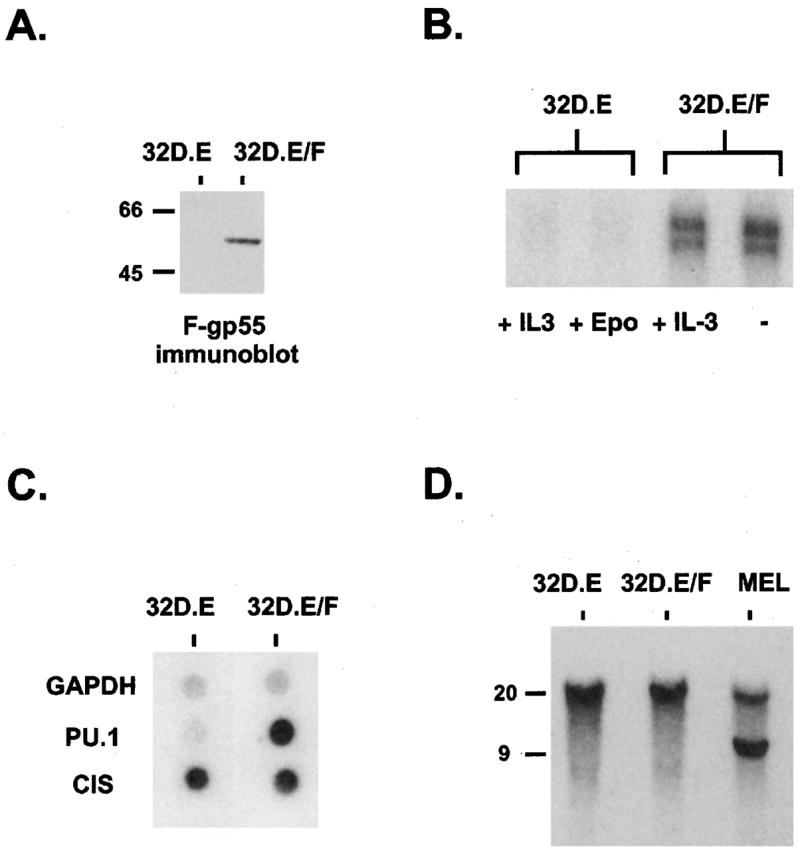
Activation of the EpoR by F-gp55, but not Epo, resulted in increased PU.1 transcription, independent of SFFV proviral integration. (a) F-gp55 immunoblot analysis for F-gp55 expression in 32D.EpoR cells (lane 1) and 32D.EpoR/F-gp55 cells (lane 2). Equal amounts of protein were loaded in each lane. The mobility of molecular size markers, in kDa, is on the left. (b) RNase protection assays for PU.1 mRNA expression in 32D.EpoR (32D.E) cells grown in IL-3 (5% WEHI culture supernatant) (lane 1) or 0.5 U/ml Epo (lane 2). 32D.EpoR/F-gp55 (32D.E/F) cells growing in IL-3-containing media (lane 3), or in the absence of any added growth factors (lane 4). (c) Nuclear run-on transcription assay for de novo transcription of GAPDH, PU.1 and CIS genes in 32D.EpoR (32D.E) cells (lane 1) and 32D.EpoR/F-gp55 (32D.E/F) cells (lane 2), as described in Materials and methods. (d) Southern blot analysis of the PU.1 gene organization in 32D.EpoR (32D.E) cells (lane 1), 32D.EpoR/F-gp55 (32D.E/F) cells (lane 2), and Friend erythroleukemia MEL cells (lane 3). Ten ug of genomic DNA was digested with EcoRV, and hybridized with probe A of the PU.1 gene (Moreau-Gachelin et al., 1989). The mobility of molecular size markers, in kilobases, is shown on the left
Since overexpression of PU.1 is required for erythroid transformation in Friend erythroleukemia we determined whether the level of PU.1 mRNA present in 32D.EpoR cells and 32D.EpoR/F-gp55 cells differed. Introduction of the EpoR alone into 32D cells did not affect PU.1 transcription as 32D.EpoR cells grown in Epo and parental 32D cells expressed the same level of PU.1 RNA (not shown). RNase protection assays were performed (Figure 2b). 32D.EpoR/F-gp55 cells cultured in IL-3-containing media or media lacking exogenous growth factors contained dramatically increased levels of PU.1 mRNA compared to 32D.EpoR cells grown in IL-3 or Epo (Figure 2b). The addition of high concentrations of Epo to 32D.EpoR/F-gp55 cells did not further increase the elevated levels of PU.1 mRNA present in these cells (data not shown).
To determine if the increased PU.1 mRNA present in 32D.EpoR/F-gp55 cells was due to de novo transcription, nuclear run-on assays were performed (Figure 2c). This analysis demonstrated that the increased PU.1 mRNA in 32D.EpoR/F-gp55 cells was indeed due to activation of transcription of the PU.1 gene (Figure 2c, row 2). The upregulation of PU.1 mRNA in 32D.EpoR/F-gp55 cells was not due to a general increase in transcription or mRNA stability as the level of glyceraldehyde-3-phosphodehydrogenase (GAPDH) mRNA was not different between the two cell lines (Figure 2c, row 1). In addition, there was minimal difference in the transcription rate of Cytokine Inducible SH2-containing protein (CIS), an Epo-induced JAK/STAT responsive gene (Yoshimura et al., 1995), indicating that in 32D.EpoR/F-gp55 cells transcriptional upregulation of PU.1 was relatively specific.
In Friend erythroleukemia cells integration of an SFFV provirus in the PU.1 gene locus is thought to activate transcription of PU.1 (Moreau-Gachelin et al., 1988; Moreau-Gachelin et al., 1989; Paul et al., 1991). To determine if this mechanism could be responsible for the increase in transcription of PU.1 observed in 32D.EpoR/F-gp55 cells, Southern blot analysis of the PU.1 gene locus in these cells was performed (Figure 2d). PU.1 gene rearrangement, as seen in the Friend erythroleukemia cells MEL (Figure 2d, lane 3) was not evident in 32D.EpoR/F-gp55 cells (Figure 2d, lane 2). Therefore, proviral integration could be excluded as a mechanism responsible for increased PU.1 transcription in 32D.EpoR/F-gp55 cells.
