

Abstract
The endoglycosidase heparanase is the predominant enzyme that degrades heparan sulfate side chains of heparan sulfate proteoglycans, activity that is strongly implicated in tumor metastasis. Apart of its well characterized enzymatic activity, heparanase was noted to exert also enzymatic-independent functions. Among these is the induction of Akt/PKB phosphorylation noted in endothelial and tumor derived cells. Protein domains of heparanase required for signaling were not identified to date, nor were heparanase binding proteins/receptors capable of transmitting heparanase signals. Here, we examined the possible function of mannose 6-phosphate receptor (MPR) and low density lipoprotein-receptor related protein (LRP), recently implicated in cellular uptake of heparanase, as heparanase receptors mediating Akt phosphorylation. We found that heparanase addition to MPR-, and LRP-deficient fibroblasts elicited Akt activation indistinguishable from control fibroblasts. In contrast, disruption of lipid rafts abrogated Akt/PKB phosphorylation following heparanase addition. These results suggest that lipid raft-resident receptor mediates heparanase signaling.
Keywords: Heparanase, Akt, phosphorylation, lipid rafts, MPR, LRP
Introduction
Heparanase is an endo-β-D-glucuronidase capable of cleaving heparan sulfate (HS) side chains at a limited number of sites [1; 2]. Traditionally, heparanase activity correlated with the metastatic potential of tumor-derived cells, attributed to enhanced cell dissemination as a consequence of HS cleavage and remodeling of the extracellular matrix (ECM) barrier [2; 3]. Similarly, heparanase activity was implicated in neovascularization, inflammation and autoimmunity, facilitating migration of vascular endothelial cells and activated cells of the immune system [1; 2; 3]. More recently, heparanase up regulation was noted in an increasing number of primary human cancers, correlating with reduced postoperative survival of cancer patients [4; 5]. In addition to the well studied catalytic feature of the enzyme, heparanase was noted to exert biological functions apparently independent of its enzymatic activity. Non enzymatic functions of heparanase include enhanced adhesion of glioma [6], lymphoma [7] and T cells [8], mediated by β1-integrin and correlated with Akt, Pyk2 and ERK activation [6; 8]. Moreover, we have demonstrated that exogenous addition of latent heparanase or its over expression in tumor-derived cells stimulate Akt phosphorylation and PI 3-kinase-dependent endothelial cell invasion and migration [9], likely supporting endothelial and tumor cell survival [10]. Protein domains of heparanase required for signaling were not identified to date, nor were identify heparanase binding proteins/receptors capable of transmitting heparanase signals. Here, we examined the possible function of mannose 6- phosphate receptor (MPR) and low-density lipoprotein-receptor related protein (LRP), recently implicated in cellular uptake of heparanase [11], as heparanase receptors mediating Akt phosphorylation. We found that heparanase addition of to MPR-, and LRP-deficient fibroblasts elicited Akt activation indistinguishable from control fibroblasts. In contrast, disruption of lipid rafts almost abrogated Akt phosphorylation following heparanase addition.
Materials and Methods
Antibodies and reagents
Antibody 1453 was raised in rabbits against the entire 65 kDa latent heparanase protein isolated from the conditioned medium of heparanase-transfected 293 cells [9; 12; 13]. Anti-Akt antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and Anti-phospho-Akt antibody was purchased from Cell Signaling Technology (Beverly, MA). Lovastatin, methyl-B-cyclodextrin (MβCD), Nycodenz, and heparin were purchased from Sigma (St. Louis, MO). Cholera toxin-β subunit-HRP conjugate was purchased from Calbiochem (San Diego, CA).
Cells and cell culture
Chinese hamster ovary (CHO) K1 cells and human cervical adenocarcinoma HeLa cells were purchased from the American Type Culture Collection (ATCC). Mouse embryonic fibroblasts (MEF) and mouse fibroblasts deficient of cell surface mannose-6-phosphate receptors (MPR300−/−) were kindly provided by Dr. Kurt von Figura (University of Göttingen, D-37075 Göttingen, Germany) [14]. Mouse fibroblasts deficient of low-density lipoprotein receptor-related protein (LRP) (PEA-13) were kindly provided by Dr. Joachim Herz (University of Texas Southwestern, Dallas, Texas 75390, USA) [14; 15]. CHO cells deficient of GPI-anchored proteins and thus lacking cell surface glypicans, were kindly provided by Dr. F. Gisou van der Goot (Department of Biochemistry, University of Geneva, Switzerland) [16]. Cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FCS and antibiotics. Mutant CHO cells (pgs A-745) deficient of xylosyltransferase and unable to initiate glycosaminoglycan synthesis, were kindly provided by Dr. J. Esko (University of California, San Diego) and grown in RPMI 1640 medium supplemented with 10% FCS and antibiotics. Transfection and evaluation of B16/BL6 mouse melanoma cells with anti-heparanase siRNA and control siRNA vectors was carried out essentially as described [17].
