

Abstract
Linkage disequilibrium (LD) may reveal much about domestication and breed history. An investigation was conducted, to analyze the extent of LD, haploblock partitioning, and haplotype diversity within haploblocks across several pig breeds from China and Europe and in European wild boar. In total, 371 single-nucleotide-polymorphisms located in three genomic regions were genotyped. The extent of LD differed significantly between European and Chinese breeds, extending up to 2 cM in Europe and up to 0.05 cM in China. In European breeds, LD extended over large haploblocks up to 400 kb, whereas in Chinese breeds the extent of LD was smaller and generally did not exceed 10 kb. The European wild boar showed an intermediate level of LD between Chinese and European breeds. In Europe, the extent of LD also differed according to genomic region. Chinese breeds showed a higher level of haplotype diversity and shared high levels of frequent haplotypes with Large White, Landrace, and Duroc. The extent of LD differs between both centers of pig domestication, being higher in Europe. Two hypotheses can explain these findings. First, the European ancestral stock had a higher level of LD. Second, modern breeding programs increased the extent of LD in Europe and caused differences of LD between genomic regions. Large White, Landrace, and Duroc showed evidence of past introgression from Chinese breeds.
LINKAGE disequilibrium (LD), which refers to nonrandom association of alleles at different loci, has received increasing attention in recent years and has gained unprecedented momentum as a result of the availability of genome sequences and large numbers of identified single-nucleotide polymorphisms (SNPs). The Human HapMap project (International Hapmap Consortium 2003, 2005) has revealed a large degree of variation of LD across the human genome and the intrinsic difficulty of analysis of genomewide LD data (Reich et al. 2001). It also showed the presence of important differences in LD among human populations, which result from differences in population history and demography (Reich et al. 2001; Ardlie et al. 2002). Furthermore, the detailed information on genomic haplotype structure was shown to be of high utility for fine mapping of genes responsible for complex multifactorial diseases (Rigby et al. 2006; Wright et al. 2006; Baessler et al. 2007; Wellcome Trust Case Control Consortium 2007).
Understanding the properties of LD in domesticated animals is important because it underlies all forms of genetic mapping (Nordborg and Tavare 2002). LD can reveal much about domestication and breed history because the distribution of LD is, in part, determined by population history and demography (Pritchard and Przeworski 2001; Tenesa et al.2007).
LD has been studied in a variety of domestic animal species, e.g., cattle (Farnir et al. 2000), sheep (McRae et al. 2002), pigs (Nsengimana et al. 2004), dogs (Lindblad-Toh et al. 2005), and chickens (Aerts et al. 2007). In some of these species, a substantial extent of LD was found over several centimorgans and exceeded the extent of LD found in humans (Reich et al. 2001). This larger extent of LD in animal species may be due to small effective population sizes in commercially held populations, and these may not be typical for the entire species. Dogs, for instance, show a large degree of variation in LD patterns, reflecting both high variability of the ancestor (wolf) and the result of low population sizes in breed formation and maintenance (Lindblad-Toh et al. 2005). In addition, most animal species are now known to have complex domestication histories (Bruford et al. 2003).
Pigs are among the most important domestic animals (Chen et al. 2007), being an important protein source. They are also an important animal model to study domestication because Chinese and European pigs' ancestors still exist (Giuffra et al. 2000). European and Chinese pigs were domesticated independently from European and Asian subspecies of wild boar (Giuffra et al. 2000; Larson et al. 2005). Studies on mitochondrial DNA suggest the occurrence of introgression of Asian domestic pigs into European breeds after domestication (Giuffra et al. 2000; Fang and Andersson 2006). More recently, Larson et al. (2007) demonstrated that domestic pigs of Near Eastern ancestry were introduced into Europe during the Neolithic. European wild boar was also domesticated by this time, possibly as a direct consequence of the introduction of Near Eastern pigs.
The possibility of using large numbers of SNPs enables the detection of nuclear haplotypes that may be associated with introgression and/or phenotypic selection that occurred during the domestication process. Analysis of the extent of useful LD (Kruglyak 1999) could provide information about sample sizes and the number of markers required to fine map genes responsible for common diseases and other phenotypic traits (Zhang et al. 2002).
Our aim was to investigate the extent of LD, LD haploblock partitioning, and haplotype diversity within haploblocks across a total of 20 pig breeds in Europe and China and in the ancestral European wild boar. With the commercial lines possibly containing the larger extent of LD in the species, we examined three genomic regions, each ∼1–3 cM, at a higher SNP density than previous studies (Nsengimana et al. 2004; Du et al. 2007). This study provides insight into the extent of useful LD across a wide range of breeds and the required sample sizes and number of markers for association studies.
MATERIALS AND METHODS
DNA samples:
DNA samples were obtained from 10 European and 10 Chinese pig breeds and from wild boar individuals from France, which came from a single reserve, were certified as 2n = 36, and are to the best of our knowledge unrelated. Sample size ranged from 15 to 25 individuals (Table 1). The material from these breeds was collected in the framework of PigBioDiv (Ollivier et al. 2005; Sancristobal et al. 2006) and PigBiodiv II (Blott et al. 2003) projects. European breeds were grouped by origin and history into local, international, and commercial breeds. Chinese breeds were grouped by lower Changjiang River basin, southwest China, central China, north China, Plateau, and south China (Zhang 1986; Fang et al. 2005; Megens et al. 2008).
TABLE 1.
