

Abstract
We develop a new method for estimating effective population sizes, Ne, and selection coefficients, s, from time-series data of allele frequencies sampled from a single diallelic locus. The method is based on calculating transition probabilities, using a numerical solution of the diffusion process, and assuming independent binomial sampling from this diffusion process at each time point. We apply the method in two example applications. First, we estimate selection coefficients acting on the CCR5-Δ32 mutation on the basis of published samples of contemporary and ancient human DNA. We show that the data are compatible with the assumption of s = 0, although moderate amounts of selection acting on this mutation cannot be excluded. In our second example, we estimate the selection coefficient acting on a mutation segregating in an experimental phage population. We show that the selection coefficient acting on this mutation is ∼0.43.
THE vast majority of analyses of selection are based on samples of molecular data obtained at a single point in time. However, in a few cases, time series of allele frequencies are available. Examples of such data are ancient DNA (aDNA) data in humans (Hummel et al. 2005), viral population data (Shankarappa et al. 1999), and data on experimentally evolved populations such as Drosophila (Buri 1956), bacterial (Woods et al. 2006), or viral/phage populations (Wichman et al. 1999, 2005; Holder and Bull 2001; Bollback and Huelsenbeck 2007). Time-series data contain much more information regarding selection than samples obtained at a single point in time, because the expected changes in allele frequencies through time are closely related to the strength of selection. The objective of this article is to develop a statistical approach for estimating selection coefficients, and testing hypotheses regarding selection coefficients, that can take advantage of the information from a time series of allele frequencies.
The method for estimating selection coefficients from allele frequency data presented here is a natural extension of existing methods for estimating the effective population size, Ne, from this type of data. A number of methods for estimating Ne, in the absence of selection, have been developed. The first such methods were moments-based estimators (Krimbas and Tsakas 1971; Nei and Tajima 1981; Pollak 1983; Waples 1989). Unfortunately, these methods suffer from a number of biases, such as an upward bias in the estimate of Ne with low-frequency alleles.
Williamson and Slatkin (1999) developed a maximum-likelihood approach for estimating Ne from changes in allele frequencies using a hidden Markov model (HMM). This approach assumes a Wright–Fisher model of neutral evolution. We can think of the model as an HMM with state space on the set of possible allele frequencies, transition probabilities among states given by the Wright–Fisher Markov chain, and with emission probabilities obtained as the sampling probabilities arising when taking a smaller sample of gene copies from the population (Anderson et al. 2000). Given such a model, the likelihood of Ne can be maximized with respect to the observed allele frequencies sampled at a number of different time points. This approach allows for samples that are irregularly spaced in time (i.e., unsampled generations), but in its original form by Williamson and Slatkin (1999), it was, for computational reasons, restricted to diallelic markers.
Anderson et al. (2000) extended the method to the case of multiple alleles using a somewhat computationally intensive Monte Carlo approach that relies on importance sampling to evaluate the likelihood. Wang (2001) further developed this approach to increase the speed of the likelihood estimation of Ne and included a simulation study showing that the behavior of likelihood-based methods is superior to that of the moments-based estimators.
More recently, Berthier et al. (2002) developed a method for estimating Ne from two time-point samples that relies on an underlying coalescent model. This method was extended to multiple time points by Beaumont (2003). This method is an improvement over previous likelihood methods in that the computation of the likelihood can be faster when many generations separate the samples. The speed of these approaches was improved considerably by Anderson (2005), who, rather than using Markov chain Monte Carlo techniques, developed a Monte Carlo importance-sampling approach. This method has the nice property that the accuracy of the estimator can more easily be established, as was not the case with the previous methods.
In this article we expand on these methods to estimate both 2Ne and the selection coefficient, s, from temporal samples of diallele frequency data. In contrast to previous approaches, we use the diffusion process as the underlying Markov process describing changes in allele frequencies. This allows the method to be computationally efficient even for large population sizes. In the following we present the theory and demonstrate the method on two common types of data that are being collected today, aDNA and experimental evolution studies.