To determine if PU.1 transcriptional upregulation occurred in primary erythroblasts expressing F-gp55, mice were infected with SFF.F-gp55 virus and TER-119+erythroblasts isolated from the spleen 1 or 2 weeks following infection (Vannucchi et al., 2000). To control for the increase in splenic erythroblasts found in SFF.F-g55-infected, some uninfected mice were treated with phenylhydrazine, which induces a hemolytic anemia and a responding increase in erythroblast production. TER-119+erythroblasts were isolated from the spleens of these mice. Total mRNA was analysed by a PU.1 Northern blot (Figure 3). There was little PU.1 mRNA present in primary erythroblasts from phenylhydrazine treated mice (Figure 3, lanes 1 and 2). In contrast, the PU.1 mRNA levels in splenic erythroblasts were increased by 1 week after infection with SFF.F-gp55, and further increased by week 2 following infection (Figure 3, lanes 3 and 4). The PU.1 mRNA levels present in splenic erythroblasts 1 – 2 weeks following SFFV infection were much less than that found in the established Friend erythroleukemia cell line, MEL, however. Furthermore, the early increase in PU.1 mRNA in SFF.F-gp55 infected splenic erythroblasts occurred before detectable clonal erythroleukemic cell lines could be established, ex vivo (Figure 1c). This result demonstrated that during the early polyclonal erythroblastosis phase of Friend erythroleukemia activation of the EpoR by F-gp55 in primary erythroblasts resulted in a small, but significant increase in PU.1 transcription, as was observed in the model 32D cell line system (Figure 2).
Figure 3.
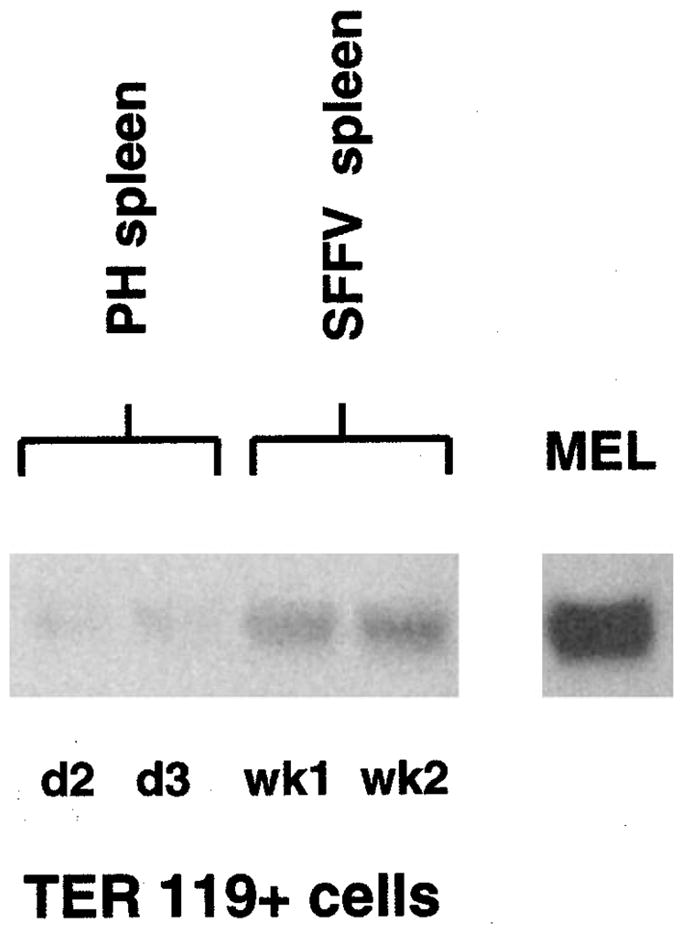
PU.1 transcription is increased in primary erythroblasts from mice infected with SFFV virus (PH). Total RNA was prepared from TER-119+primary erythroblasts isolated from the spleens of mice treated with phenylhydrazine (lane 1 – day 2 post treatment; lane 2 – day 3 post treatment) or infected with SFFV virus (lane 3 – 1 week post infection; lane 4 – 2 weeks post infection). Lane 5 is a sample from the established Friend erythroleukemia cell line MEL. 10 ug of total RNA was loaded in each lane
Next we asked whether the activity of a PU.1 promoter-reporter plasmid was enhanced in cells, whose proliferation resulted from activation of the EpoR by F-gp55. 32D.EpoR and 32D.EpoR/F-gp55 cells (Figure 4b) and Friend erythroleukemia MEL cells (Figure 4c) were transiently transfected with a minimal murine PU.1 promoter (−334, Figure 4a) driving expression of luciferase. Transfection efficiency was normalized to beta-galactosidase expression derived from a co-transfected CMV-beta-galactosidase plasmid, and results presented as relative luciferase units (RLU). As a negative control cells were also transfected with an inactive PU.1 promoter-luciferase plasmid (−7, Figure 4a). Both 32D.EpoR/F-gp55 and MEL cells exhibited a robust capacity to upregulate PU.1 transcription in trans (Figure 4b,c). In contrast, Epo-stimulated 32D.EpoR cells did not activate the PU.1 promoter.
Figure 4.
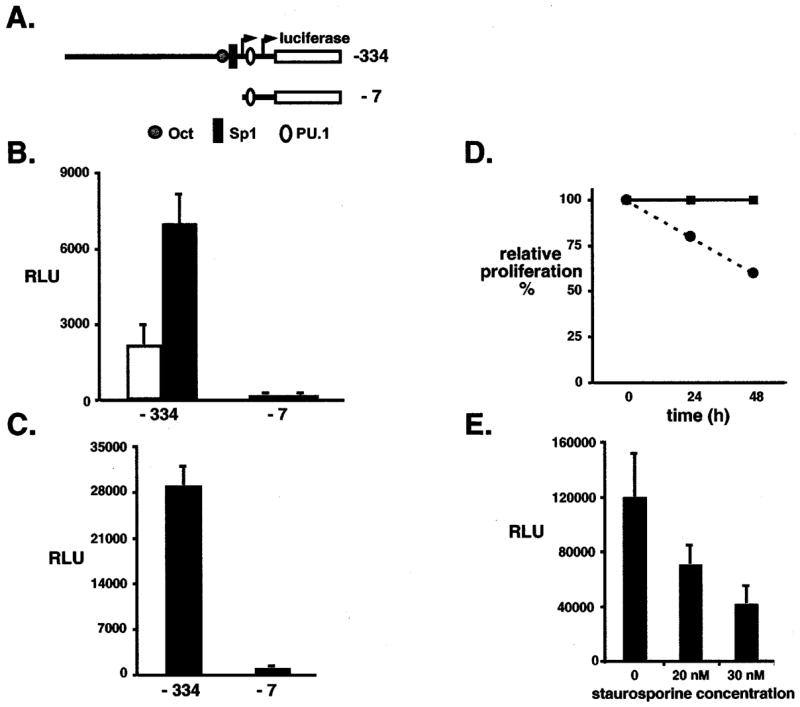
PU.1 promoter activity is increased following activation of the EpoR by F-gp55. (a) Schematic representation of the minimal murine PU.1 promoter luciferase plasmid (−334) and an inactive PU.1 promoter reporter plasmid (−7). Octamer (Oct), Sp1 and PU.1 binding sites within the promoter are highlighted. The horizontal arrows identify the location of the transcriptional start sites. The left arrow is the site utilized in 32D myeloid cells whereas the right arrow marks the site utilized in Friend erythroleukemia cells. (b) 32D.EpoR cells (white column) and 32D.EpoR/F-gp55 cells (black column) were transfected with PU.1 promoter reporter plasmid and CMV.beta-galactosidase plasmid in a 10:1 ratio. Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and the standard deviation from the mean for three to four independent experiments. (c) MEL cells were transfected with PU.1 promoter reporter plasmid and CMV.beta-galactosidase plasmid in a 10:1 ratio. Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and the standard deviation from the mean for three to four independent experiments. (d) MEL cell proliferation curve. MEL cells were grown in regular media (filled squares, solid line) or following the addition of 30 nM staurosporine (PKC inhibitor) (filled circles, dashed line). Results are presented as percent growth in regular media. (e) MEL cells were transfected with the minimal PU.1 promoter reporter plasmid (−334) and CMV.beta-galactosidase plasmid in a 10:1 ratio, then grown in the presence of increasing concentration of staurosporine for 26 h. Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and the standard deviation from the mean for three to four independent experiments
These results indicated that activation of the EpoR by F-gp55, but not Epo, resulted in transcriptional upregulation of PU.1, and that this could occur in the absence of any proviral integration within the PU.1 gene locus. This transcriptional upregulation of PU.1 was accomplished by an in trans mechanism and was observed in primary erythroblasts cells during the early phase of Friend erythroleukemia.
