

Abstract
AFLP markers are becoming one of the most popular tools for genetic analysis in the fields of evolutionary genetics and ecology and conservation of genetic resources. The technique combines a high-information content and fidelity with the possibility of carrying out genomewide scans. However, a potential problem with this technique is the lack of homology of bands with the same electrophoretic mobility, what is known as fragment-size homoplasy. We carried out a theoretical analysis aimed at quantifying the impact of AFLP homoplasy on the estimation of within- and between-neutral population genetic diversity in a model of a structured finite population with migration among subpopulations. We also investigated the performance of a currently used method (DFDIST software) to detect selective loci from the comparison between genetic differentiation and heterozygosis of dominant molecular markers, as well as the impact of AFLP homoplasy on its effectiveness. The results indicate that the biases produced by homoplasy are: (1) an overestimation of the frequency of the allele determining the presence of the band, (2) an underestimation of the degree of differentiation between subpopulations, and (3) an overestimation or underestimation of the heterozygosis, depending on the allele frequency of the markers. The impact of homoplasy is quickly diminished by reducing the number of fragments analyzed per primer combination. However, substantial biases on the expected heterozygosity (up to 15–25%) may occur with ∼50–100 fragments per primer combination. The performance of the DFDIST software to detect selective loci from dominant markers is highly dependent on the number of selective loci in the genome and their average effects, the estimate of genetic differentiation chosen to be used in the analysis, and the critical bound probability used to detect outliers. Overall, the results indicate that the software should be used with caution. AFLP homoplasy can produce a reduction of up to 15% in the power to detect selective loci.
THE amplified fragment length polymorphism (AFLP) technique (Vos et al. 1995) is becoming one of the most popular methods in the fields of conservation and evolutionary genetics and ecology (Mueller and Wolfenbarger 1999; Bensch and Akesson 2005; Bonin et al. 2007; Meudt and Clarke 2007), as it combines a high reproducibility and information content with the possibility of making genomewide screenings. Because of the anonymous nature of the fragments generated by the AFLP technique, however, one major concern is the incidence of size homoplasy due to the lack of homology of comigrating fragments. This implies that fragments of a given size migrating in a band may involve more than one locus of the genome and, therefore, the inferences obtained from the band can produce misleading conclusions.
Several empirical approaches have been used to estimate levels of homoplasy in AFLP data sets. A few studies have demonstrated the presence of homoplasy in AFLP data by sequencing comigrating fragments within the same individual or from different individuals of the same or different species (Rouppe van der Voort et al. 1997; Peters et al. 2001; El-Rabey et al. 2002; Rombauts et al. 2003; Mechanda et al. 2004; Mendelson and Shaw 2005). Hansen et al. (1999) and O'Hanlon and Peakall (2000) developed a simplified method for detecting size homoplasy comparing the AFLP banding patterns resulting from several rounds of selective amplification using PCR primers differing in the number of selective nucleotides. This method revealed that the proportion of comigrating nonhomologous fragments within single individuals of sugar beet was 13% (Hansen et al. 1999) and as high as 100% for comparisons among pairs of individuals from distantly related taxa of Carduinae thistles (O'Hanlon and Peakall 2000). Moreover, it has been shown that in interspecific studies of Echinacea (Mechanda et al. 2004), Hordeum (El-Rabey et al. 2002), and Carduinae (O'Hanlon and Peakall 2000), homoplasy increases with increasing taxonomic rank. Empirical approaches, however, are technically demanding and time-consuming, and only a small number of fragments have been examined (Meudt and Clarke 2007).
Computer simulations have allowed surveying a larger number of markers by modeling the distribution of fragment lengths and generating probabilities of comigration and homoplasy. The size distribution of AFLP fragments is strongly asymmetrical, with a much higher proportion of smaller than larger fragments, particularly so for genomes with low GC content (Innan et al. 1999; Vekemans et al. 2002; Koopman and Gort 2004; Gort et al. 2006). Thus, the possible impact of homoplasy is larger for small- than for large-size fragments. For example, Vekemans et al. (2002) analyzed AFLP fragments from samples of one population of Phaseolus lunatus and two populations of Lolium perenne and divided the observed fragments into four size categories (75–124, 125–199, 200–299, and 300–450 bp). From this experimental data and from simulated data, they found a highly significant negative correlation between fragment size and the frequency of the marker allele associated with the fragments, which suggests that there is an upward bias in the allelic frequency for the small-size fragments. From the empirical data they also showed that the estimates of nucleotide diversity within populations of each species and those of nucleotide divergence between populations of L. perenne increased with increasing fragment sizes, suggesting that homoplasy produces an underestimation of both within- and between-population diversity estimated from AFLP fragments.
Koopman and Gort (2004) found that size homoplasy determines a lack of concordance between the empirical AFLP fragment-length distributions and the predicted distributions generated in silico for the Arabidopsis thaliana genome. A general observation is that the larger the number of bands detected per primer combination of selective nucleotides, the larger the possible magnitude of homoplasy (Vekemans et al. 2002; Koopman and Gort 2004; Gort et al. 2006). This is in agreement with the observations of Althoff et al. (2007), who used a computer program that mimics the AFLP procedure to examine the homology of fragments in whole genomes from eight species representing a range of genome sizes. They found that, for a given primer combination, both the number of fragments and the degree of homoplasy increased with genome size. The average number of homoplasious fragments was 11% for small genomes (<400 Mb), but increased to 41.5% for large genomes (>2 Gb).
The empirical and simulation studies described above indicate that homoplasy is a real component of AFLP data sets. This suggests that homoplasy may produce biases in the estimation of genetic diversity from AFLP markers, but the magnitude and direction of the bias remains to be investigated in detail. In addition, the use of AFLPs to identify selective loci in genome scans is becoming an active area of research in evolutionary studies (Wilding et al. 2001; Storz and Nachman 2003; Campbell and Bernatchez 2004; Scotti-Saintagne et al. 2004; Storz et al. 2004; Storz and Dubach 2004; Acheré et al. 2005; Beaumont 2005; Nielsen 2005; Storz 2005; Bonin et al. 2006; Jump et al. 2006; Mealor and Hild 2006; Murray and Hare 2006; Miller et al. 2007; Papa et al. 2007; Nosil et al. 2008; Smith et al. 2008). Genomewide scans of selective loci generally use information on heterozygosity and genetic differentiation (Beaumont 2005; Storz 2005), and the putative biases in these parameters due to homoplasy could produce some misinterpretations. The purpose of this work was to carry out a comprehensive theoretical analysis on the impact of fragment-size homoplasy upon the estimation of genetic diversity within and between populations. We also investigated the performance of a currently used method (DFDIST software) to detect selective loci from the comparison between genetic differentiation and heterozygosis of dominant molecular markers, as well as the impact of AFLP homoplasy on its effectiveness.
ANALYTICAL AND SIMULATION PROCEDURE
An analytical procedure was used to simulate either neutral or selected AFLP fragments in a subdivided population. Genetic-diversity parameters of the population were calculated for all simulated AFLP fragments and for those that would actually be observed in an experimental setting, where fragments of the same size comigrate in the same electrophoretic band (homoplasy). This allowed us to assess the bias imposed by homoplasy on the estimation of the genetic-diversity parameters in the population. An independent method based on coalescent simulations of DNA sequences was used to check the analytical procedure. In the second part of the study, a program used to detect selective loci, based on estimates of population genetic diversity, was used to check the impact of homoplasy on the detection of selective AFLP markers. Each of these procedures is detailed below.
Allele frequency distributions for neutral loci:
A classical island model at equilibrium between migration and drift was used to obtain the frequency of neutral markers in a subdivided population. For biallelic loci the distribution of allelic frequencies φ(p) followed a beta distribution,
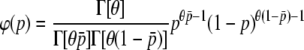 |
(1) |
(Wright 1937), where θ = 4Nm[n/(n − 1)], n is the number of subpopulations, N is the subpopulation size, m is the migration rate, 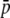 is the average allele frequency in the whole population, and
is the average allele frequency in the whole population, and 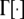 is the gamma function. Allelic frequencies sampled from the theoretical distribution (Equation 1) were assigned to each subpopulation for a number (NL) of loci. The number of subpopulations was assumed to be either n = 2 or 100, and the subpopulation size N = 500, although other subpopulation sizes (50, 1000, 5000, and 10,000) were also considered but gave similar results and are not shown. Appropriate values of θ were found so as to produce the desired levels of genetic differentiation (FST; Wright 1951) for each scenario. An illustration of the distribution of allelic frequencies for different levels of migration is given in Figure 1A.
is the gamma function. Allelic frequencies sampled from the theoretical distribution (Equation 1) were assigned to each subpopulation for a number (NL) of loci. The number of subpopulations was assumed to be either n = 2 or 100, and the subpopulation size N = 500, although other subpopulation sizes (50, 1000, 5000, and 10,000) were also considered but gave similar results and are not shown. Appropriate values of θ were found so as to produce the desired levels of genetic differentiation (FST; Wright 1951) for each scenario. An illustration of the distribution of allelic frequencies for different levels of migration is given in Figure 1A.
Figure 1.—

(A) Distribution, φ(p), of allelic frequencies (p), for a neutral allele in a subdivided population following an island model obtained from the beta distribution (Equation 1; assuming n = ∞), for different values of Nm, where N is the subpopulation size and m is the per-generation migration rate. (B) Distribution, φ(p), of allelic frequencies (p), for a selected allele in a subdivided population obtained from the transition matrix approach, for different values of the selection coefficient of the favorable allele (s), and assuming a number of migrants per generation of Nm = 1 from other (n = ∞) subpopulations where the average allelic frequency is 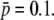 (C) Distribution, G(L), of the number of fragment lengths (L) cut by restriction enzymes EcoRI and MseI, obtained by Equation 2 of Innan et al. (1999), assuming different values of GC content (g).
(C) Distribution, G(L), of the number of fragment lengths (L) cut by restriction enzymes EcoRI and MseI, obtained by Equation 2 of Innan et al. (1999), assuming different values of GC content (g).