Sample size by breed
| Origin | Breed | Sample size | Target 1 SNPs (%)a | Target 2 SNPs (%)a | Target 4 SNPs (%)a |
|---|---|---|---|---|---|
| Europe | |||||
| Local | British Saddleback (BS01) | 25 | 72.50 | 40.35 | 38.25 |
| Large Black (LB01) | 25 | 75.00 | 62.28 | 38.25 | |
| Mangalitsa (MA01) | 23 | 42.50 | 78.95 | 52.46 | |
| Middle White (MW01) | 23 | 75.00 | 47.37 | 32.79 | |
| Tamworth (TA01) | 25 | 82.50 | 64.04 | 22.40 | |
| International | Landrace (LR01) | 15 | 72.50 | 42.11 | 31.69 |
| Pietrain (Pi03) | 24 | 47.50 | 67.54 | 23.50 | |
| Commercial | Duroc (DU02) | 25 | 47.50 | 56.14 | 80.33 |
| Hampshire (HA01) | 25 | 75.00 | 59.65 | 66.12 | |
| Large White (LW05) | 25 | 37.50 | 37.72 | 38.25 | |
| China | |||||
| Lower Changjiang River basin | Erhulian (Erhu) | 24 | 27.50 | 53.51 | 34.97 |
| Meishan (MS02) | 25 | 25.00 | 30.70 | 51.37 | |
| Southwest China | Guanling (Guan) | 24 | 32.50 | 50.00 | 49.18 |
| Neijiang (Nei) | 25 | 42.50 | 50.88 | 61.20 | |
| Central China | Jinhua (Jin) | 25 | 55.00 | 77.19 | 61.75 |
| Ningxiang (Ninx) | 23 | 37.50 | 46.49 | 48.63 | |
| North China | Mashen (Mas) | 24 | 17.50 | 64.04 | 38.80 |
| Plateau | Tibetan (Tib) | 24 | 40.00 | 52.63 | 27.87 |
| South China | Longling (Lon) | 25 | 17.50 | 40.35 | 20.22 |
| Wuzishan (Wuz) | 25 | 22.50 | 41.23 | 25.68 | |
| French wild boar (FrWb) | 25 | 45.00 | 82.46 | 57.92 | |
Minor allele frequency ≤0.05.
SNP development and selection:
The National Institutes of Health (NIH) Intramural Sequencing Center (NISC) (http://www.nisc.nih.gov) sequenced a large number of porcine BACs (supplemental Table 1). These BACs were derived from a pig BAC library developed using DNA of four crossbred male pigs (breed composition: 37.5% Yorkshire, 37.5% Landrace, and 25% Meishan) (Fahrenkrug et al. 2001). NISC grouped these BACs by targets. The porcine sequences from targets 1, 2, and 4 have previously been used to randomly identify SNPs (Jungerius et al. 2005). In our study additional SNPs were identified within these genomic regions by aligning sequences derived from overlapping BAC clones (supplemental Table 1). The list of identified SNPs and respective accession numbers is in supplemental Table 2.
SNP mapping:
BAC sequences were masked for repeat motifs using RepeatMAsker v3.1.6 and RepBase 11.06 (http://repeatmasker.org) and aligned to porcine BAC end sequences (BES) available in GenBank using Megablast v2.2.14 (Altschul et al. 1990). Positions of BES in the pig genome are available in the FPC map (http://www.sanger.ac.uk/Projects/S_scrofa/). Hits with a bit score ≥1000 were therefore selected and used to obtain the BAC position on the FPC map (FPC map of 08.10.06). The SNP position on the BAC was converted to a SNP position on the chromosome, using information on the BAC position and sequence length. SNP positions are in supplemental Table 2.
SNP genotyping:
Because only small amounts of genomic DNA were available for each Chinese breed except Meishan, whole-genome amplification (WGA) (Dean et al. 2002) was performed on these samples using the REPLI-g kit from QIAGEN (Valencia, CA), with 50–150 ng of input genomic DNA.
Genotyping was done in a 1536-plex format using the GoldenGate assay and Sentrix array matrices (Illumina, San Diego) (Fan et al. 2003). Genotyping, including data editing, was performed by the Illumina service facility. A total of 1536 SNPs were genotyped with this procedure, but only 44 located in target 1, 128 located in target 2, and 199 located in target 4 are described in this study.
Predicted decay of LD by breed:
To measure LD, pairwise r2 was calculated using Haploview version 3.2 (Barrett et al. 2005). In this study, r2 was chosen because it is very useful in the case of biallelic markers such as SNPs and because it is independent from sample size (Devlin and Risch 1995). Further, Du et al. (2007) evaluated recently how r2 and D′ are affected by several levels of minor allele frequency (MAF). Their results suggest that D′ is highly dependent on levels of MAF, whereas r2 is less.
For SNPs genotyped for 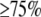 of the total samples per breed in each genomic region within breed, tests for deviations from Hardy–Weinberg (HW) equilibrium were performed and allele frequencies for all SNPs were estimated. SNPs in HW disequilibrium (P < 0.001) and/or with MAF < 0.05 were excluded.
of the total samples per breed in each genomic region within breed, tests for deviations from Hardy–Weinberg (HW) equilibrium were performed and allele frequencies for all SNPs were estimated. SNPs in HW disequilibrium (P < 0.001) and/or with MAF < 0.05 were excluded.
To assess the extent and decline of LD between breeds the equation
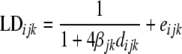 |
(1) |
was used (Sved 1971; Heifetz et al. 2005), where 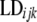 is the observed LD for marker pair i of breed j in genomic region k,
is the observed LD for marker pair i of breed j in genomic region k, 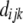 is the distance in base pairs for marker pair i of breed j in genomic region k,
is the distance in base pairs for marker pair i of breed j in genomic region k, 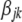 is the coefficient that describes the decline of LD with distance for breed j in genomic region k, and
is the coefficient that describes the decline of LD with distance for breed j in genomic region k, and 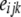 is a random residual. For each genomic region within breed
is a random residual. For each genomic region within breed 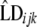 ,
, 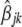 , and
, and 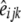 were estimated using the nonlinear fit function in R environment (http://www.r-project.org/). Graphic displays of
were estimated using the nonlinear fit function in R environment (http://www.r-project.org/). Graphic displays of 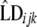 vs. distance were produced.
vs. distance were produced.