A functional F-gp55 activated EpoR signal was required for PU.1 transcriptional upregulation in Friend erythroleukemia cells
Accumulating evidence indicates that F-gp55 activated EpoR signals differ from Epo activated EpoR signals (Tarr et al., 1997; Muszynski et al., 2000; Nishigaki et al., 2000). Specifically F-gp55/EpoR activated PKC activity is required for proliferation of Friend erythroleukemia cells (Muszynski et al., 2000). To determine if a F-gp55/EpoR activated signal was required for transcriptional upregulation of PU.1, Friend erythroleukemia MEL cells were transiently transfected with the minimal PU.1 promoter-luciferase plasmid, then treated with, or without staurosporine (a PKC inhibitor), and luciferase activity determined. Staurosporine mediated inhibition of PKC activity in MEL cells resulted in a 40% reduction in proliferation rate, without any increase in cell death (Figure 4d, and not shown). Increasing concentrations of staurosporine resulted in progressively decreased PU.1 transcriptional activity in MEL cells (Figure 4e), whereas the level of F-gp55 protein was not affected by staurosporine treatment (not shown). This result indicated that a functional F-gp55/EpoR proliferative signal was required for upregulation of PU.1 transcription in Friend erythroleukemia cells.
PU.1 transcriptional regulation in Friend erythroleukemia cells
Increased PU.1 transcription following activation of the EpoR by F-gp55 prompted an analysis of PU.1 promoter activity in Friend erythroleukemia cells. Regulation of PU.1 gene expression in myeloid, and lymphoid cell lines has been previously investigated (Chen et al., 1995; Chen et al., 1996), however little is known of the mechanisms regulating PU.1 expression in MEL cells. Reporter plasmids containing deletion mutations of the PU.1 promoter (Figure 5a) were transiently transfected into MEL cells. Transfection efficiency was normalized to beta-galactosidase activity from a co-transfected CMV.beta-galactosidase plasmid. Hela cells and fibroblast cell lines, which do not express PU.1 mRNA, did not activate luciferase activity when transfected with the minimal PU.1 promoter (Chen et al., 1995 and data not shown). Removal of the −334 to −61 region of the PU.1 promoter had little effect upon transcriptional activity in MEL cells, however, further deletion of the −61 to −48 region resulted in an enhancement in transcriptional activity, suggesting the presence of repressor activity conveyed by the −334 to −61 region of the PU.1 promoter in MEL cells (Figure 5b). The major positive regulation of PU.1 transcription in MEL cells appeared to reside in the −48 to −7 region as deletion of this region resulted in a complete loss in activity (Figure 5b).
Figure 5.
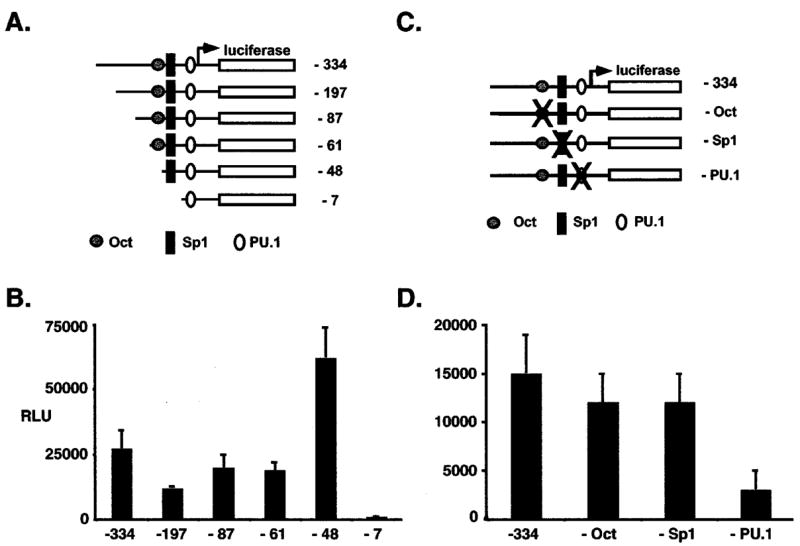
PU.1 promoter activity in Friend erythroleukemia cells. (a) Schematic representation of mouse minimal PU.1 promoter reporter plasmid (−334) and progressive deletions of the minimal promoter. (b) MEL cells were transfected with PU.1 promoter reporter plasmid and CMV.beta-galactosidase plasmid in a 10:1 ratio. Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and the standard deviation from the mean for three to four independent experiments. (c) Schematic representation of the minimal mouse PU.1 promoter reporter plasmid (−334) and promoter isoforms containing disabling point mutations (X) in the Oct (− Oct), Sp1 (− Sp1), and PU.1 (− PU.1) binding sites. (d) MEL cells were transfected as in (b). Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and the standard deviation from the mean for three to four independent experiments
To explore the contribution of specific transcription factors to the trans regulation of the PU.1 promoter in MEL cells, cells were transiently transfected with PU.1 promoter reporter plasmids containing disabling point mutations in the Octamer binding site, Sp1 binding site, and the PU.1 binding site (Figure 5c) (Chen et al., 1995). PU.1 binding site mutations reduced transcriptional activity by fivefold, whereas mutations in the Octamer and Sp1 binding sites had little effect (Figure 5d). These results indicated that PU.1 autoregulation of PU.1 transcription was important for the trans-regulation of PU.1 transcription in Friend erythroleukemia cells.