Assignment to AFLP fragments:
AFLP fragment lengths, obtained from the expected distribution derived by Innan et al. (1999), were randomly ascribed to the NL loci. For typical fragments flanked by an EcoRI adapter on one side, and an MseI adapter on the other, the expected frequency of occurrence of fragments of length L is
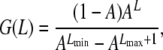 |
(2) |
where 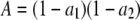 ,
, 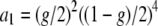 ,
, 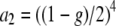 , Lmin and Lmax are the minimum and maximum fragment lengths screened, and g is the genomic GC content (Innan et al. 1999; Vekemans et al. 2002). The distribution obtained from Equation 2 also applies to fragments flanked by an EcoRI adapter on each side (Innan et al. 1999). Values of Lmin = 40 and Lmax = 440 were assumed in all simulations that correspond to PCR fragments between 72 and 472, because the typical EcoRI and MseI primers contain a 16-bp-long sequence when excluding the selective nucleotides. Values of g between 0.3 and 0.7 were investigated. Figure 1C shows the distribution of fragment lengths for the different values of g. Note that the lower the GC content assumed, the larger the number of fragments of short length.
, Lmin and Lmax are the minimum and maximum fragment lengths screened, and g is the genomic GC content (Innan et al. 1999; Vekemans et al. 2002). The distribution obtained from Equation 2 also applies to fragments flanked by an EcoRI adapter on each side (Innan et al. 1999). Values of Lmin = 40 and Lmax = 440 were assumed in all simulations that correspond to PCR fragments between 72 and 472, because the typical EcoRI and MseI primers contain a 16-bp-long sequence when excluding the selective nucleotides. Values of g between 0.3 and 0.7 were investigated. Figure 1C shows the distribution of fragment lengths for the different values of g. Note that the lower the GC content assumed, the larger the number of fragments of short length.
True loci vs. experimental fragments:
Irrespective of the length of the fragments, all simulated loci were obviously identified individually. Thus, true fragment frequencies and differentiation could be calculated assuming Hardy–Weinberg equilibrium. This group of fragments is called true fragments or loci hereafter. In an experimental setting, however, PCR fragments (bands) may correspond to different loci with the same fragment length (homoplasy). Fragments detected as different PCR bands are called experimental fragments or bands hereafter. The number of experimental bands depends on the number of true loci and the distribution of fragment sizes (Equation 2). For example, if g = 0.5, for NL = 1000 true loci there are ∼345 experimental bands, whereas for NL = 200 true loci there are ∼151 experimental bands (see also Figure 4 of Vekemans et al. 2002 for a graph showing the expected number of detectable fragments as a function of the total number of true fragments). The number of true loci considered varied from 60 to 1000.
Figure 4.—

Average allelic frequency (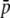 ), average expected heterozygosity (HS), and degree of genetic differentiation between subpopulations (FST) from experimental fragments (open circles) and true fragments (solid circles), for different classes of fragment lengths. The results correspond to one replicate with the following parameters: N = 500, n = 100, g = 0.5, FST = 0.1, and
), average expected heterozygosity (HS), and degree of genetic differentiation between subpopulations (FST) from experimental fragments (open circles) and true fragments (solid circles), for different classes of fragment lengths. The results correspond to one replicate with the following parameters: N = 500, n = 100, g = 0.5, FST = 0.1, and 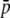 = 0.1 (left column of graphs) or
= 0.1 (left column of graphs) or 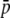 = 0.7 (right column of graphs).
= 0.7 (right column of graphs).
A comparison between true and experimental fragments allows us to understand the bias due to homoplasy when estimating allele frequencies, gene diversity, and differentiation. The frequency of the hypothetical allele that explains the incidence of the lth experimental fragment of length L in a given subpopulation was calculated as
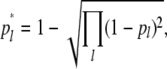 |
(3) |
where pl is the frequency of the marker allele associated to the true lth locus in the subpopulation, and the product is for all true loci with the same fragment length L. Allelic frequencies, average expected heterozygosities of subpopulations (HS), expected heterozygosities for the whole population (HT), and degree of differentiation, FST = (HT − HS)/HT (Nei 1973), where HT and HS are averages over loci, were calculated for both true and experimental fragments. Results for these parameters were generally presented as the ratio between their corresponding values for experimental and true loci, i.e., as the bias incurred by the experimental fragments with respect to the true loci in the estimation of genetic diversity.
Coalescent simulations:
To check the previous analytical procedure by a different, independent method, simulations were carried out by the retrospective coalescent approach. The program SIMCOAL 2.1.2 (Excoffier et al. 2000; http://cmpg.unibe.ch/software/simcoal2/) was used to simulate DNA sequences corresponding to two subpopulations of equal size (2N = 1000 haploid individuals) separated for a period of 500 generations and carrying out reciprocal migration at a rate m per generation. Twenty sequences were sampled from each subpopulation and cut in fragments at the motifs GAATTC (enzyme EcoRI) and TTAA (enzyme MseI). The number of nucleotides simulated was chosen so as to produce a total number of ∼1000 fragments (true loci). In particular, 34,000 nucleotides were required (distributed in four chromosomes with 8600 nucleotides each). The default transition rate of 0.333 and no recombination rate within chromosomes were used. To compare this coalescent method and the previous approach for a particular common scenario, a migration rate m = 0.0008 between the two subpopulations and a mutation rate per nucleotide u = 0.00011 were found to produce an average allelic frequency of true loci of 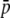 = 0.1 and a degree of differentiation between subpopulations of FST = 0.1. Allelic frequencies, average expected heterozygosities, and degree of differentiation were calculated from the samples for both true and experimental fragments. One hundred coalescent simulations and 100 analytical runs were carried out to compare the results of both methods.
= 0.1 and a degree of differentiation between subpopulations of FST = 0.1. Allelic frequencies, average expected heterozygosities, and degree of differentiation were calculated from the samples for both true and experimental fragments. One hundred coalescent simulations and 100 analytical runs were carried out to compare the results of both methods.
Selective loci:
Because Equation 1 can provide the distribution of allelic frequencies only in the case of neutral loci, a transition matrix approach was used to obtain allelic distributions for selective loci (see, e.g., Fernández et al. 2005). Assume that for a given locus with genotypes AA, Aa, and aa and genotypic frequencies p2, 2pq, and q2, the corresponding fitnesses in a given subpopulation of size N are 1 + s, 1 + s/2, and 1, respectively, with s ≥ 0. Assume also that the locus is not selected in the remaining subpopulations, where its average frequency is 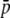 , and that every generation a fraction m of genes arrives at the considered subpopulation by migration. The model is also equivalent to an island–continent model where selection occurs exclusively in the island. To generate the distribution of frequencies for the selected gene in the considered subpopulation, taking account of selection, drift, and migration, a transition matrix A, of size (2N + 1) × (2N + 1), was constructed with terms
, and that every generation a fraction m of genes arrives at the considered subpopulation by migration. The model is also equivalent to an island–continent model where selection occurs exclusively in the island. To generate the distribution of frequencies for the selected gene in the considered subpopulation, taking account of selection, drift, and migration, a transition matrix A, of size (2N + 1) × (2N + 1), was constructed with terms
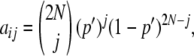 |
giving the probability that the number of copies of the gene is j at a generation, given that it was i in the previous one, where p = i/2N (0 ≤ i ≤ 2N) and
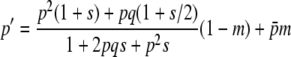 |
is the expected allele frequency after selection and migration. The state of the vector of frequencies at generation t, 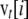 , with terms from i = 0 to 2N, was obtained from that at generation t − 1 as
, with terms from i = 0 to 2N, was obtained from that at generation t − 1 as 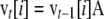 . Iterations were run until reaching the asymptotic stage. This was assumed to occur when the squared difference between two consecutive values of the sum of terms for the vector
. Iterations were run until reaching the asymptotic stage. This was assumed to occur when the squared difference between two consecutive values of the sum of terms for the vector 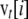 was <10−6. Selective coefficients were sampled from an exponential distribution,
was <10−6. Selective coefficients were sampled from an exponential distribution, 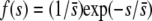 , with mean effect
, with mean effect 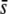 , and were assigned to randomly chosen fragments. Figure 1B illustrates the distribution of frequencies for a range of selective coefficients. The case corresponding to a neutral locus (s = 0) produced the same distribution as that obtained from the beta distribution (Equation 1), as expected (cf. line for Nm = 1 in Figure 1A and line for s = 0 in Figure 1B). For increasing values of the selection coefficient the distribution of allelic frequencies is increasingly displaced to the right, but note that for moderate values of s, the expected frequency can take a wide range of values.
, and were assigned to randomly chosen fragments. Figure 1B illustrates the distribution of frequencies for a range of selective coefficients. The case corresponding to a neutral locus (s = 0) produced the same distribution as that obtained from the beta distribution (Equation 1), as expected (cf. line for Nm = 1 in Figure 1A and line for s = 0 in Figure 1B). For increasing values of the selection coefficient the distribution of allelic frequencies is increasingly displaced to the right, but note that for moderate values of s, the expected frequency can take a wide range of values.
Identification of selective fragments by population comparisons:
To assess the impact of homoplasy on the identification of selective loci in AFLP genome scans, data obtained with the analytical approach above was analyzed using the DFDIST software (http://www.rubic.rdg.ac.uk/∼mab/stuff/), a modification for dominant markers of the software developed by Beaumont and Nichols (1996). Briefly, this program implements a Bayesian method developed by Zhivotovsky (1999) to estimate allelic frequencies from the proportion of recessive phenotypes in the sample. It also estimates the Weir and Cockerham (1984) FST between the subgroups defined in the sample. Coalescent simulations are then performed to generate an FST null sampling distribution from upon the neutral expectations. The simulated FST data are used to identify those loci that may not fit the neutral drift model given their unusually low or high FST values. The software DFDIST is the method most frequently used when using AFLPs (see Scotti-Saintagne et al. 2004; Acheré et al. 2005; Bonin et al. 2006; Jump et al. 2006; Mealor and Hild 2006; Murray and Hare 2006; Miller et al. 2007; Papa et al. 2007; Nosil et al. 2008; Smith et al. 2008).
For the scenarios to be analyzed with the DFDIST program, we considered two subpopulations of size N = 500 and a proportion of loci (between 1 and 10%) assumed to be under positive selection in one of them. For example, for the case of NL = 1000 loci, 50 selective loci and 950 neutral ones were assumed for a proportion of selective loci of 5%). Subpopulation sample frequencies for true fragments (ps) in a random sample of M individuals taken from the subpopulation were obtained from the binomial probability of sampling j copies of a given allele,
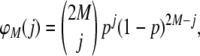 |
where p is the subpopulation allele frequency for the locus. Absolute frequencies of theoretical true PCR bands (corresponding to true fragments) and no bands in each sample were calculated as 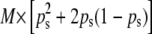 and
and 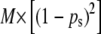 , respectively. The analogous absolute frequency of experimental PCR bands (or no bands) was obtained with the corresponding experimental allelic frequencies (
, respectively. The analogous absolute frequency of experimental PCR bands (or no bands) was obtained with the corresponding experimental allelic frequencies (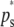 ; Equation 3). Assumed subpopulation sample sizes were M = 20, 40, 80, and 160 individuals.