Test for breed and genomic region effects in the extent of LD:
Markers were not evenly distributed within genomic regions. This can have an effect in the evaluation of LD, since pairwise calculations are not assessed at equal distances and may cause a distortion in LD values. To test for breed and genomic region effects it was necessary to correct 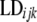 for differences in map distance when evaluating differences in LD between genomic regions.
for differences in map distance when evaluating differences in LD between genomic regions. 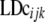 is the distance-corrected and variance-stabilized LD for marker pair i in genomic region k and breed j and it was estimated using
is the distance-corrected and variance-stabilized LD for marker pair i in genomic region k and breed j and it was estimated using  and
and 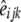 obtained with Equation 1:
obtained with Equation 1:
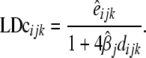 |
(2) |
Differences in LD between genomic regions (target 1, target 2, and target 4) and breeds were analyzed,
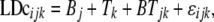 |
(3) |
where 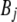 is the fixed effect of breed j,
is the fixed effect of breed j, 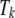 is the fixed effect of genomic region k,
is the fixed effect of genomic region k, 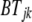 is the fixed interaction effect, and
is the fixed interaction effect, and 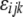 is the random residual. Equation 3 was fitted using the linear model function in the R environment (http://www.r-project.org/). Differences among all interaction levels were tested using the lsmeans function in SAS version 9.1 (SAS Institute, Cary, NC).
is the random residual. Equation 3 was fitted using the linear model function in the R environment (http://www.r-project.org/). Differences among all interaction levels were tested using the lsmeans function in SAS version 9.1 (SAS Institute, Cary, NC).
LD haploblock partitioning and haplotype diversity:
Due to the variation in local recombination rates, the breakdown of LD is often discontinuous and presents a haploblock-like structure (Daly et al. 2001; International Hapmap Consortium 2005). Therefore, it is important to analyze the haploblock structure and haplotypes that underlie LD. Analysis of haploblock partition defines the haploblock from the LD measure r2, initiating and extending a haploblock according to the pairwise and grouped r2 values (Gu et al. 2005). The algorithm starts a haploblock by selecting the pair of adjacent SNPs with the highest r2 (r2 > α) and extends the haploblock if the average r2 between an adjacent site and current haploblock members is >β and each r2 > γ. Here, α > β > γ and in this case they were α = 0.4, β = 0.3, and γ = 0.1 (Gu et al. 2005). After the first haploblock is identified, a new pair of adjacent SNPs with the highest r2 (r2 ≥ α) is used to start a new haploblock accretion process.
Haplotypes within haploblocks were obtained using an accelerated EM algorithm, similar to the partition/ligation method of Qin et al. (2002) and implemented in Haploview version 3.2 (Barrett et al. 2005). The method creates highly accurate population frequency estimates of the phased haplotypes on the basis of the maximum likelihood as determined from the unphased genotypes.
Plots of LD were generated using Haploview version 3.2 (Barrett et al. 2005). Frequencies of classes of haploblock sizes were calculated per breed. In this study, haplotype diversity was considered as the number of haplotypes found within a haploblock. Plots of haplotype diversity for each genomic region were produced.
Areas in the analyzed genomic regions that presented haploblock-like structure in all breeds were selected for study of haplotype frequency and of haplotype sharing between breeds. In these areas, a unique haploblock was forced for all breeds (supplemental Table 3) and haplotypes were generated as described above. Median-joining networks (Bandelt et al. 1999) of these haplotypes were made using Network 4.2 (http://www.fluxus-engineering.com).
RESULTS
SNP identification and selection:
Accurate estimation of the extent of LD within a selection of breeds from Europe and China required availability of genomic regions with high densities of identified SNPs. Because such information was not available at the start of the current study, we decided to analyze three genomic regions in pigs for which high-quality sequences were available. The comparative vertebrate sequencing project of NISC (http://www.nisc.nih.gov) provided the necessary porcine genome sequences and NISC target 1, target 2, and target 4 were chosen for the present study. Because the available sequenced BACs were derived from crossbred animals originally derived from three different breeds, it is likely that overlapping BACs are derived from different haplotypes. Consequently, these overlapping sequences provide a rich resource for the identification of SNPs. Alignment of these sequences identified several hundred potential SNPs of which 371 were selected for genotyping. Of these SNPs 93% yielded genotyping results for >75% of individuals for all genomic regions. In total, 40 SNPs in target 1, 114 in target 2, and 183 in target 4 remained for further analysis.
Due to the absence of a genome sequence for the pig, positions of SNPs were based on alignment of BAC sequences with BES from clones located on the porcine BAC contig map (FPC map: http://www.sanger.ac.uk/Projects/S_scrofa/). This comparison indicates that target 1 and target 4 are located within q21 of SSC18 and target 2 within q17 of SSC3 (Figure 1). The SNPs' distribution across each genomic region for SNPs with genotypes in at least 75% of the animals is shown in Figure 1. Available BACs are unevenly distributed along the chromosome and, consequently, SNPs also are unevenly distributed across the different genomic regions.
Figure 1.—

SNPs distribution by genomic region. (A) Target 1, (B) target 2, and (C) target 4. SNP positions are indicated by vertical tick marks, and numbers indicate total number of SNPs in a region. BACs are named according to NISC code and are indicated by horizontal bars. Accession numbers and clone names of the BACs are in supplemental Table 1.
Predicted decay of LD per breed:
Markers with departures from HW equilibrium were found at low frequencies in each genomic region and discarded. The proportion of markers with MAF < 0.05 is higher for most European breeds especially in target 1 and for Mangalitsa in target 2. Predicted values of LD vs. linkage distance per genomic region and per breed are in Figure 2. Most tightly linked SNP pairs have the highest r2 and average r2 rapidly decreases as linkage distance increases. Overall, there is a clear difference in the decay of LD between Chinese and European pig breeds for each of three genomic regions; r2 decreases over short distances in Chinese breeds. This difference is most prominent in target 1 and target 4. (Figures 2A and 3).