Cell lysates and protein blotting
Cell cultures were incubated for 20 h under serum-free conditions and were left untreated as control or incubated with recombinant heparanase (1µg/ml), produced and purified as described [6], for 30 min, and cell lysates were subjected to SDS polyacrylamid gel electrophoresis (SDS-PAGE) and immunoblotting, as described [6; 9].
Flotation analysis
Detergent-insoluble complexes were analyzed on flotation gradients, as described [18]. To detect the rafts-resident ganglioside GM1 on dot blots, 5 µl of each flotation fraction were blotted onto nitrocellulose paper. The paper was air dried, blocked with 5% BSA in PBS, and incubated with cholera toxin β-subunit (CTβB)-HRP (12.5 ng/ml, 2 h). Dot blots were developed with ECL.
Results
Heparanase induces Akt activation independent of MPR and LRP
Apart of its well characterized enzymatic activity, heparanase was noted to exert also enzymatic-independent functions. Among these is the induction of Akt/PKB phosphorylation noted in endothelial and tumor derived cells, facilitating cell motility [6; 9]. In order to further reveal the involvement of endogenous cellular levels of heparanase in Akt/PKB regulation, B16/BL6 mouse melanoma cells were transfected with anti-heparanase siRNA, or control siRNA vectors and heparanase expression was examined (Fig. 1A). Heparanase expression (Fig. 1A, upper panel) and activity (not shown) were significantly reduced in cells treated with anti-heparanase siRNA sequence, compared with control siRNA vector, in agreement with our previous findings utilizing this cell system [17]. Interestingly, Akt/PKB phosphorylation levels were markedly decreased in cells transfected with anti-heparanase siRNA vector (Fig. 1A, third panel), to an extent comparable with heparanase gene silencing (Fig. 1A, upper panel). This finding indicates that endogenous heparanase modulates Akt/PKB activation. Akt/PKB activation by heparanase appeared to be independent of heparan sulfate proteoglycan (HSPG) [9], suggesting its interaction with other cell surface molecules. Since MPR and LRP have recently been implicated in heparanase uptake [11], we suspected that these molecules may also be involved in heparanase signaling. In order to examine this possibility, heparanase (1 µg/ml) was added to control fibroblasts (MEF), and to fibroblasts deficient of MPR (MRP300−/−) or LRP (PEA13), and Akt/PKB phosphorylation was then evaluated by immunoblotting. Addition of heparanase to control fibroblasts induced Akt/PKB phosphorylation (Fig. 1B, MEF), in agreement with our previous findings [9]. Notably, addition of heparanase to MPR-, and LRP-deficient cells resulted in Akt/PKB phosphorylation to a similar extent (Fig. 1B, 300−/−, PEA13). To exclude the possibility of heparanase signaling mediated by other species of LRP, Akt/PKB phosphorylation was examined in the absence (Control) or presence of recombinant receptor-associated protein (RAP), an antagonist of this receptor family [19]. As demonstrated in figure 1C (upper panel), heparanase uptake by MEF was significantly lower in the presence of RAP, as judged by reduced amounts of the 65 kDa latent enzyme associated with the cell and the lack of heparanase processing, supporting the notion that LRP and related receptors mediate heparanase binding and uptake. Akt/PKB phosphorylation, however, was not affected by RAP (Fig.1C, middle panel). Similarly, treatment of PEA13 cells with RAP had no effect on Akt/PKB activation by heparanase (Fig. 1D, upper panel), excluding the possible involvement of LRP-family members and suggesting the involvement of other heparanase binding proteins that mediate Akt/PKB activation.
Figure 1.