; Equation 3). Assumed subpopulation sample sizes were M = 20, 40, 80, and 160 individuals.
The parameter conditions used in the DFDIST analyses were as follows:
The critical frequency for the most common allele was 0.99 (loci where the most frequent allele had a frequency ≥0.99 were excluded).
The scale for the Zhivotovsky (1999) parameters for estimating allele frequencies was 0.25 (it was checked that the estimation of frequencies was accurate with this parameter value).
The number of resamplings used to obtain the confidence intervals for outliers was 10,000 (several runs were made with 100,000 resamplings and results did not change).
The smoothing proportion used was 0.04.
The estimate of average FST to be used in the DFDIST simulations was obtained in three ways: (i) the average estimated FST calculated by the Ddatacal program (one of the programs of the Dfdist package); (ii) a trimmed mean FST (provided also by the Ddatacal program) obtained excluding 30% of the highest and 30% of the lowest FST values (this trimmed mean FST is supposed to be an estimate of the average “neutral” FST uninfluenced by outlier loci; Bonin et al. 2006); and (iii) the median of FST values from the file data_fst_outfile provided by the Ddatacal program, after removing those loci for which the heterozygosity is <0.1. This was the procedure used in the simulations of Beaumont and Balding (2004).
Different values of the parameter 4Neμ were used (from 0.004 to 0.1), finding no differences in the performance of the method within this range. Thus, a value of 0.04 was used throughout.
The critical probability for detecting outliers was α = 5 or 0.05%.
To make fair comparisons between true and experimental data sets, a given number of runs were pooled in each case to provide the same number of true and experimental loci (bands) for analysis with the program DFDIST (see Table 1). For example, for the case with NL = 1000 true fragments, for which only ∼345 experimental fragments become available from each run because of fragment-size homoplasy, three simulation replicates (that would be equivalent to using three different primer combinations in a PCR setting) were pooled. From each replicate, all experimental fragments (∼345) and 345 randomly taken true fragments were used. Thus, ∼345 × 3 = 1035 true or experimental loci were analyzed by the DFDIST program and compared. It should be noted that although the proportion of the genome with selective loci is 5%, the 1035 experimental fragments implied 15% of selective loci because each experimental fragment represents on average three true loci (i.e., the 1035 experimental fragments actually correspond to ∼1035 × 3 = 3105 true loci). An analogous procedure was carried out for other numbers of true loci (see Table 1).
TABLE 1.
Experimental setting for the analysis with the DFDIST program
| % of homoplasious bandsd | Total no. of bands (true or experimental)e | Expected no. of bands with at least a selective locus
|
|||||
|---|---|---|---|---|---|---|---|
| NL per primera | Selective loci (5%)b | Bands per primerc | No. of runs (primers) | True bands | Experimental bands | ||
| 1000 | 50 | 345 | 74 | 3 | 1035 | 52 | 150 |
| 200 | 10 | 151 | 26 | 7 | 1057 | 53 | 70 |
| 100 | 5 | 87 | 14 | 12 | 1044 | 52 | 60 |
| 60 | 3 | 55 | 9 | 19 | 1045 | 52 | 57 |
Number of true loci assumed per simulation replicate (equivalently to per primer combination).
Number of selective loci per primer combination (5% of NL).
Number of experimental bands observed per primer combination (average over 100 replicates assuming a GC content of g = 0.5).
Percentage of homoplasious experimental bands, i.e., percentage of experimental bands that include two or more true loci.
Product between number of bands per primer combination and number of primer combinations.
Analyses were carried out for different values of FST and mean selection coefficients (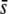 = 0, 0.001, 0.005, 0.05, and 0.5). The proportion of outliers detected by the method (loci above the 95 or 99.95% confidence limit) and the proportion of true selective loci detected were calculated both for true and experimental loci. Each scenario was replicated 10 times and results were averaged over replicates.
= 0, 0.001, 0.005, 0.05, and 0.5). The proportion of outliers detected by the method (loci above the 95 or 99.95% confidence limit) and the proportion of true selective loci detected were calculated both for true and experimental loci. Each scenario was replicated 10 times and results were averaged over replicates.
RESULTS
Impact of homoplasy on neutral gene diversity and differentiation among subpopulations:
Table 2 compares the results obtained with the analytical method and the simulation (SIMCOAL) method with the objective of checking the former. The scenario corresponds to a population subdivided into two subpopulations (n = 2) of size N = 500 each and a migration rate such that the differentiation between subpopulations is FST = 0.1. The average allelic frequency in both cases was set up to be 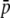 = 0.1 for the true loci. The first column for each approach refers to the true loci, the corresponding results for the experimental “loci” are shown in the second column, and the ratio between both (i.e., the amount of bias of the experimental estimates relative to the true values) are in the third column. In this particular case, homoplasy produces a 2.5-fold inflation in the average allelic frequency (
= 0.1 for the true loci. The first column for each approach refers to the true loci, the corresponding results for the experimental “loci” are shown in the second column, and the ratio between both (i.e., the amount of bias of the experimental estimates relative to the true values) are in the third column. In this particular case, homoplasy produces a 2.5-fold inflation in the average allelic frequency (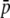 ), a 1.9-fold inflation in gene diversity (expected heterozygosity within subpopulations, HS), and a 10% reduction in the value of genetic differentiation (FST). All results were very similar for both approaches. In what follows, all results will refer to the analytical method, which was faster and easier to implement and allowed for better control of the parameters.
), a 1.9-fold inflation in gene diversity (expected heterozygosity within subpopulations, HS), and a 10% reduction in the value of genetic differentiation (FST). All results were very similar for both approaches. In what follows, all results will refer to the analytical method, which was faster and easier to implement and allowed for better control of the parameters.
TABLE 2.
Results obtained with the analytical approach (Equations 1–3) and the simulation approach (using the program SIMCOAL; see text for details) for the average number of loci, average allele frequency (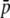 ), expected heterozygosity within subpopulations (HS), and genetic differentiation between subpopulations (FST) for true and experimental fragments
), expected heterozygosity within subpopulations (HS), and genetic differentiation between subpopulations (FST) for true and experimental fragments
| Analytical approach
|
Simulation approach (SIMCOAL)
|
|||||
|---|---|---|---|---|---|---|
| True | Experimental | Bias | True | Experimental | Bias | |
| No. loci | 1000.0 | 345.5 | 1000.5 | 351.3 | ||
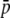 |
0.100 | 0.251 | 2.503 | 0.102 | 0.249 | 2.445 |
| HS | 0.147 | 0.286 | 1.941 | 0.140 | 0.266 | 1.894 |
| FST | 0.100 | 0.090 | 0.893 | 0.100 | 0.092 | 0.925 |
Bias is the ratio between true and experimental estimates for the different parameters. Results are based on 100 replicates for each approach. Standard errors <0.002 for 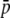 , HS, and FST and <0.4 (Analytical) and 6.1 (SIMCOAL) for the number of loci are shown. Parameters: NL = 1000, subpopulation size N = 500, number of subpopulations n = 2, genome GC content g = 0.5, average allelic frequency
, HS, and FST and <0.4 (Analytical) and 6.1 (SIMCOAL) for the number of loci are shown. Parameters: NL = 1000, subpopulation size N = 500, number of subpopulations n = 2, genome GC content g = 0.5, average allelic frequency 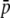 = 0.1, and neutral genetic differentiation FST = 0.1.
= 0.1, and neutral genetic differentiation FST = 0.1.
Figure 2 shows the bias for the parameters estimated with the experimental fragments relative to the true loci for a range of values of genetic differentiation between subpopulations (FST) for a scenario of NL = 1000 true loci (continuous line) and 200 loci (broken line). These correspond to ∼345 and 151 experimental fragments, i.e., observed PCR bands (see Table 1). For 1000 true fragments the biases in the average allelic frequency and heterozygosity are very substantial and almost invariable for different values of FST. The bias in the estimated level of differentiation is maximal at low values of FST and decreases at higher ones. Note, however, that this bias is a relative one, and the absolute bias (absolute difference in FST between experimental and true fragments) is higher for intermediate values of FST. All biases are substantially lower with 200 true fragments, as the degree of homoplasy is relatively small in this case.
Figure 2.—

Amount of bias of the estimates of average allelic frequency (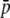 ), average expected heterozygosity (HS), and degree of genetic differentiation between subpopulations (FST) obtained from experimental fragments relative to true loci, for different values of FST. The continuous line refers to the case with NL = 1000 true loci, and the broken line to NL = 200. The average allelic frequency of true fragments is
), average expected heterozygosity (HS), and degree of genetic differentiation between subpopulations (FST) obtained from experimental fragments relative to true loci, for different values of FST. The continuous line refers to the case with NL = 1000 true loci, and the broken line to NL = 200. The average allelic frequency of true fragments is 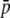 = 0.1. The default parameters are N = 500, n = 100, g = 0.5, and
= 0.1. The default parameters are N = 500, n = 100, g = 0.5, and 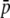 = 0.1. All results are the averages of 10 replicates.
= 0.1. All results are the averages of 10 replicates.
Figure 3 shows the bias of the experimental parameters relative to the true ones under a range of situations. The first column of graphs shows the biases for different values of the GC content (g). The continuous line refers to NL = 1000 true loci (345 bands) and the broken line to 200 loci (151 bands). The graphs show that the bias is generally larger for small values of g, as expected, but the variation is not very large. The second column of graphs shows results for different average allele frequencies in the whole set of subpopulations (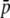 ). The lower the true average frequency, the higher is the bias for the estimates of this parameter. For the heterozygosity (HS) there is an overestimation for low values of
). The lower the true average frequency, the higher is the bias for the estimates of this parameter. For the heterozygosity (HS) there is an overestimation for low values of 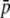 and an underestimation for intermediate or high values of
and an underestimation for intermediate or high values of 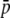 . This occurs because homoplasy always increases the allele frequencies, and this may imply a decrease in average heterozygosity for those loci whose true allele frequency is intermediate or high (note that heterozygosity for a biallelic locus is maximal for p = 0.5). The estimates of the total heterozygosity in the population (HT) are biased by a similar magnitude and direction as for the HS, so that results are not shown. For the estimates of FST, the largest underestimations occur for intermediate values of
. This occurs because homoplasy always increases the allele frequencies, and this may imply a decrease in average heterozygosity for those loci whose true allele frequency is intermediate or high (note that heterozygosity for a biallelic locus is maximal for p = 0.5). The estimates of the total heterozygosity in the population (HT) are biased by a similar magnitude and direction as for the HS, so that results are not shown. For the estimates of FST, the largest underestimations occur for intermediate values of 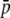 . Finally, the third column of graphs shows the biases for different numbers of true loci assuming
. Finally, the third column of graphs shows the biases for different numbers of true loci assuming 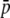 = 0.1 (without dots) or 0.2 (with dots). The larger the number of true loci, the higher the level of homoplasy is and, therefore, the higher the amount of bias in the estimates.