Figure 2.—


Predicted LD and physical distance in the three genomic regions. (A) For each genomic region, the relationship between average predicted LD (LDij) and genomic distance (bp) is shown per biogeographical region. Vertical bars represent the standard error. The relationship between predicted LD (LDij) and distance (base pairs) is shown per breed and per genomic region: target 1 (B), target 2 (C), and target 4 (D). Chinese breeds are represented by dashed lines, European breeds are represented by solid lines, and wild boar is represented by a thick solid line. In target 1, observed r2 is 1 for breeds Tamworth, Duroc, Middle White British Saddleback, and Large Black. Therefore predicted values were not estimated for these breeds and B shows observed r2.
Figure 3.—

Comparison of LD between genomic regions: T1, target 1; T2, target 2; and T4, target 4. Pairwise LD (observed r2) plots are shown for Ningxiang (Ninx) and Large White (LW05) as an example of the LD in the two main centers of pig domestication (Europe and China) and for the European wild boar representing the ancestral European population: white areas, r2 = 0; shades of gray, 0 < r2 < 1; and black areas, r2 = 1.
In target 1, observed r2 = 1 for breeds Tamworth, Duroc, Middle White, British Saddleback, and Large Black. Therefore predicted values were not estimated for these breeds and Figure 2B shows observed r2.
For all genomic regions, LD decays more rapidly in Chinese breeds than in European breeds, indicating that in these breeds the extent of LD is smaller than in European breeds. LD in the European wild boar (FrWb) is in between European and Chinese breeds for target 2 and target 4 (Figure 2, C and D).
In European breeds, different patterns of decay of LD were observed across the three analyzed genomic regions between local, international, and commercial groups of breeds (Table 1, Figure 2). International and commercial breeds present a larger extent of LD than local European breeds, but there are some exceptions. For example, Landrace, an international breed, which shows a large extent of LD in target 1 and in target 4, presents a rapid LD decay in target 2. These results showed that besides differences in the pattern of decay of LD between breeds, differences between genomic regions also exist.
Differences were also observed among Chinese breeds. Breeds Ningxiang, Wuzishan, Tibetan, and Neijang had the lowest levels of LD than other Chinese breeds in all genomic regions, whereas breeds Mashen, Meishan, and Guanling had the highest levels of LD of Chinese breeds. Differences in LD do not appear to be correlated to geographic area of origin.
Test for breed and genomic region effects in the extent of LD:
Plotting predicted LD values vs. genetic distance reveals clear differences between breeds as well as between genomic regions. Results of fitting the linear model (Equation 3) indicated that differences among breed and genomic region were significant (Table 2). Also, interaction between these two main effects was significant (Table 2). Differences among levels of interactions (breed j vs. region k) were tested and a P-values matrix is shown in supplemental Table 3. The extent of LD was significantly different between Chinese and European breeds (P < 0.001). Within the Chinese group, the interaction effect between breed and genomic region was nonsignificant (P > 0.05) for most breeds. Most Chinese breeds and European wild boar do not have significantly different levels of extent of LD. A clear exception is breed MS02, which showed an extent of LD significantly different from all the other Chinese breeds for each genomic region. Ningxiang and Tibetan showed significantly different levels of extent of LD in target 1 (P < 0.001).
TABLE 2.
ANOVA of main effects and their interaction on LDcijk
| d.f. | SS | MS | F-value | P-value | |
|---|---|---|---|---|---|
| Breed | 19 | 29.26 | 1.54 | 70.57 | <0.001 |
| Target | 2 | 2.32 | 1.16 | 79.98 | <0.001 |
| Breed × target | 33 | 23.86 | 0.72 | 33.429 | <0.001 |
| Residuals | 134,373 | 2,795.16 | 0.02 |
LDcijk is the distance-corrected and variance-stabilized LD for marker pair i in genomic region k and breed j.
Contrary to Chinese breeds, European breeds showed high levels of significant differences in the extent of LD within the group (P < 0.01) across all genomic regions. For example, Mangalitsa, a local breed, presented a significantly different level of extent of LD when compared with other European breeds and wild boar (P < 0.001) and with Chinese breeds (P < 0.05) in target 2. In target 4, Mangalitsa was shown to be different from all breeds and European wild boar at a high significance level (P < 0.001).
Haploblock partitioning:
Haploblocks were defined using r2 (Gu et al. 2005). The pattern of haploblock partitioning showed similarities across genomic regions for Chinese breeds (Figure 3 and supplemental Figure 1). A large number of haploblocks with size up to 10 kb were generally present (supplemental Figure 2). Among European breeds and wild boar, the overall pattern of haploblock partitioning consisted of SNPs grouped in lower numbers of haploblocks, ranging from 50 to >200 kb in target 1 and target 4 (Figure 3 and supplemental Figure 1). In target 2, larger haploblocks were also present but the proportion of haploblocks up to 10 kb was higher (supplemental Figure 2).
The average number of haplotypes found in haploblocks with an equal number of SNPs for Chinese and European breeds and wild boar by genomic region is shown in Figure 4. For haploblocks with an equal number of SNPs, Chinese breeds had a higher number of haplotypes than wild boar and European breeds.
Figure 4.—

Comparison of haplotype diversity per genomic region. The number of haplotypes per haploblock is plotted against the number of SNPs in each haploblock for each genomic region (target 1, target 2, and target 4) for Europe and China and for the European wild boar.
For each genomic region, areas characterized by the existence of larger haploblocks in one or several breeds and smaller haploblocks in the remaining breeds were selected to investigate haplotype sharing between European and Chinese breeds and European wild boar. SNPs included in this analysis are presented in supplemental Table 4. Supplemental Figure 3 shows the overall view of haploblock partitioning per genomic region.