In Friend erythroleukemia cells PU.1 transcriptional regulation differed depending upon the level of PU.1 protein
During the preleukemic stage of Friend erythroleukemia PU.1 levels are low and cells retain the capacity for terminal differentiation, whereas in the leukemic stage Friend erythroleukemia cells express high levels of PU.1 and are blocked in their ability to differentiate. During both stages the EpoR is activated by F-gp55. MEL cells are representative of the leukemic stage of disease. Our analysis of PU.1 transcriptional regulation in MEL cells indicated that PU.1 autoregulated its own transcription. Therefore, we asked whether the transcriptional regulation of the PU.1 gene in MEL cells was affected by the level of PU.1 protein present in these cells. A short period (17 h) of DMSO treatment decreased PU.1 levels in MEL cells without any alteration in proliferation rate, survival, or differentiation (Figure 6b, right panel, and not shown). MEL cells were transiently transfected with the minimal PU.1 promoter-luciferase plasmid or a PU.1 promoter-luciferase plasmid containing a disabling point mutation in the PU.1 binding site (Figure 6a), then treated for 17 h with DMSO, and the relative luciferase activity determined (Figure 6b). The mimimal PU.1 promoter showed similar levels of activity in the absence or presence of DMSO, indicating that PU.1 protein levels did not influence its transcription rate. In contrast, transcription from the mutant promoter was increased following DMSO treatment and concomitant downregulation of PU.1 protein levels (Figure 6b: from 17% activity of the minimal promoter to 58% activity).
Figure 6.
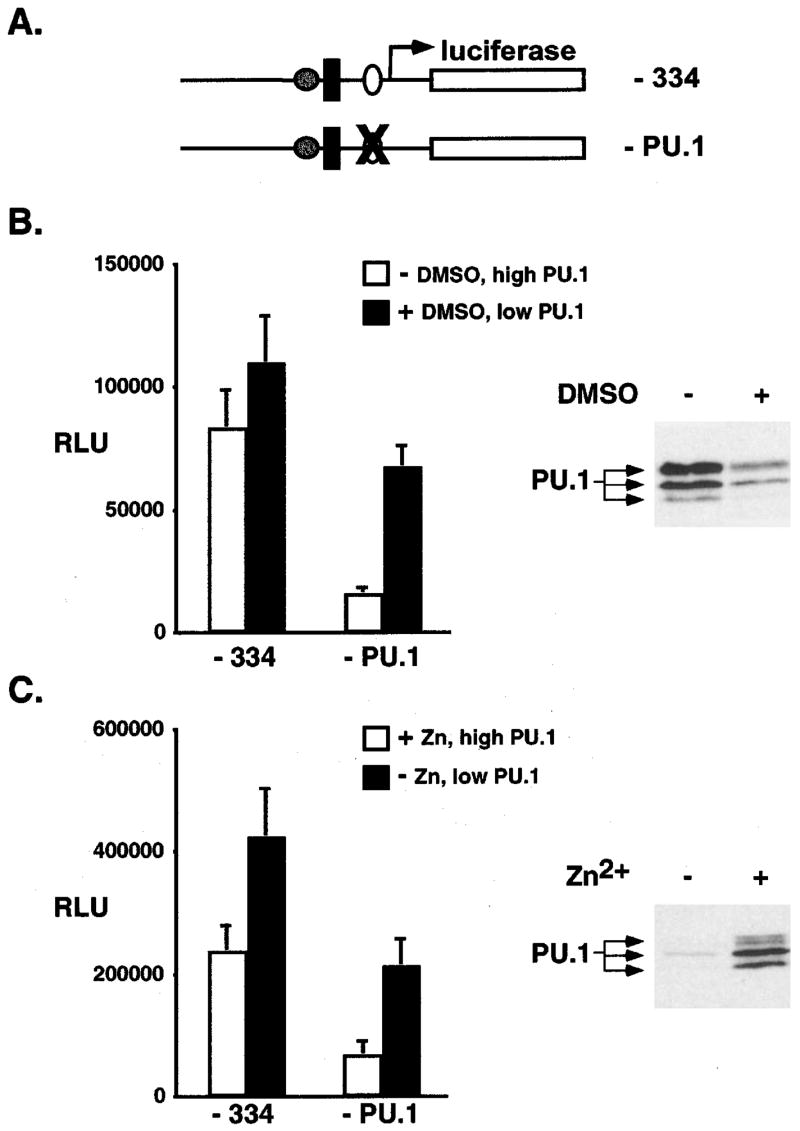
PU.1 promoter regulation in Friend erythroleukemia cells differed depending upon the level of PU.1 protein present. (a) Schematic representation of minimal PU.1 promoter (−334) and a disabling point mutation (X) in the PU.1 binding site (− PU.1). (b) MEL cells were transfected with −334 or −PU.1 promoter reporter plasmid and CMV.beta-galactosidase plasmid in a 10:1 ratio, then treated with (+) 1.5% DMSO or without (−) for 17 h. Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and standard deviation from the mean for three to four independent experiments. White columns represent −DMSO treatment. Black columns represent+DMSO treatment. Panel on the right is an immunoblot analysis for PU.1 protein levels in cells following DMSO treatment (+) or without DMSO treatment (−). Equal amounts of total protein were loaded in each lane. (c) MEL-B8/3 cells were transfected as described in (b) and then treated with 100 uM ZnCl2(+) or not (−) for 17 h. Results were normalized to beta-galactosidase activity and protein levels and reported as the relative luciferase units (RLU). Results represent the mean and standard deviation from the mean for three to four independent experiments. White columns represent+Zn treatment. Black columns represent −Zn treatment. Panel on the right is an immunoblot analysis for PU.1 protein levels in cells following Zn treatment (+) or without Zn treatment (−). Equal amounts of total protein were loaded in each lane
In a second approach we made use of another Friend erythroleukemia cell line, MEL-B8/3. This particular erythroleukemic cell line contains low basal levels of PU.1 protein, that can be increased following the introduction of a Zn-inducible PU.1 plasmid (Figure 6c, right panel) (Yamada et al., 1997). A similar analysis of PU.1 promoter activity was then performed in these cells (Figure 6c). As observed in MEL cells, in MEL-B8/3 cells containing low levels of PU.1 the transcriptional activity of the PU.1 binding site mutant promoter was increased relative to cells expressing high levels of PU.1 (from 25% activity of the minimal promoter to 50% activity). These results suggested that under conditions of low PU.1 levels (analogous to the early stage of Friend erythroleukemia) PU.1 autoregulation is not a major regulatory mechanism. Thus, the mechanisms regulating PU.1 transcription may differ during the early and late stages of Friend erythroleukemia disease.