= 0.1 (without dots) or 0.2 (with dots). The larger the number of true loci, the higher the level of homoplasy is and, therefore, the higher the amount of bias in the estimates.
Figure 3.—

Amount of bias of the estimates of average allelic frequency (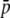 ), average expected heterozygosity (HS), and degree of genetic differentiation between subpopulations (FST) obtained from experimental fragments relative to true loci, for different values of GC content (g), average true allelic frequency (
), average expected heterozygosity (HS), and degree of genetic differentiation between subpopulations (FST) obtained from experimental fragments relative to true loci, for different values of GC content (g), average true allelic frequency (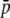 ), and number of true loci (NL). In the first two columns the solid line refers to the case with NL = 1000 true loci and the dashed line to NL = 200. In the third column the solid line refers to
), and number of true loci (NL). In the first two columns the solid line refers to the case with NL = 1000 true loci and the dashed line to NL = 200. In the third column the solid line refers to 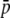 = 0.1, and the dotted line refers to
= 0.1, and the dotted line refers to 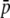 = 0.2. The default parameters are N = 500, n = 100, g = 0.5,
= 0.2. The default parameters are N = 500, n = 100, g = 0.5, 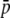 = 0.1, and FST = 0.1. All results are the averages of 10 replicates.
= 0.1, and FST = 0.1. All results are the averages of 10 replicates.
Because small-length fragments are more frequent (see Figure 1B), the bias in the estimates provided by the experimental fragments is larger the shorter the fragment length. This is illustrated in Figure 4, where the estimates of parameters from experimental fragments (open circles) and true loci (solid circles) are shown for fragments of different lengths. The left column of graphs corresponds to an average allelic frequency of 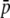 = 0.1, for which experimental estimates of HS are overestimations and those of FST are underestimations. The right column of numbers refers to an average of
= 0.1, for which experimental estimates of HS are overestimations and those of FST are underestimations. The right column of numbers refers to an average of 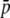 = 0.7, which implies underestimations of both HS and FST.
= 0.7, which implies underestimations of both HS and FST.
The previous results are somewhat conservative because they refer to genomes with a GC content of g = 0.5. For many species, however, the overall genome GC content is lower, e.g., rice (0.43), humans (0.41), Arabidopsis (0.35) (Yu et al. 2002), Caenorhabditis (0.35) (Stein et al. 2003), Drosophila (0.43) (Akashi et al. 1998), and the biases incurred can be larger. For example, for g = 0.35 and assuming 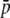 = 0.1 and FST = 0.1, for 100 true loci (implying 77 experimental bands), the average frequency would be overestimated by 27% and the expected heterozygosity by 20%, whereas FST would be underestimated by only 2%. Even with 60 true loci (51 experimental bands) the average frequency and heterozygosity are overestimated by ∼15%. Thus, in terms of estimation of neutral genetic differentiation, homoplasy has a substantial impact only when > ∼200 experimental bands per primer are scored. However, in terms of estimation of heterozygosity, a substantial impact may occur even for 50–75 experimental bands scored per primer combination.
= 0.1 and FST = 0.1, for 100 true loci (implying 77 experimental bands), the average frequency would be overestimated by 27% and the expected heterozygosity by 20%, whereas FST would be underestimated by only 2%. Even with 60 true loci (51 experimental bands) the average frequency and heterozygosity are overestimated by ∼15%. Thus, in terms of estimation of neutral genetic differentiation, homoplasy has a substantial impact only when > ∼200 experimental bands per primer are scored. However, in terms of estimation of heterozygosity, a substantial impact may occur even for 50–75 experimental bands scored per primer combination.
Detection of selective loci:
Assessment of the efficiency of the DFDIST software for detecting selective loci:
We first assessed the efficiency of the DFDIST software for detecting selective loci considering only the true loci, i.e., without yet introducing the problem of homoplasy. Figure 5 shows results for a variable proportion of selective loci assumed in the genome (from 0 to 10%), considering an average selection coefficient for the selective genes of 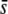 = 0.5 or 0.005, a critical probability value α = 5%, and an average neutral FST = 0.025. Note that this latter (as before, and thereafter, unless indicated) refers to Nei's (1973) estimate of FST [the corresponding Weir and Cockerham's (1984) estimate used by the DFDIST software is 0.037]. The left column of graphs in Figure 5 represents the percentage of outliers detected by the method. This is ∼4% when the nontrimmed (NT) FST mean is assumed, ∼10% when the trimmed (T) FST mean is assumed, and ∼15% when the median (M) FST is assumed, and these results are almost independent of the proportion of selective loci simulated and the average selection coefficient. The middle column of graphs in Figure 5 shows the percentage of outliers that are in fact selective loci. This percentage is substantial only for a high proportion of selective loci in the genome, a large
= 0.5 or 0.005, a critical probability value α = 5%, and an average neutral FST = 0.025. Note that this latter (as before, and thereafter, unless indicated) refers to Nei's (1973) estimate of FST [the corresponding Weir and Cockerham's (1984) estimate used by the DFDIST software is 0.037]. The left column of graphs in Figure 5 represents the percentage of outliers detected by the method. This is ∼4% when the nontrimmed (NT) FST mean is assumed, ∼10% when the trimmed (T) FST mean is assumed, and ∼15% when the median (M) FST is assumed, and these results are almost independent of the proportion of selective loci simulated and the average selection coefficient. The middle column of graphs in Figure 5 shows the percentage of outliers that are in fact selective loci. This percentage is substantial only for a high proportion of selective loci in the genome, a large 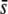 and the use of the trimmed or nontrimmed mean FST. Finally, the right column of graphs in Figure 5 shows the percentage of selective loci that are actually detected by the method. This is very high for selective loci with
and the use of the trimmed or nontrimmed mean FST. Finally, the right column of graphs in Figure 5 shows the percentage of selective loci that are actually detected by the method. This is very high for selective loci with 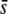 = 0.5 but on the order of ∼10–20% for small
= 0.5 but on the order of ∼10–20% for small 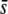 , almost irrespective of the proportion of selective loci in the genome.
, almost irrespective of the proportion of selective loci in the genome.
Figure 5.—

Percentage of outliers (A, D, G, J, M, and P), percentage of outliers that correspond to truly selective loci (B, E, H, K, N, and Q), and percentage of selective loci that are detected as outliers (C, F, I, L, O, and R) by the program DFDIST after analyzing ∼1000 (true) fragments, for a variable proportion of selective loci in the genome with average effect 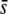 . The critical probability threshold value for detecting outliers is α = 5%. The average estimate of the FST used for the DFDIST simulations is the nontrimmed mean (NT), a trimmed mean (T), and the median (M). Default parameters (see text): N = 500, NL = 1000, n = 2, g = 0.5,
. The critical probability threshold value for detecting outliers is α = 5%. The average estimate of the FST used for the DFDIST simulations is the nontrimmed mean (NT), a trimmed mean (T), and the median (M). Default parameters (see text): N = 500, NL = 1000, n = 2, g = 0.5, 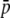 = 0.1, M = 40, and FST = 0.025. All results are the average of 10 replicates.
= 0.1, M = 40, and FST = 0.025. All results are the average of 10 replicates.
Figure 6 presents results analogous to those of Figure 5 but using a critical probability value of α = 0.05% to detect outliers. The percentage of outliers detected gives an approximately right estimate of the true proportion of selective loci in the genome only for scenarios with a large 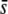 , a large proportion of selective loci (≥3%), and using a trimmed mean or median FST (Figure 6, G and M). Otherwise, the prediction is rather deficient. The percentage of selective loci present in outliers is very high for
, a large proportion of selective loci (≥3%), and using a trimmed mean or median FST (Figure 6, G and M). Otherwise, the prediction is rather deficient. The percentage of selective loci present in outliers is very high for 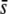 = 0.5 in most cases, except for a large proportion of selective loci (10%) and a nontrimmed mean FST (Figure 6B; for which no outliers are in fact detected) and when the proportion of selective loci is small (1%) with a trimmed or median FST (Figure 6, H and N). The percentage of selective loci in outliers is rather small for
= 0.5 in most cases, except for a large proportion of selective loci (10%) and a nontrimmed mean FST (Figure 6B; for which no outliers are in fact detected) and when the proportion of selective loci is small (1%) with a trimmed or median FST (Figure 6, H and N). The percentage of selective loci in outliers is rather small for 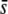 = 0.005, with a better performance of the mean FST (Figure 6, E and K) than the median FST (Figure 6Q). The percentage of selective loci detected is large only for high
= 0.005, with a better performance of the mean FST (Figure 6, E and K) than the median FST (Figure 6Q). The percentage of selective loci detected is large only for high 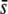 using the trimmed mean or the median FST (Figure 6, I and O).
using the trimmed mean or the median FST (Figure 6, I and O).
Figure 6.—

Percentage of outliers (A, D, G, J, M, and P), percentage of outliers that correspond to truly selective loci (B, E, H, K, N, and Q), and percentage of selective loci that are detected as outliers (C, F, I, L, O, and R) by the program DFDIST after analyzing ∼1000 (true) fragments, for a variable proportion of selective loci in the genome with average effect 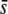 . The critical probability threshold value for detecting outliers is α = 0.05%. All other parameters and definitions are as in Figure 5.
. The critical probability threshold value for detecting outliers is α = 0.05%. All other parameters and definitions are as in Figure 5.