The selected areas were in target 1, block 1 (4 kb) and block 2 (149 kb); in target 2, block 1 (8 kb) and block 2 (2 kb); and in target 4, block 1 (293 kb). In general, smaller haploblocks were found in Chinese breeds, resulting in a larger number of haplotypes. The haplotype diversity and respective frequencies are represented by a median-joining network for the 8-kb region of target 2 (Figure 5); for the remaining genomic regions these data are available in supplemental Figure 4.
Figure 5.—

Median-joining network of the SNP-based haplotypes from target 2, block 1. In each circle (identified as a solid area), haplotype frequencies in European and Chinese breeds and in European wild boar are shown. The circle area is proportional to frequency and branch length is proportional to the number of mutations. Each line is annotated with its corresponding mutational change (corresponding to the SNP ID in supplemental Table 2).
Wild boar shared most haplotypes with European breeds, although one haplotype is also shared with Chinese breeds. Some haplotypes that occur at high frequency in Chinese breeds are shared by Large White Duroc, Landrace, Pietrain, and Mangalitsa. Meishan shares haplotypes that occur at high frequency in European breeds.
DISCUSSION
Differences of LD between European and Chinese breeds:
In this study, comparison between pig breeds from two major areas of domestication (China and Europe) across three genomic regions revealed large and significant differences in the extent and pattern of LD. The observed pattern of decay of LD is similar to what has been observed in previous studies in humans (Daly et al. 2001), cattle (Farnir et al. 2000), chickens (Aerts et al.2007), and pigs (Du et al. 2007).
In Chinese breeds, LD is mostly organized in haploblocks of up to 10 kb, while in European breeds, LD haploblocks may be up to 400 kb in size. Haplotype diversity within the haploblocks was higher in Chinese breeds. Chinese breeds also showed a lower percentage of SNPs with MAF < 0.05. Higher haplotype diversity and a lower proportion of fixed markers is an indication of higher genetic diversity in Chinese breeds. This has also been reported by Fang et al. (2005) and Megens et al. (2008) using microsatellites and by Fang and Andersson (2006) studying mitochondrial DNA.
European wild boar showed levels of LD and haplotype diversity in between the values found in the European and Chinese breeds, partitioned in a low number of haploblocks of up to 200 kb. Further analysis showed that European breeds share the most frequent haplotypes with the studied population of European wild boar.
Two hypotheses for the existence of significant differences of LD between European and Chinese pig breeds can be considered: (a) differences are due to differences in ancestral stock; and (b) modern breeding practices in Europe resulted in small effective population size.
The first hypothesis is supported by the fact that the European wild boar had a higher level of LD and lower genetic diversity compared to Chinese breeds. European and Chinese breeds were domesticated from different ancestors that might have had different levels of genetic diversity and LD. This hypothesis is also supported by the results obtained by Larson et al. (2005), which showed that European wild boars have lower genetic diversity in mitochondrial DNA compared to Asian wild boars. In addition, these authors suggest that domestication of pig breeds in Asia involved several lineages of wild boar, suggesting that the original gene pool for domestication was more diverse when compared to that in Europe. However, our findings regarding the level of LD in European wild boar cannot be generalized for the species since the European wild boar is distributed widely (from Western Europe and the Mediterranean basin to Russia) and this study analyzed only one wild boar population from France. European wild boar populations suffered a serious decrease, resulting in the extinction in the British Isles and in parts of Northern Europe. Only recently the population started to increase, and areas such as Sweden, Finland, and Estonia were recolonized (Erkinaro et al. 1982; Leaper et al. 1999), suggesting that a recent bottleneck could have resulted in an increased level of LD. To validate this hypothesis a wider geographic sampling of both European and Asian wild boar is necessary, which was beyond the scope of this study.
Animal domestication is the process by which captive animals adapt to humans and to the captive environment. This involves directed changes in the gene pool occurring over generations, during which domesticated animals will differ from their wild counterparts (Price 2002), diminishing effective population sizes, increasing inbreeding, and consequently increasing LD (Lindblad-Toh et al. 2005). This takes us to our second hypothesis, which relates the increase of LD to an effect of small population sizes and modern breeding practices. Domestication of pigs in Europe occurred independently from the pig domestication in Asia (Giuffra et al. 2000; Larson et al. 2005) but may have started due to the introduction of Near Eastern pigs during the Neolithic (Larson et al. 2007). Nowadays, Europe has 37% of world pig breeds (Scherf 2000). In this study, several local breeds and international and commercial lines were analyzed. Tamworth is listed as endangered breed (breeds with <1000 breeding females and/or ≤20 breeding males). British Saddleback, Large Black, Mangalitsa, and Middle White are listed as endangered maintained breeds (breeds with <1000 breeding females and/or ≤20 breeding males but that are maintained by an active conservation program) (Scherf 2000). Therefore these local breeds are characterized by having small effective population sizes, which severely affects their diversity, as shown by the high levels of LD and the high proportion of SNPs with fixed alleles. This study analyzed international breeds Landrace and Pietrain and commercial lines Duroc, Hampshire, and Large White, and these were the ones that showed the highest levels of LD. These are the breeds used by the pig industry to produce pork meat and related products. Modern breeding practices started in the middle of the previous century and the introduction of BLUP selection after 1990 allowed a rapid increase of genetic gain (Merk 2000). At the same time it is likely that this resulted in decreased effective population sizes by limiting the genetic inflow into commercial breeding lines (Jones 1998).
A founder effect on LD of populations seems to be evident in the case of the Meishan line, which was brought to Europe 25 years ago and started from a small number of individuals and has since been kept in a small population. The level of LD in this line is higher compared to that in the other Chinese breeds, although still lower than that in most European breeds.