MATERIALS AND METHODS
Theory:
The trajectory of an allele through time can be modeled as a Markov process (see, e.g., Ewens 2004). One set of models assumes discrete time and overlapping (e.g., Moran models) or nonoverlapping (e.g., Wright–Fisher models) generations. In the limit of large population sizes, all of these models can be described by a common diffusion process, X(t) ∈ [0, 1], with transition probabilities described by the backward Kolmogorov equations
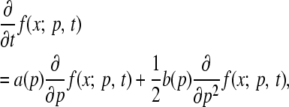 |
(1) |
where f(x; p, t) = p(X(t) = x | X(0) = p) is the density of the allele frequency t time units after it had frequency p, and a(p) = sNep(1 − p) and b(p) = p(1 − p). The model is parameterized in terms of the selection coefficient, s, acting on the mutations (assuming codominance for a diploid population) and Ne, the effective population size. Time is measured in terms of 2Ne generations. Without recurrent mutation, the diffusion process has exit barriers at X(t) = 0 and X(t) = 1, and absorption probabilities
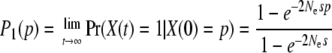 |
(2) |
and probability of loss given by P0(p) = 1 − P1(p). In one application of the model we consider only paths that have led to fixation of the mutation [absorption at X(t) = 1] as the mutation in this case reaches fixation and is known to be beneficial. In practice this assumes that the mutation is beneficial and will not be appropriate for data sets in which the mutation is not, a priori, known to be beneficial and does not reach fixation during the sampling period. The conditional transition probabilities of the process are then, for 0 < X(t) < 1, redefined as
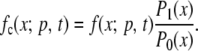 |
(3) |
On the basis of this model we wish to calculate the joint sampling probabilities of allele frequencies sampled at different times. Conceptually, we can think of this as a hidden Markov process (HMM) problem in continuous time and with a continuous state space, where the hidden Markov process is given by Equation 1. The emission probabilities of the process are given by the binomial sampling probabilities
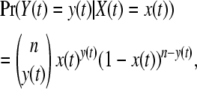 |
(4) |
where Y(t) is the number of alleles of the mutant type in a sample of size n, but n has been suppressed in the notation on the left-hand side of the equation. Assume that samples have been obtained at k time points, t1, t2, …, tk. The recursive function
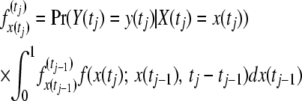 |
(5) |
then gives the joint sampling distribution of x(tj) and the observations before time tj and
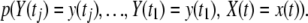 |
(6) |
Equation 5 is the product of the transition probability along the diffusion, integrated over all possible states at the previous time, and multiplied by the sampling probability of the observation at the current time step. It is similar to the recursive equation that is the basis for the dynamic programming algorithm known as the forward algorithm for HMMs (see, e.g., Durbin et al. 1998, p. 58). The only difference is that the summation has been replaced by an integral due to the fact that the hidden Markov process is defined on a continuous-state space. Equations 5 and 6 imply that the joint sampling probability is given by
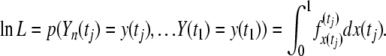 |
(7) |
To fully specify the system, we use the initial condition
 |
(8) |
This corresponds to using a uniform [0, 1] prior for x(t0). An alternative approach might have been to assume that x(t0) is sampled from the stationary distribution under recurrent mutation. In such an approach much of the information regarding s would come from its initial frequency. If the allele initially is common, that would provide evidence in favor of large values of s. However, particularly in the example regarding experimental evolution, it is clear that assumptions of initial stationarity are not met. We have, therefore, instead chosen the use of a uniform prior, which has the effect that the initial allele frequency does not enter into the calculation of the likelihood function and that the only information regarding initial population frequency of the allele entering into the calculations is its initial sample frequency.
Using this approach, the likelihood function for the parameters s and Ne can then be calculated for a time series in linear time using standard dynamic programming algorithms, if the integral on the right-hand side of Equation 5 can be solved.