Figure 7 extends the above results assuming a trimmed mean FST and 5% of selective loci in the genome for a wider range of average selection coefficients, a different value of neutral FST (0.1), and different sample sizes (M). The percentage of outliers detected with a critical probability of α = 5% is again ∼10% for FST = 0.025 (Figure 7A), irrespective of the average selection coefficient, and this value is reduced to ∼7% for FST = 0.1 (Figure 7D). Runs corresponding to a higher value of FST = 0.2 actually produce even lower proportions of outliers (data not shown). For a critical probability of α = 0.05 (Figure 7, G and J) the correct proportion of selective loci (5%) is predicted only for a large average selection coefficient (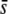 = 0.5) and a small level of genetic differentiation (FST = 0.025) but not for smaller values of
= 0.5) and a small level of genetic differentiation (FST = 0.025) but not for smaller values of 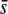 or a larger value of FST = 0.1. The percentage of selective loci found in the outliers is >80% only for large values of
or a larger value of FST = 0.1. The percentage of selective loci found in the outliers is >80% only for large values of 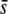 (0.05–0.5) and a critical probability of α = 0.05% (Figure 7, H and K). The percentage of selective loci detected generally increases with the average selection coefficient, as would be expected. Finally, increasing the sample size does not have any impact on the proportion of outliers detected or the efficiency of detection (Figure 7, M–O) beyond a sample size of M = 40.
(0.05–0.5) and a critical probability of α = 0.05% (Figure 7, H and K). The percentage of selective loci detected generally increases with the average selection coefficient, as would be expected. Finally, increasing the sample size does not have any impact on the proportion of outliers detected or the efficiency of detection (Figure 7, M–O) beyond a sample size of M = 40.
Figure 7.—

Percentage of outliers (A, D, G, J, and M), percentage of outliers that correspond to truly selective loci (B, E, H, K, and N), and percentage of selective loci that are detected as outliers (C, F, I, L, and O) by the program DFDIST after analyzing ∼1000 (true) fragments, for a variable average effect of selective loci (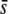 ) or sample size (M). The critical probability threshold value for detecting outliers is α. FST, degree of neutral differentiation. A trimmed mean average estimate of FST is used for the DFDIST simulations. Default parameters (see text): N = 500, NL = 1000, n = 2, g = 0.5,
) or sample size (M). The critical probability threshold value for detecting outliers is α. FST, degree of neutral differentiation. A trimmed mean average estimate of FST is used for the DFDIST simulations. Default parameters (see text): N = 500, NL = 1000, n = 2, g = 0.5, 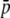 = 0.1, and M = 40. All results are the average of 10 replicates.
= 0.1, and M = 40. All results are the average of 10 replicates.
Impact of homoplasy on the detection of selective loci:
Because the levels of heterozygosity and FST are affected by homoplasy (Figures 2–4), and these are the parameters used for the detection of selective loci from molecular data, we investigated the impact of homoplasy on the detection of selective loci by comparing the analyses of true and experimental fragments. Table 3 gives average parameters from true loci and estimated parameters from experimental fragments in a case where 5% of the true loci (i.e., 50 of 1000) have an average selection coefficient of 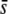 = 0.005. The first part of the table shows parameters including all loci (50 selective and 950 neutral loci). Because of the presence of selective loci, the average allelic frequency (
= 0.005. The first part of the table shows parameters including all loci (50 selective and 950 neutral loci). Because of the presence of selective loci, the average allelic frequency (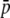 ), average heterozygosity (HS), and average FST are increased with respect to the purely neutral case (cf. Tables 1 and 3). The biases incurred by the experimental fragments are slightly smaller (for
), average heterozygosity (HS), and average FST are increased with respect to the purely neutral case (cf. Tables 1 and 3). The biases incurred by the experimental fragments are slightly smaller (for 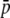 and HS) or larger (for FST) than for the neutral case (Table 1). The right side of Table 2 shows the biases exclusively for the selected loci. The highest bias from experimental loci occurs as a severe underestimation of FST, which comes down to about one-half of the true parameter value.
and HS) or larger (for FST) than for the neutral case (Table 1). The right side of Table 2 shows the biases exclusively for the selected loci. The highest bias from experimental loci occurs as a severe underestimation of FST, which comes down to about one-half of the true parameter value.
TABLE 3.
Average number of loci, average allele frequency (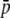 ), expected heterozygosity within subpopulations (HS), and genetic differentiation between subpopulations (FST) for true and experimental fragments
), expected heterozygosity within subpopulations (HS), and genetic differentiation between subpopulations (FST) for true and experimental fragments
| All loci
|
Selected loci
|
|||||
|---|---|---|---|---|---|---|
| True | Experimental | Bias | True | Experimental | Bias | |
| No. loci | 1000.0 | 346.2 | 50.0 | 46.3 | ||
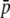 |
0.211 | 0.454 | 2.148 | 0.405 | 0.657 | 1.620 |
| HS | 0.255 | 0.338 | 1.325 | 0.224 | 0.273 | 1.218 |
| FST | 0.134 | 0.110 | 0.824 | 0.434 | 0.228 | 0.525 |
Five percent of the loci (50) are selective with a selection coefficient sampled from an exponential distribution with average 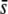 = 0.005; bias is the true/experimental ratio for the different parameter estimates. Results are based on 10 replicates. SEs <0.011 for
= 0.005; bias is the true/experimental ratio for the different parameter estimates. Results are based on 10 replicates. SEs <0.011 for 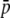 , HS, and FST and <1.6 for NL are shown. The default parameters are NL = 1000, N = 500, n = 2, g = 0.5,
, HS, and FST and <1.6 for NL are shown. The default parameters are NL = 1000, N = 500, n = 2, g = 0.5, 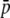 = 0.1, and FST = 0.1 (see Table 2).
= 0.1, and FST = 0.1 (see Table 2).
To assess the overall impact of homoplasy, Figure 8 presents results analogous to those of Figures 5–7, but comparing true and experimental fragments. The solid bars refer to the analysis of a set of ∼1000 fragments that are true loci, whereas the open bars refer to the corresponding analysis of ∼1000 fragments that are experimental fragments, hypothetically obtained with a variable number of primer combinations. Thus, for example, the first open bar in each graph (55 bands per primer for g = 0.5 or 50 for g = 0.35) would refer to a case where there are 19 primers producing 55 (or 50) experimental bands each (see Table 1). The second bar (87 or 76 bands per primer depending on g) corresponds to a case where there are 12 primers producing 87 (or 76) experimental bands each, and so on. Higher levels of homoplasy, corresponding to larger numbers of experimental bands, imply a slight overestimation of the percentage of outliers (Figure 8, A, D, and G) and an underestimation of the percentage of selective loci that are detected (Figure 8, C, F, and I). Homoplasy has no appreciable impact on the percentage of outliers carrying true selective loci (Figure 8, B, E, and H). The overestimation of the percentage of outliers detected can be understood from the information presented in Table 1. Note that a larger number of experimental bands implies a progressively larger number of true loci (NL). Thus, for example, for NL = 1000 (implying 345 experimental bands for g = 0.5 or 252 for g = 0.35), the 1000 experimental bands analyzed correspond to ∼3000 true loci, from which ∼150 are selected (5%), as in this case each band includes an average of three true loci of the same fragment length. Thus, ∼15% of the experimental fragments could include at least one selective true fragment. This explains why the percentage of outliers is larger for the experimental fragments than for the true loci.
Figure 8.—

Percentage of outliers (A, D, and G), percentage of outliers that correspond to truly selective loci (B, E, and H), and percentage of selective loci that are detected as outliers (C, F, and I) by the program DFDIST after analyzing ∼1000 fragments, against number of bands per primer combination. Solid bars correspond to true loci whereas open bars refer to experimental (observed) fragments, which correspond to 19 primer combinations (55 or 50 bands per primer for g = 0.5 or g = 0.35, respectively), 12 primer combinations (87 or 76 bands), 7 primer combinations (151 or 124 bands), and 3 primer combinations (345 or 252 bands; see Table 1). 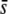 , average selection coefficient. g, genome GC content. Other default parameters (see text): N = 500, n = 2,
, average selection coefficient. g, genome GC content. Other default parameters (see text): N = 500, n = 2, 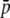 = 0.1, M = 40, and FST = 0.1. All results are the averages of 10 replicates.
= 0.1, M = 40, and FST = 0.1. All results are the averages of 10 replicates.
DISCUSSION
AFLP homoplasy and gene diversity and differentiation among subpopulations:
Our results demonstrate that the biases due to fragment homoplasy can be rather high for the estimates of 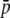 and HS, whereas generally modest for the estimates of FST (Figures 2 and 3). These biases depend not only on technical aspects that can be modified during the assay (number of loci scored per primer and fragment size), but also on factors usually unknown a priori in nonmodel organisms, such as the GC genomic composition or the demographic history and structure of populations under study.
and HS, whereas generally modest for the estimates of FST (Figures 2 and 3). These biases depend not only on technical aspects that can be modified during the assay (number of loci scored per primer and fragment size), but also on factors usually unknown a priori in nonmodel organisms, such as the GC genomic composition or the demographic history and structure of populations under study.
Our simulations indicate that the bias associated with size homoplasy rapidly increases with the number of true loci. Vos et al. (1995) recommended that, to avoid size homoplasy, the number of fragments or bands in an AFLP profile must be between 50 and 100, whereas Gort et al. (2006) suggested that even band numbers as low as 20 are not a guarantee of the absence of band homoplasy. These results are in line with an empirical study on the sugar beet by Hansen et al. (1999), who scored 456 bands in 16 AFLP lanes, giving an average of 28.5 bands per line. They reported that 13.2% of the bands were likely to be homoplasious. In our study, the comparison between the biases associated with the estimation of population parameters with a number of true loci ranging from 60 to 1000 indicates that ∼100 experimental fragments per primer combination may represent an upper limit to fragment homology among bands. From a literature review of 90 AFLP surveys on diverse animal, plant, fungi, and bacteria species (in the period 2004–2006), we found that the average number of fragments detected per primer combination ranges from 4 (Enjalbert et al. 2005) to 453 (Kalita and Malek 2006). If we take the median value of 58.8 as the more common number of detected fragments per primer combination, biases associated with the estimation of population parameters such as heterozygosity, FST and allele frequency are almost negligible in most studies. However, 18 (20%) studies showed an average number of fragments per primer combination >100 and are expected to have a high risk of biases due to size homoplasy (e.g., Laitung et al. 2004; Snäll et al. 2004; Albach et al. 2006; Fang et al. 2006; Kalita and Malek 2006; Kidd et al. 2006; Troell et al. 2006). Furthermore, as shown above, the overestimation of the expected heterozygosity can be substantial (15–25%) even for as low as 50–75 fragments per primer combination.