Analysis of nuclear haplotypes using SNPs allows the detection of introgression and phenotype selection (Zhang et al. 2002). In this study, haplotype sharing between these European breeds (Large White, Landrace, and Duroc) and Chinese breeds was found in all genomic regions. These results add support to the hybrid origin of these European breeds, reported by historical documentation (Porter 1993) and by studies of mitochondrial haplotypes (Giuffra et al. 2000; Fang and Andersson 2006). One of the shared haplotypes was also shared at a low frequency by European wild boar. This may be due to recent introgression of pig genes into European wild boar (Giuffra et al. 2000); however, more samples of European wild boar should be analyzed.
Differences of LD between regions:
The extent of LD and haploblock pattern varies significantly between genomic regions. Differences of extent of LD between genes located in different chromosomes were observed by Reich et al. (2001) in human populations. These authors found levels of LD extending up to 160 kb in some genomic regions while in others the LD extended only up to 40 kb.
Nsengimana et al. (2004) assessed LD in five populations of commercial pigs (Large White, Landrace, Duroc/Large White, and Yorkshire/Large White) in two chromosomal regions, one on SCC4 (33 cM) and another on SCC7 (48 cM), using 15 microsatellites with an interval spacing of 5 cM. The region on SSC7 presented a significantly larger extent of LD compared to SSC4. Since SSC7 harbors QTL associated with growth rate and fat deposition, Nsengimana et al. (2004) suggested that these differences in the extent of LD were due to selection.
A likely cause of the observed differences in the extent of LD between genomic regions located in chromosomes SSC18 and SSC3 is selection. In region q21 of SSC18, previous studies identified a number of genes that are obvious candidates to be under selection [e.g., INSIG1 (Qiu et al. 2005), LEP (Campbell et al. 2001), and GHRHR (Sun et al. 1997)]. Further, several QTL have been mapped to this region as well (pH, cook loss, and feed-conversion ratio) (Hu et al. 2005). In contrast, region q17 on SSC3 contains mainly genes involved in general cellular processes such as DNA transcription, transduction, and cell differentiation, which are not likely candidate genes to be under selection, and no major QTL have been described for this region (Hu et al. 2005). This hypothesis is also supported by the similarity in the extent of LD across genomic regions in the European wild boar population. The effect of selection on the extent of LD in other domestic animals has been reported in other studies [cattle (Farnir et al. 2000) and sheep (McRae et al. 2002)].
Assessing the extent of useful LD:
The threshold for useful LD that was chosen in this study was the same as previously used in LD studies of pig populations using r2 as a measure of LD (Jungerius et al. 2005; Du et al. 2007). With a threshold of 0.3, and considering that on average 1 cM is equivalent to 1 Mb, LD extended in the European breeds over 0.5–2 cM on SSC18 and 0.1–1 cM on SSC3. In the case of Chinese breeds LD ranged between 0.005 and 0.05 cM in the studied genomic regions.
This study shows that LD for European pig breeds is higher compared to that in human populations (Reich et al. 2001; Ardlie et al. 2002). Higher values of the extent of LD in domestic animals have been reported in previous studies [e.g., cattle (Farnir et al. 2000) and sheep (McRae et al. 2002)]. Previous reports on the extent of LD in European pigs (Nsengimana et al. 2004; Du et al. 2007) also showed large levels of LD exceeding the values obtained in our study. As described above, Nsengimana et al. (2004) studied the extent of LD using microsatellites, and LD was measured using D′. These authors concluded using the threshold for useful LD of 0.5 that LD ranges from 3 to 10 cM. However, these conclusions were based on a low number of distantly spaced markers. Du et al. (2007) also studied LD using commercial lines (Pietrain, Duroc, Landrace, and Large White). LD was assessed using SNPs across 18 chromosomes with an average of 330 markers per chromosome, with a maximum length of 100 cM and a marker spacing on average of 0.44 cM. LD was measured using r2 and 0.3 as a threshold for useful LD. These authors suggest that LD extends to 1–3 cM in these pig populations. This is only somewhat higher than values obtained in the current study, which could be due to the greater marker spacing used by Du et al. (2007).
By contrast, the present study aimed at assessing the extent of LD with high definition, and the trade-off was to have high SNP frequencies (0.02 cM) preferably covering regions of a few centimorgans in size. The size of target 1, however, was only 1 cM and in the case of many European breeds this length was too small to observe the decay of the LD. In targets 2 and 4 the decay of LD can be observed in detail with maxima ranging from 1 cM in target 2 to 2 cM in target 4. This experimental design allowed us to study LD in a large set of breeds, from Europe and China. The most important difference between our study and that of Nsengimana et al. (2004) and Du et al. (2007) is the high marker density used and wider sampling of pig populations. In Chinese breeds, the results of decay of LD would have been inconclusive if the same smaller marker spacing had been used as by Du et al. (2007). Our results indicate that we would have found a very steep decay in LD and would not have been able to precisely identify the point at which LD drops below 0.3. For Chinese breeds, this is the first study that aimed to assess LD. The level of extent of LD is very small, 0.005–0.05 cM, which is similar to the extent of LD observed in human populations (Reich et al. 2001; Ardlie et al. 2002; International Hapmap Consortium 2005). Populations with a shorter extent of LD are more suitable for fine mapping of genes responsible for phenotypic traits (Ardlie et al. 2002). Therefore, Chinese pig breeds may be useful to fine map QTL; however, the QTL alleles that have an effect on the phenotypic trait have to be segregating in these breeds.
For future populationwide studies with a whole-genome approach, our results indicate that, assuming a threshold of 0.3 for r2, the SNP spacing for European pig breeds should be ∼0.1 cM. This implies the use of 30,000 SNPs per individual, using the same sample sizes as in this study and assuming that all SNPs are informative (with a MAF > 0.05). For Chinese breeds in a study with a similar sample size a SNP spacing of 0.005 cM and the use of ∼500,000 SNPs per individual would be required.