Numerical approximations:
We numerically evaluate 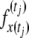 to calculate the sampling probability (Figure 1). The numerical approximation consists of two steps. First, the transition probabilities of the process are evaluated by numerically solving Equation 1 using the Crank–Nicolson method (Crank and Nicolson 1947). Briefly, derivatives are approximated by finite differencing, and implicit and explicit time steps are alternated, leading to an easily soluble sparse linear system of equations and giving both numerical stability and accuracy, which is second order in the size of the time step. We then evaluate the integral in Equation 6, using numerical integration based on quadrature using the midpoint rule. The same grid of values used for the Crank–Nicolson approximation is used for the numerical integration. To ensure a smooth likelihood surface we use a fixed grid for all parameter values.
to calculate the sampling probability (Figure 1). The numerical approximation consists of two steps. First, the transition probabilities of the process are evaluated by numerically solving Equation 1 using the Crank–Nicolson method (Crank and Nicolson 1947). Briefly, derivatives are approximated by finite differencing, and implicit and explicit time steps are alternated, leading to an easily soluble sparse linear system of equations and giving both numerical stability and accuracy, which is second order in the size of the time step. We then evaluate the integral in Equation 6, using numerical integration based on quadrature using the midpoint rule. The same grid of values used for the Crank–Nicolson approximation is used for the numerical integration. To ensure a smooth likelihood surface we use a fixed grid for all parameter values.
Figure 1.—

Graph of the hidden Markov process model (HMM) showing the calculation of 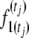 . Transition probabilities, px,i = f(x; p, t) from x to 1 are numerically solved using the Crank–Nicholson method (Crank and Nicolson 1947) on a grid of size n.
. Transition probabilities, px,i = f(x; p, t) from x to 1 are numerically solved using the Crank–Nicholson method (Crank and Nicolson 1947) on a grid of size n.
Grid size and spacing:
The adequacy of the numerical approximations to Equation 1 will depend on the grid sizes used and the spacing between points—when the density is concentrated at the exit barriers, X(t) = 0 and X(t) = 1, the approximation may be poor. Therefore, we used exponentially spaced grid points to increase the number of grid points near the boundaries, while decreasing the number of central points, when the numerical solutions did not converge. The position of the n grid points can be computed in the following way. First, the points from i = 1, i = 2, …, i = n/2, starting near the boundary at zero, can be calculated as
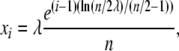 |
(9) |
where λ is the spacing parameter. Second, the position of the remaining points, [0.5, 1), is simply the reflection of the points calculated in Equation 9. To avoid spurious differences in the likelihood calculations, due to the choice of grid points, we used a fixed grid for all time points and parameter values.
CCR5-Δ32:
In the first application we use a data set consisting of time-series allele frequency data for the CCR5-Δ32 locus (see Figure 2; Hummel et al. 2005). Briefly, Hummel et al. (2005) determined the frequency of this mutation from ancient human remains and an extant representative population of northern Europeans: samples were collected at five time points dating back to 900 B.C. (Figure 2). In these analyses we assume that a human generation is 20 years. We used 500 grid points in the numerical approximations with an exponential spacing (λ = 0.005) to determine the midpoints.
Figure 2.—

Time-series data for the CCR5-Δ32 data (Hummel et al. 2005). Binomial confidence intervals are shown as shaded bars.
Bacteriophage MS2:
In the second application we utilize frequency trajectory data from a recent experimental study of adaptation of the bacteriophage MS2 by Bollback and Huelsenbeck (2007). Briefly, this study selected populations of MS2 for growth at elevated temperatures. The authors determined the frequency of a number of beneficial mutations throughout the time course of the experiment (every ≈10 passages). We apply our method to one of the nonsynonymous mutations in their experimental line 2: C206U (Figure 3). We make the assumption that each selective passage in their study consisted of 2.5 generations; C206U was tracked for ∼100 generations, or 40 serial passages. We used 500 grid points in the numerical approximation. Due to the fairly small number of generations and little time spent at the boundaries of the process, unequal spacing was not needed and equal spacing on the grid was used.
Figure 3.—

Time-series data for the bacteriophage C206U mutation (Bollback and Huelsenbeck 2007). Binomial confidence intervals are shown as shaded bars. A predicted smoothed (shaded) curve fitted projection is shown.
RESULTS AND DISCUSSION
Numerical convergence and grid point spacing:
Determining the number of grid points and the spacing of those points affects the precision of the numerical approximations. To this end we have evaluated a simple case in which we were able to analytically calculate the log likelihood and compare this to the numerical approximation. We performed the check using a number of different grid points (n = 10, 100, 200, 500, 1000, 2000) and two methods of grid point spacing (equal and unequal). Our evaluations of convergence under two types of spacing (Figure 4) show that at low numbers of grid points (n = 10 and n = 100) the difference between the expected and the observed value is large regardless of the method of spacing (7.1–9.4% and 0.7–1.1%, respectively). As the number of grid points increases, the difference declines dramatically to <0.5% and reaches an error rate of 0.04% at the largest number of points. For the test data set little difference was observed for different values of λ. As the number of grid points increases, the numerical routines for solving the set of differential equations become burdensome so a choice of grid points for a particular analysis will be a trade-off between computational burden and precision. For these reasons the applications of our method used values of 400 or 500 as the error rate was deemed to be sufficiently small for these values.