Vekemans et al. (2002) demonstrated that smaller-sized fragments are more likely to be homoplasious. Indeed, it is possible that the high degree of monomorphism often observed for small (<125 bp) fragments could be due to elevated levels of comigration of nonhomologous fragments (Vekemans et al. 2002; Bussel et al. 2005). Consistent with this, our results show that the bias due to size homoplasy increases in the small-size classes (Figure 4), although there is no clear cutoff point. Thus, shorter fragments have lower quality data. Inclusion of fragment-length information in the estimation of heterozygosity, 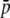 , and FST would likely result in more reliable estimates. Indeed, this approach has been efficiently used to obtain weighted similarity coefficients between unrelated genotypes (Koopman and Gort 2004).
, and FST would likely result in more reliable estimates. Indeed, this approach has been efficiently used to obtain weighted similarity coefficients between unrelated genotypes (Koopman and Gort 2004).
An additional difficulty is that the relationship between fragment size and homoplasy bias is highly dependent on the average allele frequency, leading to either overestimates or underestimates of heterozygosity. For example, Vekemans et al. (2002) deduced that, for small-size fragments, homoplasy would produce underestimation of both within- and between-population genetic diversity. This is in agreement with the results observed in Figure 4 with high 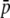 = 0.7, where it can be seen that homoplasy produces underestimations of both HS and FST. In fact, the average frequency of the allele producing a band in the empirical data analyzed by Vekemans et al. (2002) was 0.714 and 0.444 for the two species studied (see Table 2 in their article). However, if the average frequency had been much smaller, heterozygosity would have been overestimated instead of underestimated (as shown in the scenario with
= 0.7, where it can be seen that homoplasy produces underestimations of both HS and FST. In fact, the average frequency of the allele producing a band in the empirical data analyzed by Vekemans et al. (2002) was 0.714 and 0.444 for the two species studied (see Table 2 in their article). However, if the average frequency had been much smaller, heterozygosity would have been overestimated instead of underestimated (as shown in the scenario with 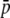 = 0.1 in Figure 4).
= 0.1 in Figure 4).
Our analysis demonstrates a moderate influence of the genome GC content on the homoplasy bias. The enzymes employed in this study are a frequent cutter (MseI) and a rare cutter (EcoRI) commonly used in AFLP surveys. Because MseI cuts are more frequent than EcoRI cuts, the average AFLP fragment size will be determined mainly by the frequency of MseI cuts. The restriction motif of MseI contains no G + C nucleotides, thus increasing the contribution of the small-sized class to the proportion of nonhomologous comigrating bands at low GC contents. This effect has also been observed with experimental data and probably results from the isochore structure of the genome in combination with the restriction motif recognized by the enzymes (i.e., MseI will preferably cut in A + T-rich isochores; Koopman and Gort 2004). Vekemans et al. (2002) found that the substitution of the commonly used MseI enzyme by a restriction enzyme recognizing a sequence motif with a balanced GC content would make the AFLP size distribution insensitive to variation in GC content. Hence, the effect of GC content on the homoplasy bias will depend on the choice of the restriction enzyme and may be avoided by using enzymes containing a balanced GC content for the shortest restriction motif, such as, for example, TaqI (Vekemans et al. 2002). However, the impact of homoplasy cannot be removed altogether even if a set of enzymes is used that guarantees a flat distribution of fragment lengths. This can be seen in Figure 3. When g = 0.7, the distribution of fragment lengths is almost uniform for the EcoRI–MseI tandem (see Figure 1C) but, even in this case, it is predicted to overestimate 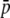 and HS, and underestimate FST (see Figure 3; left graphs).
and HS, and underestimate FST (see Figure 3; left graphs).
The model investigated assumes that the level of homoplasy depends only on the number of fragments evaluated (see Table 1) and, therefore, is independent of the level of differentiation between populations or the average allele frequency of the markers. It is however possible that increasing differentiation is associated with increasing homoplasy, as has been repeatedly observed empirically (O'Hanlon and Peakall 2000; El-Rabey et al. 2002; Mechanda et al. 2004). O'Hanlon and Peakall (2000) showed a positive correlation of the proportion of nonhomologous bands with species divergence and a negative relationship between the number of comigrating fragments and DNA sequence divergence. For distantly related species, many of the fragments tend to be unique to a species rather than shared among species, and a portion of these would be homoplasious. Thus, as the number of comigrating AFLP bands decreased, the chance that they were nonhomologous increased. If this can be generalized, the impact of homoplasy on the bias of population genetic diversity among distantly related species could be of much higher magnitude than that among conspecific populations.
To avoid the effects of homoplasy on the estimates of neutral variation, Vekemans et al. (2002) suggested (i) avoiding fragments in the small-size classes, (ii) using fragments of known map position that should have documented Mendelian inheritance when possible, and (iii) increasing the number of selective nucleotides in the primer sequences to reduce the number of fragments and thus the probability of nonhomology. However, although the exclusion of fragments of small size (i.e., <125 bp) would help to reduce levels of homoplasy by approximately one-third (Gort et al. 2006), there could still be fragments of larger size that are homoplasious. Alternatively, mapping studies are technically demanding, time-consuming, and out of the scope of many AFLP surveys (Meudt and Clarke 2007). The reduction of the number of scored bands per AFLP profile is also in conflict with the consideration that very small numbers of bands are undesirable in the sense that they reduce the information content of the AFLP profile. Therefore, even for a low number of bands per lane, a considerable percentage of comigrating bands are still nonhomologous (Vekemans et al. 2002; Gort et al. 2006).
Detection of selective loci from gene diversity and differentiation:
On the basis of the classical Lewontin and Krakauer (1973) multilocus neutrality test, several related approaches have recently been proposed for the detection of selective loci in genome scans (Bowcock et al. 1991; Beaumont and Nichols 1996; Vitalis et al. 2001; Schlötterer 2002; Beaumont and Balding 2004). One of the most popular methods, and for which software to analyze dominant data is available (DFDIST), is that from Beaumont and Nichols (1996). Beaumont and Balding (2004) investigated, by computer simulations, the performance of the codominant version of the method (FDIST program) in comparison with a hierarchical-Bayesian approach, in a structured population scenario for a set of biallelic codominant loci predominantly neutral but some of them subject to directional or balancing (heterozygote advantage) selection. They concluded that both methods could identify loci subject to directional selection when the selection coefficient was at least five times the migration rate, but that neither method was able to identify loci subject to balancing selection. We extended the analysis of Beaumont and Balding (2004) by evaluating the performance of the DFDIST software, considering dominant markers, a distribution of variable selective effects, and a scenario of two subpopulations with different levels of neutral genetic differentiation, where a variable proportion of the loci (that was set up from 1 to 10% of the genome) are subject to directional selection in one subpopulation. We assessed a number of alternatives used in different empirical or theoretical studies:
A high critical probability threshold level of 5% vs. a much lower value of 0.05%: Most authors have considered critical values of 5% (Scotti-Saintagne et al. 2004; Acheré et al. 2005; Bonin et al. 2006; Mealor and Hild 2006; Miller et al. 2007; Papa et al. 2007; Nosil et al. 2008; Smith et al. 2008) or 1% (Jump et al. 2006; Murray and Hare 2006; Papa et al. 2007; Nosil et al. 2008).
The mean FST used to build the null model neutral distribution was a trimmed mean (used by Bonin et al. 2006; Miller et al. 2007; Nosil et al. 2008; Smith et al. 2008), the raw (nontrimmed) mean (used by Scotti-Saintagne et al. 2004; Mealor and Hild 2006), or the median FST (used in simulations by Beaumont and Balding 2004).
We found that the potentiality of the method to estimate the proportion of the genome subject to selection through the proportion of outliers detected has some reliability only for the case where (i) the selection coefficients are very large (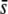 = 0.5), (ii) the trimmed mean or median FST are used, (iii) the critical probability level is low (α = 0.05%), and (iv) the level of differentiation is also low (FST = 0.025) (Figures 5–7). In all other situations, such as for α = 5% (Figure 5), nontrimmed mean or low
= 0.5), (ii) the trimmed mean or median FST are used, (iii) the critical probability level is low (α = 0.05%), and (iv) the level of differentiation is also low (FST = 0.025) (Figures 5–7). In all other situations, such as for α = 5% (Figure 5), nontrimmed mean or low 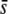 (Figure 6), or a higher level of differentiation (FST = 0.1; Figure 7), the method does not provide a right inference of the proportion of selective loci in the genome. Even in the most favorable scenarios above, the percentage of outliers does not always predict the true proportion of selective loci in the genome. For example, using the median FST and α = 0.05%, for
(Figure 6), or a higher level of differentiation (FST = 0.1; Figure 7), the method does not provide a right inference of the proportion of selective loci in the genome. Even in the most favorable scenarios above, the percentage of outliers does not always predict the true proportion of selective loci in the genome. For example, using the median FST and α = 0.05%, for 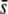 = 0.5 (Figure 6M), the proportion of outliers is >3% whereas the true proportion of selective loci is 1%. In accordance with our results, Beaumont and Balding (2004) found a proportion of outliers (as we deduced from their Table 3) close to the true proportion of selective loci in the genome (8% in their case) when considering a high constant coefficient of selection s = 0.2. However, the percentage of outliers detected was ≪8% for selective loci of small effect (s = 0.02–0.05).
= 0.5 (Figure 6M), the proportion of outliers is >3% whereas the true proportion of selective loci is 1%. In accordance with our results, Beaumont and Balding (2004) found a proportion of outliers (as we deduced from their Table 3) close to the true proportion of selective loci in the genome (8% in their case) when considering a high constant coefficient of selection s = 0.2. However, the percentage of outliers detected was ≪8% for selective loci of small effect (s = 0.02–0.05).