Discussion
In this report we demonstrate that the env gene, F-gp55, of SFFV cooperates with PU.1 to accelerate erythroleukemia induction in mice. Both the time to the development of erythroblastosis (preleukemic first stage) and erythroleukemic transformation (leukemic second stage) were shortened, relative to mice infected with a retrovirus expressing F-gp55 alone. This was not due to increased F-gp55/PU.1 virus proliferation, in vivo, as the rate of virus replication or spread was not significantly different than that found in mice infected with F-gp55 virus (not shown).
To determine whether F-gp55 activated EpoR signals affected PU.1 activity we first developed model mouse cell lines, using 32D cells, in which the only difference was the mode of activation of the EpoR (either F-gp55 or Epo). In these cells, we found that activation of the EpoR by F-gp55, but not Epo, resulted in increased transcription of PU.1 through a trans regulatory mechanism, and this occurred in the absence of an integrated SFFV provirus within the PU.1 gene locus. In support of these conclusions: RNase protection and nuclear run-on transcription assays indicated that increased transcription of PU.1 occurred in 32D cells in which the EpoR was activated by F-gp55, but not by Epo. By Southern blot analysis of the PU.1 gene locus in these cells there was no evidence of an integrated SFFV provirus. Results of transient PU.1 promoter reporter gene analysis indicated that the increase in PU.1 transcription was regulated in trans.
Importantly, transcriptional upregulation of PU.1 was also evident in primary splenic erythroblasts isolated from early stage Friend virus infected mice (prior to the detection of clonal erythroleukemic cells), thereby confirming the observations from the model 32D cell line in a physiologically relevant context. In addition, the minimal PU.1 promoter was also active in Friend erythroleukemia cells, demonstrating trans regulation of PU.1 also occurs in erythroleukemic cells. Finally, a functional proliferative signal generated by F-gp55 activation of the EpoR was required for PU.1 promoter trans activation in Friend erythroleukemia cells as inhibition of PKC activity blocked this response.
The transregulation of transcription of the PU.1 oncogene by a pathway involving activation of EpoR by F-gp55 suggests a mechanism to explain the observed cooperativity of these oncogenes in Friend erythroleukemia induction, in vivo. Other examples of transregulation of transcription during retroviral oncogenesis have been described. Bovine leukosis virus (BLV) and human T-cell lymphotropic virus (HTLV) do so via the virally encoded transactivator protein tax (Rosen et al., 1985) (Sodroski et al., 1984). The U3 region of the LTRs of Moloney MuLV LTR and Feline leukemia virus give rise to an RNA product that appears to regulate transcription of a number of genes in trans (Choi and Faller, 1995) (Ghosh and Faller, 1999). These latter viruses are slow transforming viruses whereas Friend virus is an acute transforming virus. Since F-gp55 is a transmembrane protein that activates a mitogenic signal through an interaction with the EpoR at the cell surface (Li et al., 1995), signals emanating from an F-gp55 activated EpoR, not direct regulation of nuclear transcription (as described for BLV, HTLV, and M-MuLV), lead to increased PU.1 transcription.
The increase in transcription of PU.1 following activation of the EpoR by F-gp55 was specific to PU.1 as there was minimal difference in the transcriptional rate of Cytokine Inducible SH2-containing protein (CIS), an Epo-induced JAK/STAT responsive gene (Yoshimura et al., 1995) in 32D EpoR/F-gp55 cells versus 32D EpoR cells grown in Epo. This suggests that an EpoR activated signaling pathway other than, or in addition to, the JAK/STAT pathway is required for PU.1 induction. Accumulating evidence indicates that the signaling pathways activated following interaction of the EpoR with F-gp55 differ from those activated following engagement of the EpoR by Epo. The cell surface organization of the EpoR differs when activated by F-gp55 compared to activation by Epo (Tarr et al., 1997). While constitutive activation of Stat proteins, Ras/Raf-1-MAPK pathway, PI3K pathway, the lipid phosphatase SHIP, and PKC occur in SFFV infected erythroid cells, tyrosine phosphorylation of the EpoR and activation of JAK2 appear to not be required for proliferation of SFFV infected cells (Muszynski et al., 1998; Muszynski et al., 2000; Nishigaki et al., 2000). Activation of PI3K signaling pathways in SFFV infected cells occurs through tyrosine phosphorylated Insulin Receptor Substrate-Related (IRS) adapter proteins (Nishigaki et al., 2000). The protein kinase responsible for phosphorylating these IRS adapter proteins, in SFFV-infected cells, are unknown but a recently identified truncated isoform of Stem Cell Kinase (Stk) or RON kinase as being responsible for conferring sensitivity to Friend disease makes this an attractive candidate (Persons et al., 1999). Possibly as a result of these differences novel signaling pathways, or differing activities of common signaling pathways, contribute to SFFV induced Epo-independent erythroid proliferation.
Most Friend erythroleukemia cell lines contain an SFFV provirus integrated 5′ of the PU.1 gene, in a reverse orientation relative to the PU.1 promoter. This observation led to the hypothesis that enhancer elements present in the LTR of SFFV activates PU.1 transcription, although direct experimental support for this model is lacking (Moreau-Gachelin et al., 1988; Moreau-Gachelin et al., 1989; Paul et al., 1989; Paul et al., 1991). This model is typical of many leukemogenic retroviruses with long latency periods (e.g., 6 – 12 months) (Jonkers and Berns, 1996). SFFV is an acutely transforming retrovirus, however, resulting in leukemia within 5 – 6 weeks following infection. In the myeloid cell line 32D, F-gp55 activated EpoR signals stimulated PU.1 transcription in the absence of any proviral element within the PU.1 gene locus. However, myeloid cells differ from erythroid progenitors (i.e., the target cell for transformation by SFFV) in that the PU.1 gene is already transcriptional active (Chen et al., 1995). In early proerythroblasts a low level of PU.1 protein is present. As these cells further mature PU.1 production is shut off, suggesting that persistence of PU.1 protein is not compatible with erythroid differentiation (Hromas et al., 1993). Possibly SFFV proviral insertion within the PU.1 gene locus during the evolution of Friend erythroleukemia may, in part, alter chromatin structure allowing for PU.1 transcription to persist. F-gp55 activated EpoR signals may then contribute, in part, to the increase in PU.1 transcription necessary to transform erythroblasts. In support of this possibility we observed that trans regulation of PU.1 transcription was also present in MEL cells, a fully transformed Friend erythroleukemia cell line containing an integrated SFFV provirus in the PU.1 gene locus.