Figure 4.—

Adequacy of the numerical approximations as a function of the number of grid points. Two different grid point spacings are used, equal (open circles) and exponential (solid diamonds) with a spacing parameter of λ = 0.5.
CCR5-Δ32:
The CCR5 protein is a chemokine receptor. This receptor is a coreceptor target for human immunodeficiency virus (HIV) and simian immunodeficiency virus, and possibly other related viruses (Mummidi et al. 2000; Paterlini 2002). An allele with a 32-amino-acid deletion, named CCR5-Δ32, has been determined to be at low frequency in the human population and its origin has been estimated to be at least 700–3500 years ago (Stephens et al. 1998) with empirical observations of at least 2900 years ago (Hummel et al. 2005). Because of the age of the mutation it has been argued that it is extremely unlikely to be a neutral mutation. As a result, CCR5-Δ32 has been hypothesized to have been under selection from the bubonic plague or smallpox with the low frequency being explained by intermittent temporal selection or balancing selection (for reviews see De Silva and Stumpf 2004; Stumpf and Wilkinson-Herbots 2004; Galvani and Novembre 2005). Recently, it has been demonstrated that homozygous individuals are completely resistant to HIV infection (heterozygous individuals exhibit lower infection rates and longer disease progressions) (McNicholl et al. 1997). However, the recent origin of HIV is unlikely to explain its persistence over such a long period of time although the ongoing epidemic may affect the frequency in the future. Novembre et al. (2005), using the current allelic distribution of CCR5-Δ32 in Europe, modeled the historical spread of this mutation to determine its origin, rate of spread, and selective value. They found that, depending on whether selection is uniform or varying across Europe, the most likely origin of the allele was in the north or northwest, the rate of spread exceeded 100 km per generation, and the intensity of selection was >10% (Novembre et al. 2005).
We have applied our method to estimate s from an ancient DNA data set (Hummel et al. 2005) consisting of samples gathered from multiple time points in Europe dating from 2900 years ago to present. Our estimates of s for CCR5-Δ32 had a 95% confidence interval of −0.09–0.01 with a maximum value very close to zero (s ≈ −0.0005), suggesting that the mutation is either neutral or at best slightly beneficial. The upper end of the confidence interval is close to the lower end of the confidence interval for previous estimates based on the analysis of frequency data and linkage disequilibrium (s ≈ 0.05–0.35; Stephens et al. 1998; Slatkin 2001). However, Novembre et al. (2005) found support for s ≪ 0.02 when the dispersal rate was <75 km, which is more consistent with studies of historical and modern dispersal in Europe. These values are also consistent with our estimates of s (Figure 5).
Figure 5.—

Profile likelihood of the selection coefficient for CCR5-Δ32 (Hummel et al. 2005).
We should warn against a too strong interpretation of our results because the samples are clearly not obtained from an idealized panmictic population that has gene flow with other populations. In addition, there are the usual caveats regarding human aDNA data. A full discussion of problems regarding human aDNA is beyond the scope of this article (for review, see Gilbert et al. 2005).
Bacteriophage MS2:
Experimental evolution studies of microbial populations typically follow the change in beneficial mutations through time as a matter of course (e.g., Wichman et al. 1999; Holder and Bull 2001; Bollback and Huelsenbeck 2007). The data from these types of studies are ideal for the method presented here for a number of reasons. First, they are performed in a controlled manner in which the population size is known, kept fairly constant, and generally large. Second, they are able to sample mutations throughout the bout of selection with relative ease. Third, the mutations are more often than not known to be under selection. Finally, the selective conditions are kept constant through time.