It is highly remarkable that when a high critical probability threshold (α = 5%) is applied, the proportion of outliers detected is basically independent of the proportion of selective loci in the genome (Figure 5) as well as of the value of 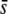 (Figure 7), which indicates that for relatively low values of
(Figure 7), which indicates that for relatively low values of 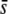 or low proportions of selective loci, the majority of outliers are false positives. This produces an obvious sense of lack of reliability in the method. In theory, the trimmed mean FST or the median FST should correct for the presence of selective loci and, therefore, they should remain invariable for increasing values of
or low proportions of selective loci, the majority of outliers are false positives. This produces an obvious sense of lack of reliability in the method. In theory, the trimmed mean FST or the median FST should correct for the presence of selective loci and, therefore, they should remain invariable for increasing values of 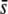 or the proportion of selective loci in the genome. However, this is not the case, and both parameters increase correspondingly, although to a lower extent than the uncorrected mean FST. For example, for FST = 0.025 and 5% of selective loci in the genome, the trimmed mean Weir and Cockerham's (1984) FST is 0.016, 0.018, and 0.038, and the median FST is 0.011, 0.013, and 0.016 for
or the proportion of selective loci in the genome. However, this is not the case, and both parameters increase correspondingly, although to a lower extent than the uncorrected mean FST. For example, for FST = 0.025 and 5% of selective loci in the genome, the trimmed mean Weir and Cockerham's (1984) FST is 0.016, 0.018, and 0.038, and the median FST is 0.011, 0.013, and 0.016 for 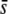 = 0, 0.005 and 0.5, respectively. This increase, inherent to any correction method, may be the cause of an approximately constant proportion of outliers irrespective of the number and effects of selective loci.
= 0, 0.005 and 0.5, respectively. This increase, inherent to any correction method, may be the cause of an approximately constant proportion of outliers irrespective of the number and effects of selective loci.
In most studies the main objective of the genome scans is to detect true selective loci among the outliers. Our results suggest that a low critical probability (α = 0.05%) is necessary (Figures 5–7) for this objective to be achieved successfully, but the method is not reliable for small values of 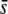 or low proportions of selective loci in the genome. For example, for a proportion of selective loci of 1%, the percentage of selective loci present among the outliers is ∼10–20% for
or low proportions of selective loci in the genome. For example, for a proportion of selective loci of 1%, the percentage of selective loci present among the outliers is ∼10–20% for 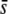 = 0.005 and 40–60% for
= 0.005 and 40–60% for 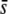 = 0.5 when the trimmed mean FST or the median FST are used (Figure 6). The use of the nontrimmed mean FST cannot be recommended in this context. For a proportion of selective loci between 1 and 5%, the percentage of selective loci present among the outliers is 100%, denoting a perfect success (Figure 6B). However, when the proportion of selective loci is 10%, no outliers are detected at all (Figure 6A) because the mean FST without any correction is too large.
= 0.5 when the trimmed mean FST or the median FST are used (Figure 6). The use of the nontrimmed mean FST cannot be recommended in this context. For a proportion of selective loci between 1 and 5%, the percentage of selective loci present among the outliers is 100%, denoting a perfect success (Figure 6B). However, when the proportion of selective loci is 10%, no outliers are detected at all (Figure 6A) because the mean FST without any correction is too large.
Beaumont and Balding (2004) simulations indicated an appreciable discrimination for selective loci when the selective effect is larger than about five times the migration rate. Thus, they found that ∼60% of outliers were truly selective loci considering selection coefficients of s = 0.02, and this rate of success was improved up to 80–100% for selective effects between 0.05 and 0.2 (results deduced from their Table 3). This result is in agreement with our observations. For a neutral FST = 0.025 the migration rate was m = 0.006, so that selective effects larger than 5m = 0.03 should be more likely detected. Figure 7H indicates that the percentage of selective loci in outliers is close to 100% for 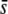 = 0.05–0.5, but <30% for
= 0.05–0.5, but <30% for 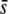 = 0.005. Analogously, for a neutral FST = 0.1 the migration rate was m = 0.00112, so that selective effects >0.006 should be more likely detected. Figure 7K shows that the percentage of selective loci in outliers is >70% for
= 0.005. Analogously, for a neutral FST = 0.1 the migration rate was m = 0.00112, so that selective effects >0.006 should be more likely detected. Figure 7K shows that the percentage of selective loci in outliers is >70% for 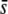 > 0.005, but only ∼20% for
> 0.005, but only ∼20% for  = 0.001. This agreement between both studies is very reassuring considering that the two models differ in a number of aspects: (i) Beaumont and Balding (2004) analyzed codominant markers using the FDIST program whereas we studied dominant markers with the DFDIST software, (ii) the number of subpopulations considered was n = 6 whereas in our case n = 2, (iii) the selective coefficients were constant instead of variable, and (iv) the model of directional selection implied divergent selection whereas in our study only one of the two subpopulations was assumed to be under selection.
= 0.001. This agreement between both studies is very reassuring considering that the two models differ in a number of aspects: (i) Beaumont and Balding (2004) analyzed codominant markers using the FDIST program whereas we studied dominant markers with the DFDIST software, (ii) the number of subpopulations considered was n = 6 whereas in our case n = 2, (iii) the selective coefficients were constant instead of variable, and (iv) the model of directional selection implied divergent selection whereas in our study only one of the two subpopulations was assumed to be under selection.
We also found that subpopulation sample size has some impact on the detection of selective loci, but this is limited beyond M = 40 individuals or so (Figure 7, M–O). This is in agreement with the conclusion of Beaumont and Nichols (1996) that there is little improvement in the performance of the FDIST method by having sample sizes larger than 25 diploids per subpopulation.
We can conclude that the detection of selective loci among outliers depends enormously on the conditions of the analysis and the proportion and effects of the selective loci. Using low critical probability threshold values (e.g., α = 0.05%) and a trimmed or the median FST seem the most appropriate procedures. We have not investigated the performance of an iterative process to obtain the mean FST of the null neutral model, suggested by Beaumont and Nichols (1996) (see Beaumont and Balding 2004), whereby the raw mean FST is first calculated, then the outliers removed, and the process repeated until no outliers are detected (see, e.g., Papa et al. 2007 for an empirical application). However, we have seen that a high degree of success is accomplished only when the proportion of selective loci in the genome is large and their average effect is also large. If these circumstances do not hold, the number of false positives can be very substantial and, therefore, the sequential removal of outliers is unlikely to work successfully. Another more reliable way to detect truly selective loci may be by combining the results of different tests (Vasemägi et al. 2005; Bonin et al. 2006), but this has yet to be checked by further analyses and simulations.
Impact of homoplasy on the detection of selective loci:
The impact of homoplasy on the identification of selective loci was investigated assuming different numbers of loci and, consequently, of experimental fragments analyzed per primer combination (Figure 8). The main observation is that homoplasy causes a slight overestimation in the proportion of outliers and a more substantial underestimation in the proportion of selective loci detected. This latter can reach up to 15% when the number of experimental bands per primer combination is on the order of ≥75–100 (Figure 8, C, F, and I). Thus, this aspect inherent to AFLP data should be taken into consideration in genomewide scans of selective loci.
Two additional important issues should be taken into account when using AFLP fragments in the detection of selective loci. First, in the cases of substantial homoplasy, some bands detected as outliers, even if they really correspond to true selective genes, do not necessarily carry a single selective fragment but they can also carry one or more neutral fragments. Therefore, a further experimental analysis of these bands should be carried out to disentangle selective and neutral comigrating fragments. The second issue is that the above threshold range refers to the number of experimental bands observed at the population level. If sample sizes are very small and the frequency of the allele producing the band is also low, the number of experimental bands observed can be much lower than the real one in the population, producing a false impression that the impact of homoplasy in the study will be low. For example, assuming genotypic frequencies at Hardy–Weinberg equilibrium and being 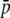 the average frequency of the marker allele associated to the experimental bands, the ratio of the number of population bands to the number of observed bands is
the average frequency of the marker allele associated to the experimental bands, the ratio of the number of population bands to the number of observed bands is 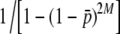 if M individuals are sampled in the population. If this number is larger than 10 the ratio is close to one, but if few individuals are sampled the ratio could be very large. For example, if M = 1 (e.g., in phylogenetic studies), the observation of 60 bands per primer combination corresponds, in fact, to 315.8 bands per primer combination in the population. Analogously, if M = 5, the observation of 60 bands in this sample corresponds to 92.1 bands per primer combination in the population. Therefore, the amount of homoplasy in these cases can be much larger than that inferred from the observed number of bands (60).
if M individuals are sampled in the population. If this number is larger than 10 the ratio is close to one, but if few individuals are sampled the ratio could be very large. For example, if M = 1 (e.g., in phylogenetic studies), the observation of 60 bands per primer combination corresponds, in fact, to 315.8 bands per primer combination in the population. Analogously, if M = 5, the observation of 60 bands in this sample corresponds to 92.1 bands per primer combination in the population. Therefore, the amount of homoplasy in these cases can be much larger than that inferred from the observed number of bands (60).
Acknowledgments
We thank Juan Galindo and two anonymous referees for useful comments on the manuscript. This work was funded by the Ministerio de Educación y Ciencia and Fondos Feder (CGL2006-13445-C02/BOS, CGL2004-03920/B0S), Plan Estratégico del Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (CPE03-004-C2), and Xunta de Galicia.