Our analyses of transcriptional regulation of the PU.1 promoter in established Friend erythroleukemia cell lines suggests that, it is regulated from two sites dependent upon the level of PU.1 protein present. When PU.1 protein levels are high, PU.1 promoter auto regulation was apparent. In cells containing low levels of PU.1 protein PU.1-site dependent auto regulation of PU.1 promoter was significantly diminished. In the setting of low levels of PU.1, possible modulation of the repressive activity associated with the −334 to −61 region of the minimal PU.1 promoter may contribute to the persistent activity of the PU.1 promoter.
Thus, in the early, preleukemic stage of Friend erythroleukemia the PU.1 promoter may be activated in a PU.1-independent manner by an EpoR/F-gp55 signal. As the disease progresses clones containing an integrated SFFV provirus in the PU.1 gene locus expand and the level of PU.1 protein present reaches a threshold after which PU.1 autoregulates PU.1 transcription, thereby maintaining high levels of PU.1 protein, necessary to maintain the transformed phenotype. The importance of the PU.1 site to the regulation of PU.1 transcription in myeloid cells has been previously proposed (Chen et al., 1995), however, the contribution of the PU.1 site to the regulation of PU.1 transcription in Friend erythroleukemic cells was more pronounced than in myeloid cells. Determining the relative contributions of cis (e.g., SFFV proviral insertion) and trans (e.g., F-gp55/EpoR signals) regulation of PU.1 transcription in Friend erythroleukemia induction is clearly important.
PU.1 contributes to erythroid transformation through an interaction with the erythroid determining factor GATA-1, inhibiting its DNA-binding capacity (Rekhtman et al., 1999; Zhang et al., 1999). The relative levels of PU.1 and GATA-1 are important for this effect. We found that PU.1 protein stability is significantly decreased in Friend erythroleukemia cells compared to non-transformed myeloid progenitors (data not shown). Since a reduction of PU.1 protein levels in Friend erythroleukemia cells results in terminal differentiation (Schuetze et al., 1992), cis (proviral insertion) and trans (activation of the EpoR by F-gp55) transcriptional regulation of the PU.1 gene may both be required to maintain high levels of PU.1 protein necessary for the erythroid differentiation arrest and promotion of their survival. Our results suggest that the action of F-gp55 contributes to both early and late stages of Friend erythroleukemia and that persistence of F-gp55 expression may be required not only to initiate erythroleukemia but also to maintain erythroleukemia following Friend virus infection.
Materials and methods
Cell line
The derivation of 32D.EpoR cells has been described (Beckman et al., 1999) and these cells were maintained in RPMI-1640 containing 10% Fetal bovine serum (FBS) (RPMI/10%FBS), supplemented with human Epo (0.5 to 1.0 U/ml). Infecting 32D.EpoR cells with SFF.F-gp55 retrovirus and selecting for all cells that grew in media lacking Epo generated 32D EpoR/F-gp55 cells. MEL-B8/3 cells containing a Zn-inducible expression plasmid of PU.1 (Yamada et al., 1997) were maintained in RPMI/10% FBS media. MEL-745 cells were grown in Dulbecco’s Modified Eagle’s Media (DMEM) with 10% FBS, and 44 mM sodium carbonate (Sigma, St. Louis, MO, USA). All cells were grown at 37°C in 5% CO2.
Retrovirus production, infection of mice, and analysis
The F-gp55 env gene fragment from a polycythemic strain of SFFV was subcloned into the pSFF vector (Bestwick et al., 1988) to generate pSFF.F-gp55. An internal ribosomal entry site (IRES) myc-tagged murine PU.1 cassette was subcloned into the EcoR1 site of pSFF to generate pSFF.PU.1. To generate pSFF.F-gp55/PU.1, the polycythemic F-gp55 env gene from SFFV was subcloned into an Xho1 site, 5′ to the IRES-PU.1 cassette, in pSFF.PU.1. The sequences of all subcloned fragments were confirmed by DNA sequencing. The preparation of retroviruses was carried out as previously described (Longmore and Lodish, 1991). Six week old, female NIH/Swiss mice were infected, intravenously, with the above retroviruses (including replication competent, nonpathogenic, helper virus, R-MuLV) as previously described (Longmore and Lodish, 1991) (Longmore et al., 1994). Following infection blood hematocrit were determined each week for 8 weeks. For tumorigenic assays, mice were sacrificed at various times following infection, the spleen isolated, and a mononuclear splenic cell population generated. Spleen cells were then cultured in murine methylcellulose media (Stem Cell Technologies, Vancouver, Canada), without any additional cytokines. After 7 to 10 days in culture, transformed erythroid colonies were identified and counted. To confirm the erythroid nature of these colonies, cytospins were performed, stained with Wright-Giemsa (Sigma, St. Louis MO, USA), and examined microscopically. In vivo rates of viral replication were determined, and compared, by titering serum from infected mice. Viral titer was determined by measuring the efficiency of each virus to transduce F-gp55 into fibroblasts.
Cell purification
Mice were treated with phenylhydrazine (PHZ) 60 mg/kg intraperitoneally on day −2 and day −1, and on day 2 or day 3 mice were sacrificed and the enlarged spleens isolated. Other mice were infected with SFF.F-gp55 virus, and sacrificed 1 or 2 weeks post infection and the spleens isolated. Splenic mononuclear light density cells were isolated on Ficoll-Paque gradients, adherent cells discarded, B and T cells depleted with Dynabeads (Dynal AS, Oslo, Norway), and TER-119 splenic erythroblasts isolated (Vannucchi et al., 2000).
RNase protection assays
Total RNA was prepared from exponentially growing cultures using TriReagent (Molecular Research Center, Inc.). RNase protection probes corresponding to the murine PU.1 coding region or nucleotides 9-105 of the murine GAPDH coding region were synthesized in the presence of 32P-UTP (800 m Ci/mmol; Amersham) using the Maxiscript kit (Ambion), following the manufacturer’s instructions, and purified. RNase protection assays were performed with 10 ug RNA using the RPA III Ribonuclease protection assay (Ambion) following the manufacturer’s instructions. The results were analysed by electrophoresis on 5% acrylamide gels containing 8 M urea followed by autoradiography.