We apply our method to the experimental MS2 bacteriophage data of Bollback and Huelsenbeck (2007). Using the trajectory for the mutation C206U (Figure 3) we evaluate 2Nes. We performed the numerical HMM integration over a reasonable set of population sizes (N = 1 × 107–2 × 108) that included the experimental value (5 × 107; Bollback and Huelsenbeck 2007) and selection coefficients (2Nes ≈ 0–1 × 109; s ≈ 0–5). Figure 6 shows the likelihood surface for C206U, plotting the log of the population size, 2N, against the log of 2Ns. The maximum observed value (shown as a plus sign in Figure 6) indicates a population size of 3.89 × 107 and a selection coefficient of 0.427 (95% C.I.: 0.386–0.819). The best supported population size estimate is very close to the experimental values (N = 5 × 107; Bollback and Huelsenbeck 2007). However, because of the extremely large population sizes and strong selection, the mutation's trajectory is strongly deterministic and little information exists to estimate the population effective size; the 95% confidence interval spans all of the values evaluated as expected. The estimate of s is reasonable considering the fitness gains (w − 1 = 3) and number of beneficial mutations (n = 4) observed in the experimental population (Bollback and Huelsenbeck 2007): our estimate of s suggests that C206U accounts for 13–27% of the total fitness gain in the population.
Figure 6.—

Likelihood surface for the bacteriophage C206U mutation (Bollback and Huelsenbeck 2007). The 95% confidence interval is shown in black with the maximum point on the surface depicted by a plus (+) sign.
Practical limitations and assumptions:
Two assumptions of our method merit discussion. First, in the applications presented, we ignore recurrent mutation. However, recurrent mutation is not likely to significantly affect allele frequencies in most cases, except those with weak selection, a high mutation rate, and large populations. The MS2 populations, for example, meet two of these conditions (Bollback and Huelsenbeck 2007)—high mutation rate and large population size—but the strong selection experienced is expected to overwhelm any input from mutation. Second, our method assumes that the samples are taken from a panmictic population; highly structured populations will clearly have an effect on the estimation of 2Nes, particularly for recessive mutations. Unfortunately, sampling from structured populations cannot easily be accommodated in the current framework and merits future work.
Conclusions:
We show here that estimation of 2Nes is possible from time series of allele frequency data. The nature of the data naturally presents some limitations. The estimates of 2Ne and s will in many cases be very correlated. Additionally, as Ne becomes large, the trajectory of the allele frequency through time becomes approximately deterministic and there should be little or no power to estimate Ne (see Figure 6). Nonetheless, even in such cases the method will provide estimates of s that take into account the uncertainty associated with the estimation of population allele frequencies from sample allele frequencies. The greatest strength of the method, however, is in the cases where 2Nes is moderate and joint estimates of Ne and s can be done. In such cases, the method can also be used to test the hypothesis of s = 0. An example of such an application was given for the CCR5-Δ32 data.
Acknowledgments
This work was supported by grants from the Danish Natural Science Research Council (FNU no. 272-06-0316) to J.P.B., the Danish Medical Research Council (FSS no. 271-05-0599) to R.N. and J.P.B., and Danmarks Grundsforskningsfond to R.N.
References
- Anderson, E. C., 2005. An efficient Monte Carlo method for estimating Ne from temporally spaced samples using a coalescent-based likelihood. Genetics 170 955–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E. C., E. G. Williamson and E. A. Thompson, 2000. Monte Carlo evaluation of the likelihood for Ne from temporally spaced samples. Genetics 156 2109–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont, M. A., 2003. Estimation of population growth or decline in genetically monitored populations. Genetics 164 1139–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthier, P., M. A. Beaumont, J.-M. Cornuet and G. Luikart, 2002. Likelihood-based estimation of the effective population size using temporal changes in allele frequencies: a genealogical approach. Genetics 160 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollback, J. P., and J. P. Huelsenbeck, 2007. Clonal interference is alleviated by high mutation rates in large populations. Mol. Biol. Evol. 24 1397–1406. [DOI] [PubMed] [Google Scholar]
- Buri, P., 1956. Gene frequency in small populations of mutant Drosophila. Evolution 10 367–402. [Google Scholar]
- Crank, J., and P. Nicolson, 1947. A practical method for numerical evaluation of solutions of partial differential equations of the heat conduction type. Proc. Camb. Philos. Soc. 43 50–67. [Google Scholar]
- de Silva, E., and M. P. Stumpf, 2004. HIV and the CCR5-Δ32 resistance allele. FEMS Microbiol. Lett. 241 1–12. [DOI] [PubMed] [Google Scholar]
- Durbin, R., S. Eddy, A. Krogh and G. Mitchison, 1998. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK.