Acknowledgments
European pig DNA samples, other than provided by the authors, were provided by Agence de la Sélection Porcine, France; Georg-August University, Göttingen, Germany; Rare Breeds Survival Trust, United Kingdom; Roslin Institute, United Kingdom; and PIC International Group, United Kingdom. European wild boar samples were provided by Alain Ducos (Unité Mixte de Recherche, Institut National de la Recherche Agronomique–ENVT 898, Cytogénétique des Populations Animales), and Chinese breeds were provided by Ning Li (China Agricultural University, National Laboratories for Agrobiotechnology). This work was funded by European Union grant QLRT-2001-01059. This work was conducted as part of the SABRETRAIN Project, funded by the Marie Curie Host Fellowships for Early Stage Research Training, as part of the 6th Framework Programme of the European Commission.
References
- Aerts, J., H. J. Megens, T. Veenendaal, I. Ovcharenko, R. Crooijmans et al., 2007. Extent of linkage disequilibrium in chicken. Cytogenet. Genome Res. 117 338–345. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Ardlie, K. G., L. Kruglyak and M. Seielstad, 2002. Patterns of linkage disequilbrium in the human genome. Nat. Rev. Genet. 3 299–309 (erratum: Nat. Rev. Genet. 3: 566. [DOI] [PubMed] [Google Scholar]
- Baessler, A., M. Fischer, B. Mayer, M. Koehler, S. Wiedmann et al., 2007. Epistatic interaction between haplotypes of the ghrelin ligand and receptor genes influence susceptibility to myocardial infarction and coronary artery disease. Hum. Mol. Genet. 16 887–899. [DOI] [PubMed] [Google Scholar]
- Bandelt, H. J., P. Forster and A. Rohl, 1999. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16 37–48. [DOI] [PubMed] [Google Scholar]
- Barrett, J. C., B. Fry, J. Maller and M. J. Daly, 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263–265. [DOI] [PubMed] [Google Scholar]
- Blott, S., L. Andersson, M. Groenen, M. Sancristobal, C. Chevalet et al., 2003. Characterisation of genetic variation in the pig breeds of China and Europe—the pigbiodiv2 project. Arch. Zootec. 52 207. [Google Scholar]
- Bruford, M. W. M. W., D. G. D. G. Bradley and G. G. Luikart, 2003. DNA markers reveal the complexity of livestock domestication. Nat. Rev. Genet. 4 900–910. [DOI] [PubMed] [Google Scholar]
- Campbell, E. M. G., S. C. Fahrenkrug, J. L. Vallet, T. P. L. Smith and G. A. Rohrer, 2001. An updated linkage and comparative map of porcine chromosome 18. Anim. Genet. 32 375–379. [DOI] [PubMed] [Google Scholar]
- Chen, K. K., T. T. Baxter, W. M. W. M. Muir, M. A. M. A. Groenen and L. B. L. B. Schook, 2007. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int. J. Biol. Sci. 3 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly, M. J., J. D. Rioux, S. E. Schaffner, T. J. Hudson and E. S. Lander, 2001. High-resolution haplotype structure in the human genome. Nat. Genet. 29 229–232. [DOI] [PubMed] [Google Scholar]
- Dean, F. B., S. Hosono, L. H. Fang, X. H. Wu, A. F. Faruqi et al., 2002. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 99 5261–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin, B., and N. Risch, 1995. A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29 311–322. [DOI] [PubMed] [Google Scholar]
- Du, F. X., A. C. Clutter and M. M. Lohuis, 2007. Characterizing linkage disequilibrium in pig populations. Int. J. Biol. Sci. 3 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkinaro, E., K. Heikura, E. Pullianen and S. Sulkava, 1982. Occurrence and spread of the wild boar (Sus scrofa) in eastern Fennoscandia. Memo. Flora Fauna Fennoscand. 58 39–47. [Google Scholar]
- Fahrenkrug, S. C., G. A. Rohrer, B. A. Freking, T. P. L. Smith, K. Osoegawa et al., 2001. A porcine BAC library with tenfold genome coverage: a resource for physical and genetic map integration. Mamm. Genome 12 472–474. [DOI] [PubMed] [Google Scholar]
- Fan, J. B., A. Oliphant, R. Shen, B. G. Kermani, F. Garcia et al., 2003. Highly parallel SNP genotyping. Cold Spring Harbor Symp. Quant. Biol. 68 69–78. [DOI] [PubMed] [Google Scholar]
- Fang, M., X. Hu, T. Jiang, M. Braunschweig, L. Hu et al., 2005. The phylogeny of Chinese indigenous pig breeds inferred from microsatellite markers. Anim. Genet. 36 7–13. [DOI] [PubMed] [Google Scholar]
- Fang, M. Y., and L. Andersson, 2006. Mitochondrial diversity in European and Chinese pigs is consistent with population expansions that occurred prior to domestication. Proc. R. Soc. B Biol. Sci. 273 1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnir, F., W. Coppieters, J. J. Arranz, P. Berzi, N. Cambisano et al., 2000. Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 10 220–227. [DOI] [PubMed] [Google Scholar]
- Giuffra, E., J. M. H. Kijas, V. Amarger, O. Carlborg, J. T. Jeon et al., 2000. The origin of the domestic pig: independent domestication and subsequent introgression. Genetics 154 1785–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, S., A. J. Pakstis and K. K. Kidd, 2005. HAPLOT: a graphical comparison of haplotype blocks, tagSNP sets and SNP variation for multiple populations. Bioinformatics 21 3938–3939. [DOI] [PubMed] [Google Scholar]
- Heifetz, E. M., J. E. Fulton, N. O'Sullivan, H. Zhao, J. C. M. Dekkers et al., 2005. Extent and consistency across generations of linkage disequilibrium in commercial layer chicken breeding populations. Genetics 171 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Z. L. Z.-L., S. S. Dracheva, W. W. Jang, D. D. Maglott, J. J. Bastiaansen et al., 2005. A QTL resource and comparison tool for pigs: PigQTLDB. Mamm. Genome 16 792–800. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium, 2003. The International HapMap Project. Nature 426 789–796. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium, 2005. A haplotype map of the human genome. Nature 437 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G. F., 1998. Aspects of domestication, common breeds and their origin, pp. 17–50 in The Genetics of the Pig, edited by M. F. Rothschild and A. Ruvinsky. CAB International, New York.