Nuclear run-on transcription assays
Nuclei were isolated from 108 cells in exponential growth phase. Cells were lysed in buffer containing 0.5% Triton X-100, 10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 and incubated on ice for 5 min. Nuclei were pelleted by microcentrifugation for 1 min and resuspended in an equal volume of storage buffer (40% glycerol, 50 mM Tris-HCl pH 8.0, 5 mM MgCl2, 0.1 mM EDTA). For run-on transcription assays, 100 ul of nuclei (@ 3.3 × 107) were mixed with 100 ul 2 × reaction mix (50 mM HEPES pH 7.4, 5 mM MgCl2, 5mM DTT, 150 mM KCl, 10% glycerol, 0.7 mM ATP, 0.7 mM CTP, 0.7 mM GTP, 0.8 uM UTP) containing 2 m Ci/ml 32P-UTP (3000 Ci/mmol; Amersham) and incubated for 25 min at room temperature. Genomic DNA was then digested by incubation with 60 ug/ml RNase-free DNase (specific activity=37500 u/ml; Worthington Biochemicals) for 20 min at 37°C. Reactions were terminated by the addition of an equal volume of 2 × stop buffer (2% SDS, 7 M Urea, 10 mM Tris-HCl pH 8.0, 0.35 M LiCl, 1 mM EDTA). 32P-labeled RNA was purified by proteinase K (500 ug/ml) digestion for 1 h at 42°C, followed by phenol:chloroform extraction and TCA precipitation using 50 ug tRNA as carrier. RNA samples were resuspended in 100 ul of buffer containing 10 mM Tris-HCl pH 7.4, 1 mM EDTA and 0.5% SDS. Samples were denatured for 10 min at 65°C and added to prehybridized nitrocellulose filters containing immobilized cDNAs. Hybridization was performed at 42°C for 84 h in buffer containing 50% formamide, 6 × SSC, 5 × Denhardt’s solution, 0.1% SDS and 50 ug/ml tRNA. Filters were washed once with 6 × SSC/0.2% SDS, once with 2 × SSC/0.2% SDS and twice with 0.2 × SSC/0.2% SDS for 30 min each at 65°C. Filters were air-dried and exposed to x-ray film at −80°C.
Southern blot analysis
Total genomic DNA was isolated by the DNA Isolation Kit (Boehringer Mannheim) following the manufacturer’s instructions. Ten ug of genomic DNA was digested overnight with EcoRV and electrophoresed in a 0.8% agarose gel. DNA was then transferred to GeneScreen Plus membranes (NEN Life Science, Boston, MA, USA), prehybridized in DIG Easy Hyb solution (Roche Molecular Biochemicals), and then hybridized overnight at 45°C with 32P-labeled PU.1 genomic probe A (Moreau-Gachelin et al., 1989). The Membrane was then rinsed once in 2 × SSC and washed for 20 min in 2 × SSC at room temperature followed by a 0.2 × SSC/0.1% SDS wash at 60°C for 30 min. The membrane was then exposed to x-ray film at −80°C.
PU.1 promoter analyses
Expression plasmids containing the mouse PU.1 promoter driving expression of luciferase were kindly provided by D Tenen (Harvard Medical School, Boston MA, USA) (Chen et al., 1995). 32D cells were transiently transfected by electroporation (BTX, Electro Cell Manipulator 600). MEL cells were transiently transfected using cationic lipid reagent Tf × 50 (Promega), as described (Elnitski and Hardison, 1999). Transfection efficiency was normalized to b-galactosidase activity by co-transfection with a CMV.b-galactosidase reporter plasmid. The ratio between PU.1 promoter luciferase and CMV.b-galactosidase plasmids was 10 : 1. Following electroporation cells were kept on ice for 10 min and resuspended in RPMI/10% FBS with or without Epo. Cells were harvested after 17 h and luciferase activity (Luciferase Reporter Assay System; Promega) and b-galactosidase activity (b-galactosidase Enzyme Assay System, Promega, or b-Gal Kit, Clontech) determined, following the manufacturer’s instructions. Results were reported as the relative luciferase units (RLU) and represent the mean and the standard deviation from the mean for three to four independent experiments.
Protein analyses
For immunoblots, exponentially growing cells were pelleted, washed once with ice cold PBS, and lysed with RIPA buffer (containing protease inhibitors). Following clarification of the cell extract protein determinations were performed (BioRad protein determination kit, Pierce, Rockford IL, USA). Samples were fractionated on 10% SDS – PAGE gels, under reducing conditions, transferred to PVDF membrane (Amersham Pharmacia Biotech), and then incubated with primary antibodies. Antibodies used included rabbit polyclonal anti-PU.1 antibody (SC-352; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rat monoclonal anti-F-gp55 antibody (Dr Sandra Ruscetti, NCI Frederick), and rabbit polyclonal anti-myc antibody (Dr A Shaw, Washington University, St. Louis MO, USA). Bound antibody was detected using Horse radish peroxidase coupled secondary antibodies and ECL reagents (SuperSignal West Dura Trial Kit; Pierce, Rockford, IL, USA).
Acknowledgments
We would like to thank Dr Dan Tenen (Harvard Medical School, Boston MA, USA) for generously providing mouse PU.1 promoter luciferase plasmids, and helpful comments. Drs J Ghysdael and S Ruscetti for helpful comments. Dr T Yamada, Sasaki Institute, Tokyo for providing us with MEL-B8/3 cells. This work was supported by the NIH (R01 CA77447) and the Gillson Longenbaugh foundation (to SSW), and from the Association for International Cancer Research, the American Heart Association (9940116N), the Edward Mallinckrodt Jr. Foundation, and the NIH (RO1 CA75315) (to GD Longmore). GD Longmore is an Established Investigator of the American Heart Association.