References
- Acheré, V., J. M. Favre, G. Nesnard and S. Jeandroz, 2005. Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol. Ecol. 14 3191–3201. [DOI] [PubMed] [Google Scholar]
- Akashi, H., R. M. Kliman and A. Eyre-Walker, 1998. Mutation pressure, natural selection, and the evolution of base composition in Drosophila. Genetica 102/103 49–60. [PubMed] [Google Scholar]
- Albach, D. C., P. Schönswetter and A. Tribsch, 2006. Comparative phylogeography of the Veronica alpina complex in Europe and North America. Mol. Ecol. 15 3269–3286. [DOI] [PubMed] [Google Scholar]
- Althoff, D. M., M. A. Gitzendanner and K. A. Segraves, 2007. The utility of amplified fragment length polymorphisms in phylogenetics: a comparison of homology within and between genomes. Syst. Biol. 56 477–484. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., 2005. Adaptation and speciation: What can FST tell us? Trends Ecol. Evol. 20 435–440. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and D. J. Balding, 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13 969–980. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and R. A. Nichols, 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B 263 1619–1626. [Google Scholar]
- Bensch, S., and M. Akesson, 2005. Ten years of AFLP in ecology and evolution: Why so few animals? Mol. Ecol. 14 2899–2914. [DOI] [PubMed] [Google Scholar]
- Bonin, A., P. Taberlet, C. Miaud and F. Pompanon, 2006. Explorative genome scan to detect candidate loci for adaptation along a gradient of altitude in the common frog (Rana temporaria). Mol. Biol. Evol. 23 773–783. [DOI] [PubMed] [Google Scholar]
- Bonin, A., D. Ehrich and S. Manel, 2007. Statistical analysis of amplified fragment length polymorphism data: a toolbox for molecular ecologists and evolutionists. Mol. Ecol. 16 3737–3758. [DOI] [PubMed] [Google Scholar]
- Bowcock, A. M., J. R. Kidd, J. L. Mountain, J. M. Hebert, L. Carotenuto et al., 1991. Drift, admixture, and selection in human evolution. Proc. Natl. Acad. Sci. USA 88 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussel, J. D., M. Waycott and J. A. Chappill, 2005. Arbitrarily amplified DNA markers for phylogenetic inference. Perspect. Plant Ecol. Evol. Syst. 7 3–26. [Google Scholar]
- Campbell, D., and L. Bernatchez, 2004. Generic scan using AFLP markers as a means to assess the role of directional selection in the divergence of sympatric whitefish ecotypes. Mol. Biol. Evol. 21 945–956. [DOI] [PubMed] [Google Scholar]
- El-Rabey, H. A., A. Badr, R. Schaffer-Pregl, W. Martin and F. Salamini, 2002. Speciation and species separation in Hordeum L. (Poaceae) resolved by discontinuous molecular markers. Plant Biol. 4 567–575. [Google Scholar]
- Enjalbert, J., X. Duan, M. Leconte, M. S. Hovmøller and C. de Vallavielle-pope, 2005. Genetic evidence of local adaptation of wheat yellow rust (Puccina striiformis f. sp. tritici) within France. Mol. Ecol. 14 2065–2073. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., J. Novembre and S. Schneider, 2000. SIMCOAL: a general coalescent program for the simulation of molecular data in interconnected populations with arbitrary demography. J. Hered. 91 506–509. [DOI] [PubMed] [Google Scholar]
- Fang, J., J. Tao and C. T. Chao, 2006. Genetic diversity in fruiting-mei, apricot, plum and peach revealed by AFLP analysis. J. Hortic. Sci. Biotechnol. 81 898–902. [Google Scholar]
- Fernández, B, A. García-Dorado and A. Caballero, 2005. The effect of antagonistic pleiotropy on the estimation of the average coefficient of dominance of deleterious mutations. Genetics 171 2097–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gort, G, W. J. M. Koopman and A. Stein, 2006. Fragment length distributions and collision probabilities for AFLP markers. Biometrics 62 1107–1115. [DOI] [PubMed] [Google Scholar]
- Hansen, M., T. Kraft, M. Christiansen and N.-O. Nilsson, 1999. Evaluation of AFLP in Beta. Theor. Appl. Genet. 98 845–852. [Google Scholar]
- Innan, H., R. Terauchi, G. Kahl and F. Tajima, 1999. A method for estimating nucleotide diversity from AFLP data. Genetics 151 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jump, A. S., J. M. Hunt, J. A. Martínez-Izquierdo and J. Peñuelas, 2006. Natural selection and climate change: temperature-linked spatial and temporal trends in gene frequency in Fagus sylvatica. Mol. Ecol. 15 3469–3480. [DOI] [PubMed] [Google Scholar]
- Kalita, M., and W. Malek, 2006. Application of the AFLP method to differentiate Genista tinctoria microsymbionts. J. Gen. Appl. Microbiol. 52 321–328. [DOI] [PubMed] [Google Scholar]
- Kidd, M. R., C. E. Kidd and T. D. Kocher, 2006. Axes of differentiation in the bower-building cichlids of lake Malawi. Mol. Ecol. 15 459–478. [DOI] [PubMed] [Google Scholar]
- Koopman, W. J. M., and G. Gort, 2004. Significance tests and weighted values for AFLP similarities, based on Arabidopsis in silico AFLP fragment length distributions. Genetics 167 1915–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitung, B., E. Chauvet, N. Feau, K. Fève, L. Chikhi et al., 2004. Genetic diversity in Tetrechaetum elegans, a mitosporic aquatic fungus. Mol. Ecol. 13 1679–1692. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., and J. Krakauer, 1973. Distribution of gene frequency as a test of the theory of the selected neutrality of polymorphism. Genetics 74 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealor, B. A., and A. L. Hild, 2006. Potential selection in native grass populations by exotic invasion. Mol. Ecol. 15 2291–2300. [DOI] [PubMed] [Google Scholar]
- Mechanda, S. M., B. R. Baum, D. A. Johnson and J. T. Arnason, 2004. Sequence assessment of comigrating AFLP™ bands in Echinacea—implications for comparative biological studies. Genome 47 15–25. [DOI] [PubMed] [Google Scholar]
- Mendelson, T. C., and K. L. Shaw, 2005. Use of AFLP markers in surveys of arthropod diversity. Methods Enzymol. 395 161–177. [DOI] [PubMed] [Google Scholar]
- Meudt, H. M., and A. C. Clarke, 2007. Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci. 12 106–117. [DOI] [PubMed] [Google Scholar]
- Miller, N. J., M. Ciosi, T. W. Sappington, S. T. Ratcliffe, J. L. Spencer et al., 2007. Genome scan of Diabrotica virgifera virgifera for genetic variation associated with crop rotation tolerance. J. Appl. Entomol. 131 378–385. [Google Scholar]
- Mueller, U. G., and L. L. Wolfenbarger, 1999. AFLP genotyping and fingerprinting. Trends Ecol. Evol. 14 389–394. [DOI] [PubMed] [Google Scholar]
- Murray, M. C., and M. P. Hare, 2006. A genome scan for divergent selection in a secondary contact zone between Atlantic and Gulf of Mexico oysters, Crassostrea virginica. Mol. Ecol. 15 4229–4242. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 2005. Molecular signatures of natural selection. Annu. Rev. Genet. 39 197–218. [DOI] [PubMed] [Google Scholar]
- Nosil, P., S. P. Egan and D. J. Funk, 2008. Heterogeneous genomic differentiation between walking-stick ecotypes: “isolation by adaptation” and multiple roles for divergent selection. Evolution 62 316–336. [DOI] [PubMed] [Google Scholar]
- O'Hanlon, P. C., and R. Peakall, 2000. A simple method for the detection of size homoplasy among amplified fragment length polymorphism fragments. Mol. Ecol. 9 815–816. [DOI] [PubMed] [Google Scholar]
- Papa, R., E. Bellucci, M. Rossi, S. Leonardi, D. Rau et al., 2007. Tagging the signatures of domestication in common bean (Phaseolus vulgaris) by means of pooled DNA samples. Ann. Botany 100 11039–11051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J. L., H. Constandt, P. Neyt, G. Cnops, J. Zethof et al., 2001. A physical amplified fragment-length polymorphism map of Arabidopsis. Plant Physiol. 127 1575–1589. [PMC free article] [PubMed] [Google Scholar]
- Rombauts, S., Y. van de Peer and P. Rouzé, 2003. AFLP in silico, simulating AFLP fingerprints. Bioinformatics 19 776–777. [DOI] [PubMed] [Google Scholar]
- Rouppe van der Voort, J. N., P. van Zandvoort, H. J. van Eck, R. T. Folkertsma, R. C. Hutten et al., 1997. Use of allele specificity of comigrating AFLP markers to align genetic maps from different potato genotypes. Mol. Gen. Genet. 255 438–447. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 2002. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti-Saintagne, C., S. Mariette, I. Porth, P. G. Goicoechea, T. Barreneche et al., 2004. Genome scanning for interspecific differentiation between two closely related oak species [Querqus robur L. and Q. petrea (Matt.) Liebl.]. Genetics 168 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T. B., B. Milá, G. F. Grether, H. Slabbekoorn, I. Sepil et al., 2008. Evolutionary consequences of human disturbance in a rainforest bird species from Central Africa. Mol. Ecol. 17 58–71. [DOI] [PubMed] [Google Scholar]
- Snäll, T., J. Fogelqvist, P.J. Ribeiro and M. Lascoux, 2004. Spatial genetic structure in two congeneric epiphytes with different dispersal strategies analysed by three different methods. Mol. Ecol. 13 2109–2119. [DOI] [PubMed] [Google Scholar]
- Stein, L. D., Z. Bao, D. Blasiar, T. Blumenthal, M. R. Brent et al., 2003. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1 166–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz, J. F., 2005. Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol. Ecol. 14 671–688. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., and J. M. Dubach, 2004. Natural selection drives altitudinal divergence at the albumin locus in deer mice, Peromyscus maniculatus. Evolution 58 1342–1352. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., and M. W. Nachman, 2003. Natural selection on protein polymorphism in the rodent genus peromyscus: evidence from interlocus contrasts. Evolution 57 2628–2635. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., B. A. Payseur and M. W. Nachman, 2004. Genome scans of DNA variability in human reveal evidence for selective sweeps outside of Africa. Mol. Biol. Evol. 21 1800–1811. [DOI] [PubMed] [Google Scholar]
- Troell, K., A. Engstrom, D. A. Morrison, J. G. Mattsson and J. Hoglund, 2006. Global patterns reveal strong population structure in Haemonchus controtus, a nematode parasite of domesticated ruminants. Int. J. Parasitol. 36 1305–1316. [DOI] [PubMed] [Google Scholar]
- Vasemägi, A., J. Nilsson and C. R. Primmer, 2005. Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic salmon (Salmo salar L.). Mol. Biol. Evol. 22 1067–1076. [DOI] [PubMed] [Google Scholar]
- Vekemans, X., T. Beauwens, M. Lemaire and I. Roldán-Ruiz, 2002. Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol. Ecol. 11 139–151. [DOI] [PubMed] [Google Scholar]
- Vitalis, R., K. Dawson and P. Boursot, 2001. Interpretation of variation across loci as evidence of selection. Genetics 158 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos, P., R. Hogers, M. Bleeker, T. van de Lee, M. Hornes et al., 1995. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 21 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B., and C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wilding, C. S., R. K. Butlin and J. Grahame, 2001. Differential gene exchange between parapatric morphs of Littorina saxatilis detected using AFLP markers. J. Evol. Biol. 14 611–619. [Google Scholar]
- Wright, S., 1937. The distribution of gene frequencies in populations. Proc. Natl. Acad. Sci. USA 23 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1951. The genetic structure of populations. Ann. Eugen. 15 323–354. [DOI] [PubMed] [Google Scholar]
- Yu, J., S. Hu, J. Wang, G. K.-S. Wong, S. Li et al., 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. Indica). Science 296 79–92. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky, L. A., 1999. Estimating population structure in diploids with multilocus dominant DNA markers. Mol. Ecol. 8 907–913. [DOI] [PubMed] [Google Scholar]