- Ewens, W. J., 2004. Mathematical Population Genetics, Ed. 2. Springer-Verlag, New York.
- Galvani, A. P., and J. Novembre, 2005. The evolutionary history of the CCR5-Δ32 HIV-resistance mutation. Microbes Infect. 7 302–309. [DOI] [PubMed] [Google Scholar]
- Gilbert, M. T., H. J. Bandelt, M. Hofreiter and I. Barnes, 2005. Assessing ancient DNA studies. Trends Ecol. Evol. 20 541–544. [DOI] [PubMed] [Google Scholar]
- Holder, K. K., and J. J. Bull, 2001. Profiles of adaptation in two similar viruses. Genetics 159 1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel, S., D. Schmidt, B. Kremeyer, B. Herrmann and M. Oppermann, 2005. Detection of the CCR5-Δ32 HIV resistance gene in Bronze Age skeletons. Genes Immun. 6 371–374. [DOI] [PubMed] [Google Scholar]
- Krimbas, C. B., and S. Tsakas, 1971. The genetics of Dacus oleae. V. Changes of esterase polymorphism in a natural population following insecticide control—Selection or drift? Evolution 25 454–460. [DOI] [PubMed] [Google Scholar]
- McNicholl, J. M., D. K. Smith, S. H. Qari and T. Hodge, 1997. Host genes and HIV: the role of the chemokine receptor gene CCR5 and its allele (Δ32 CCR5). Emerg. Infect. Dis. 3 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummidi, S., M. Bamshad, S. S. Ahuja, E. Gonzalez, P. M. Feuillet et al., 2000. Evolution of human and non-human primate CC chemokine receptor 5 gene and mRNA. Potential roles for haplotype and mRNA diversity, differential haplotype-specific transcriptional activity, and altered transcription factor binding to polymorphic nucleotides in the pathogenesis of HIV-1 and simian immunodeficiency virus. J. Biol. Chem. 275 18946–18961. [DOI] [PubMed] [Google Scholar]
- Nei, M., and F. Tajima, 1981. Genetic drift and estimation of effective population size. Genetics 98 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre, J., A. P. Galvani and M. Slatkin, 2005. The geographic spread of the CCR5 Δ32 HIV-resistance allele. PLoS Biol. 3 e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterlini, M. G., 2002. Structure modeling of the chemokine receptor CCR5: implications for ligand binding and selectivity. Biophys. J. 83 3012–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak, E., 1983. A new method for estimating the effective population size from allele frequency changes. Genetics 104 531–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch et al., 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73 10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 2001. Simulating genealogies of selected alleles in a population of variable size. Genet. Res. 78 49–57. [DOI] [PubMed] [Google Scholar]
- Stephens, J. C., D. E. Reich, D. B. Goldstein, H. D. Shin, M. W. Smith et al., 1998. Dating the origin of the CCR5-Δ32 AIDS-resistance allele by the coalescence of haplotypes. Am. J. Hum. Genet. 62 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, M. P., and H. M. Wilkinson-Herbots, 2004. Allelic histories: positive selection on a HIV-resistance allele. Trends Ecol. Evol. 19 166–168. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2001. A pseudo-likelihood method for estimating effective population size from temporally spaced samples. Genet. Res. 78 243–257. [DOI] [PubMed] [Google Scholar]
- Waples, R. S., 1989. A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichman, H., J. Millstein and J. Bull, 2005. Adaptive molecular evolution for 13,000 phage generations: a possible arms race. Genetics 170 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichman, H. A., M. R. Badgett, L. A. Scott, C. M. Boulianne and J. J. Bull, 1999. Different trajectories of parallel evolution during viral adaptation. Science 285 422–424. [DOI] [PubMed] [Google Scholar]
- Williamson, E. G., and M. Slatkin, 1999. Using maximum likelihood to estimate population size from temporal changes in allele frequencies. Genetics 152 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, R., D. Schneider, C. L. Winkworth, M. A. Riley and R. E. Lenski, 2006. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc. Natl. Acad. Sci. USA 103 9107–9112. [DOI] [PMC free article] [PubMed] [Google Scholar]