References
- Barnache S, Wendling F, Lacombe C, Denis N, Titeux M, Vainchecker W, Moreaeu-Gachelin F. Oncogene. 1998;16:2989–2995. doi: 10.1038/sj.onc.1202095. [DOI] [PubMed] [Google Scholar]
- Beckman DL, Lin LL, Quinones ME, Longmore GD. Blood. 1999;94:2667–2675. [PubMed] [Google Scholar]
- Ben-David Y, Bernstein A. Cell. 1991;66:831–834. doi: 10.1016/0092-8674(91)90428-2. [DOI] [PubMed] [Google Scholar]
- Bestwick RK, Kozak SL, Kabat D. Proc Natl Acad Sci (USA) 1988;85:5404–5408. doi: 10.1073/pnas.85.15.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ray-Gallet D, Zhang P, Hetherington CJ, Gonzalez DA, Zhang D-E, Moreau-Gachelin F, Tenen DG. Oncogene. 1995;11:1549–1560. [PubMed] [Google Scholar]
- Chen H, Zhang P, Radomska HS, Hetherington C, Zhang D-E, Tenen DG. J Biol Chem. 1996;271:15743–15752. doi: 10.1074/jbc.271.26.15743. [DOI] [PubMed] [Google Scholar]
- Choi S-Y, Faller DV. J Virol. 1995;69:7054–7060. doi: 10.1128/jvi.69.11.7054-7060.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnitski L, Hardison R. Blood Cells. 1999;25:299–304. doi: 10.1006/bcmd.1999.0257. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Ghosh SK, Faller DV. J Virol. 1999;73:4931–4940. doi: 10.1128/jvi.73.6.4931-4940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoatlin ME, Kabat D. Trends in Microbiology. 1995;3:51–57. doi: 10.1016/s0966-842x(00)88875-7. [DOI] [PubMed] [Google Scholar]
- Howard JC, Yousefi S, Cheong G, Bernstein A, Ben-David Y. Oncogene. 1993;8:2721–2729. [PubMed] [Google Scholar]
- Hromas R, Orazi A, Meiman RS, Maki R, Van Beveran C, Moore J, Klemscz M. Blood. 1993;82:2998–3004. [PubMed] [Google Scholar]
- Jonkers J, Berns A. Biochem Biophys Acta. 1996;1287:29–57. doi: 10.1016/0304-419x(95)00020-g. [DOI] [PubMed] [Google Scholar]
- Kelley LL, Hicks GG, Hsieh FF, Prasher JM, Green WF, Miller MD, Eide EJ, Ruley HE. Oncogene. 1998;17:1119–1130. doi: 10.1038/sj.onc.1202037. [DOI] [PubMed] [Google Scholar]
- Li J-P, Bestwick RK, Spiro C, Kabat D. J Virol. 1987;61:2782–2792. doi: 10.1128/jvi.61.9.2782-2792.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-P, D’Andrea A, Lodish H, Baltimore D. Nature. 1990;343:762–764. doi: 10.1038/343762a0. [DOI] [PubMed] [Google Scholar]
- Li JP, Hu HO, Niu QT, Fang C. J Virol. 1995;69:1714–1719. doi: 10.1128/jvi.69.3.1714-1719.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longmore GD, Lodish HF. Cell. 1991;67:1089–1102. doi: 10.1016/0092-8674(91)90286-8. [DOI] [PubMed] [Google Scholar]
- Longmore GD, Pharr PN, Lodish HF. Mol Cell Biol. 1994;14:2266–2277. doi: 10.1128/mcb.14.4.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager D, Mak TW, Bernstein A. Proc Natl Acad Sci USA. 1981;78:1703–1707. doi: 10.1073/pnas.78.3.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau-Gachelin F, Ray D, Mattei M-G, Tambourin P, Tavitian A. Oncogene. 1989;4:1449–1456. [PubMed] [Google Scholar]
- Moreau-Gachelin F, Tavatian A, Tambourin P. Nature. 1988;331:277–280. doi: 10.1038/331277a0. [DOI] [PubMed] [Google Scholar]
- Moreau-Gachelin F, Wendling F, Molina T, Denis N, Titeux M, Grimber G, Briand P, Vainchenker W, Tavitian A. Mol Cell Biol. 1996;16:2453–2463. doi: 10.1128/mcb.16.5.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat M, Cheng A, Kimura N, Bernstein A, Benchimol S. Nature. 1985;314:633–636. doi: 10.1038/314633a0. [DOI] [PubMed] [Google Scholar]
- Muszynski KM, Thompson D, Hanson C, Lyons R, Spadaccini A, Ruscetti SK. J Virology. 2000;74:8444–8451. doi: 10.1128/jvi.74.18.8444-8451.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszynski KW, Ohashi T, Hanson C, Ruscetti SK. J Virology. 1998;72:919–925. doi: 10.1128/jvi.72.2.919-925.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigaki K, Hanson C, Ohashi T, Thompson D, Muszynski K, Ruscetti SK. J Virology. 2000;74:3037–3045. doi: 10.1128/jvi.74.7.3037-3045.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Scheutze S, Kozak SL, Kozak CA, Kabat D. J Virol. 1991;65:464–467. doi: 10.1128/jvi.65.1.464-467.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Schuetze S, Kozak S, Kabat D. J Virol. 1989;63:4958–4961. doi: 10.1128/jvi.63.11.4958-4961.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira R, Raingeaud J, Pironin M, Ghysdael J, Tran Quang C. Oncogene. 2000;19:5106–5110. doi: 10.1038/sj.onc.1203886. [DOI] [PubMed] [Google Scholar]
- Persons DA, Paulson RF, Loyd MR, Herley MT, Bodner SM, Bernstein A, Correll PH, Ney PA. Nature Genetics. 1999;23:159–165. doi: 10.1038/13787. [DOI] [PubMed] [Google Scholar]
- Rekhtman N, Radparvar F, Evans T, Skoultchi AI. Genes Dev. 1999;13:1398–1411. doi: 10.1101/gad.13.11.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen CA, Sodroski JG, Kettman R, Burny A, Haseltin WA. Science. 1985;227:320–322. doi: 10.1126/science.2981432. [DOI] [PubMed] [Google Scholar]
- Ruscetti SK. Int J Biochem Cell Biol. 1999;31:1089–1109. doi: 10.1016/s1357-2725(99)00074-6. [DOI] [PubMed] [Google Scholar]
- Schuetze S, Paul R, Gliniak BC, Kabat D. Mol Cell Biol. 1992;12:2967–2975. doi: 10.1128/mcb.12.7.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetze S, Stenberg PE, Kabat D. Mol Cell Biol. 1993;13:5670–5678. doi: 10.1128/mcb.13.9.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodroski JG, Rosen C, Haseltine W. Science. 1984;225:381–385. doi: 10.1126/science.6330891. [DOI] [PubMed] [Google Scholar]
- Tarr K, Watowich SS, Longmore GD. J Biol Chem. 1997;272:9099–9107. doi: 10.1074/jbc.272.14.9099. [DOI] [PubMed] [Google Scholar]
- Tran Quang C, Wessely O, Pironin M, Beug H, Ghysdael J. EMBO J. 1997;16:5639–5653. doi: 10.1093/emboj/16.18.5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucchi AM, Paoletti F, Linari S, Cellai C, Caporale R, Ferrini P, Sanchez M, Migliaccio G, Migliaccio AR. Blood. 2000;95:2559–2568. [PubMed] [Google Scholar]
- Weinberg RA. In: Oncogenes and the molecular origins of cancer. Weinberg RA, editor. Cold Spring Harbor; Cold Spring Harbor Laboratory Press: 1989. pp. 307–327. [Google Scholar]
- Wolff L, Ruscetti S. Science. 1985;228:1549–1554. doi: 10.1126/science.2990034. [DOI] [PubMed] [Google Scholar]
- Yamada T, Kondoh N, Matsumoto M, Yoshida M, Maekawa A, Oikawa T. Blood. 1997;89:1383–1393. [PubMed] [Google Scholar]
- Yoshimura A, Ohkubo T, Kiguchi T, Jenkins N, Gilbert DJ, Copeland NG, Hara T, Miyajima A. EMBO J. 1995;14:2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Behre G, Pan J, Iwama A, Wara-aswapati N, Radomska HS, Auron PE, Tenen DG, Sun Z. Proc Natl Acad Sci USA. 1999;96:8705–8710. doi: 10.1073/pnas.96.15.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]