- Jungerius, B. J., J. J. Gu, R. Crooijmans, J. J. Van Der Poel, M. A. M. Groenen et al., 2005. Estimation of the extent of linkage disequilibrium in seven regions of the porcine genome. Anim. Biotechnol. 16 41–54. [DOI] [PubMed] [Google Scholar]
- Kruglyak, L., 1999. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 22 139–144. [DOI] [PubMed] [Google Scholar]
- Larson, G., K. Dobney, U. Albarella, M. Y. Fang, E. Matisoo-Smith et al., 2005. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307 1618–1621. [DOI] [PubMed] [Google Scholar]
- Larson, G., U. Albarella, K. Dobney, P. Rowley-Conwy, J. Schibler et al., 2007. From the cover: ancient DNA, pig domestication, and the spread of the Neolithic into Europe. Proc. Natl. Acad. Sci. USA 104 15276–15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaper, R., G. Massei, M. L. Gorman and R. Aspinall, 1999. The feasibility of reintroducing wild boar (Sus scofa) to Scotland. Mammal Rev. 29 239–259. [Google Scholar]
- Lindblad-Toh, K., C. M. Wade, T. S. Mikkelsen, E. K. Karlsson, D. B. Jaffe et al., 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438 803–819. [DOI] [PubMed] [Google Scholar]
- McRae, A. F., J. C. McEwan, K. G. Dodds, T. Wilson, A. M. Crawford et al., 2002. Linkage disequilibrium in domestic sheep. Genetics 160 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megens, H.-J., R. P. M. A. Crooijmans, M. Sancristobal, X. Hui, N. Li et al., 2008. Biodiversity of pig breeds from China and Europe estimated from pooled DNA samples: differences in microsatellite variation between two areas of domestication. Genet. Sel. Evol. 40 103–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merks, J. W. M., 2000. One century of genetic changes in pigs and future needs, pp. 8–19 in The Challenge of Genetic Change in Animal Production, edited by W. G. Hill, S. C. Bishop, B. McQuirk, J. C. McKay, G. Simm and A. J. Webb. British Society of Animal Science Occasional Publication, Edinburgh, UK.
- Nordborg, M., and S. Tavare, 2002. Linkage disequilibrium: what history has to tell us. Trends Genet. 18 83–90. [DOI] [PubMed] [Google Scholar]
- Nsengimana, J., P. Baret, C. S. Haley and P. M. Visscher, 2004. Linkage disequilibrium in the domesticated pig. Genetics 166 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollivier, L., L. Alderson, G. C. Gandini, J. L. Foulley, C. S. Haley et al., 2005. An assessment of European pig diversity using molecular markers: partitioning of diversity among breeds. Conserv. Genet. 6 729–741. [Google Scholar]
- Porter, V., 1993. Pigs, A Handbook to the Breeds of the World. Helm Information, London.
- Price, E. O., 2002. Animal Domestication and Behavior. CABI International, New York.
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Z. H. S., T. H. Niu and J. S. Liu, 2002. Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am. J. Hum. Genet. 71 1242–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, H., T. Xia, X. D. Chen, L. Gan, S. Q. Feng et al., 2005. Characterization of pig INSIG1 and assignment to SSC18. Anim. Genet. 36 284–286. [DOI] [PubMed] [Google Scholar]
- Reich, D. E., M. Cargill, S. Bolk, J. Ireland, P. C. Sabeti et al., 2001. Linkage disequilibrium in the human genome. Nature 411 199–204. [DOI] [PubMed] [Google Scholar]
- Rigby, R. J., M. M. A. Fernando and T. J. Vyse, 2006. Mice, humans and haplotypes—the hunt for disease genes in SLE. Rheumatology 45 1062–1067. [DOI] [PubMed] [Google Scholar]
- Sancristobal, M., C. Chevalet, C. S. Haley, R. Joosten, A. P. Rattink et al., 2006. Genetic diversity within and between European pig breeds using microsatellite markers. Anim. Genet. 37 189–198. [DOI] [PubMed] [Google Scholar]
- Scherf, B. D., 2000. World Watch List for Domestic Animal Diversity, Ed. 2. Food and Agriculture Organization, Rome.
- Sun, H. S., C. Taylor, L. Wang, M. F. Rothschild, C. K. Tuggle et al., 1997. Mapping of growth hormone releasing hormone receptor to swine chromosome 18. Anim. Genet. 28 351–353. [DOI] [PubMed] [Google Scholar]
- Sved, J. A., 1971. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 2 125–141. [DOI] [PubMed] [Google Scholar]
- Tenesa, A., P. Navarro, B. J. Hayes, D. L. Duffy, G. M. Clarke et al., 2007. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium, 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, W. T., I. S. Young, D. P. Nicholls, C. Patterson, K. Lyttle et al., 2006. SNPs at the APOA5 gene account for the strong association with hypertriglyceridaemia at the APOA5/A4/C3/A1 locus on chromosome 11q23 in the Northern Irish population. Atherosclerosis 185 353–360. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., 1986. Pig Breeds in China. Shanghai Scientific and Technical Publishers, Shanghai, China.
- Zhang, K., M. H. Deng, T. Chen, M. S. Waterman and F. Z. Sun, 2002. A dynamic programming algorithm for haplotype block partitioning. Proc. Natl. Acad. Sci. USA 99 7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]