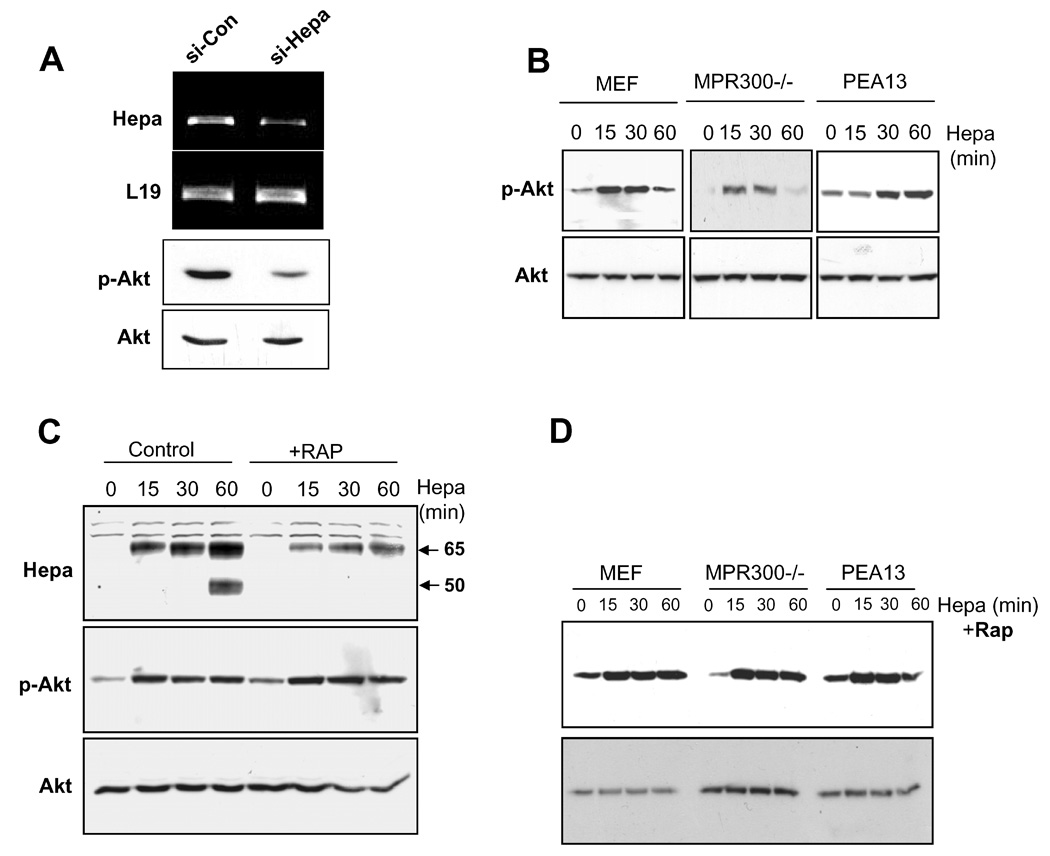
A. Enhanced Akt phosphorylation following heparanase addition is not mediated by MPR or LRP. A. Heparanase down regulation. B16/BL6 mouse melanoma cells were transfected with anti-heparanase (si-Hepa) or control (si-Con) si-RNA vectors, and heparanase expression was examined by RT-PCR analysis (upper panel). The expression of L19 (second panel) was used as an internal control. Corresponding cell lysates were subjected to immunoblotting applying anti-phospho-Akt (p-Akt, third panel) and total Akt (lower panel) antibodies. B. Control mouse embryonic fibroblasts (MEF), and fibroblasts deficient for MPR (MPR300−/−) or LRP (PEA13) were left untreated (0) or incubated (37°C) with heparanase (1 µg/ml) for the time indicated (min). Cell lysates were then prepared and subjected to immunoblotting with anti phospho-Akt (p-Akt, upper panels) and anti-Akt (Akt, lower panels) antibodies. C. RAP treatment. MEF or were left untreated (Control) or incubated with RAP (10 µg/ml, +RAP) for 15 min. Heparanase (1 µg/ml) was then added for the time indicated and cell lysates were subjected to immunoblotting with anti-heparanase (Hepa, upper panel), anti-phospho Akt (middle panel) and total Akt (lower panel) antibodies. D. MEF, MPR300−/−, and PEA13 cells were treated with RAP as above and heparanase (1 µg/ml) was then added for the time indicated (min). Total cell lysates were subjected to immunoblotting with anti-phospho Akt (upper panel) and anti-Akt (lower panel) antibodies.
Akt/PKB activation by heparanase is mediated by raft component(s)
Lipid rafts are membrane microdomains that are predominantly enriched in sphingolipids and cholesterol, serving as a platform for several signaling pathways, including Akt/PKB [20; 21]. To examine whether Akt/PKB activation by heparanase involves lipid rafts, we utilized cholesterol depletion with methyl-β-cyclodextrin (MβCD) or inhibition of cholesterol biosynthesis by lovastatin treatment, to impair rafts integrity (Fig. 2A). Raft disruption by lovastatin significantly reduced Akt/PKB phosphorylation by heparanase, and MβCD treatment abrogated phospho Akt/PKB induction almost completely (Fig. 2A, upper panel), suggesting that Akt/PKB activation by heparanase is mediated by lipid rafts resident component(s).
Figure 2.
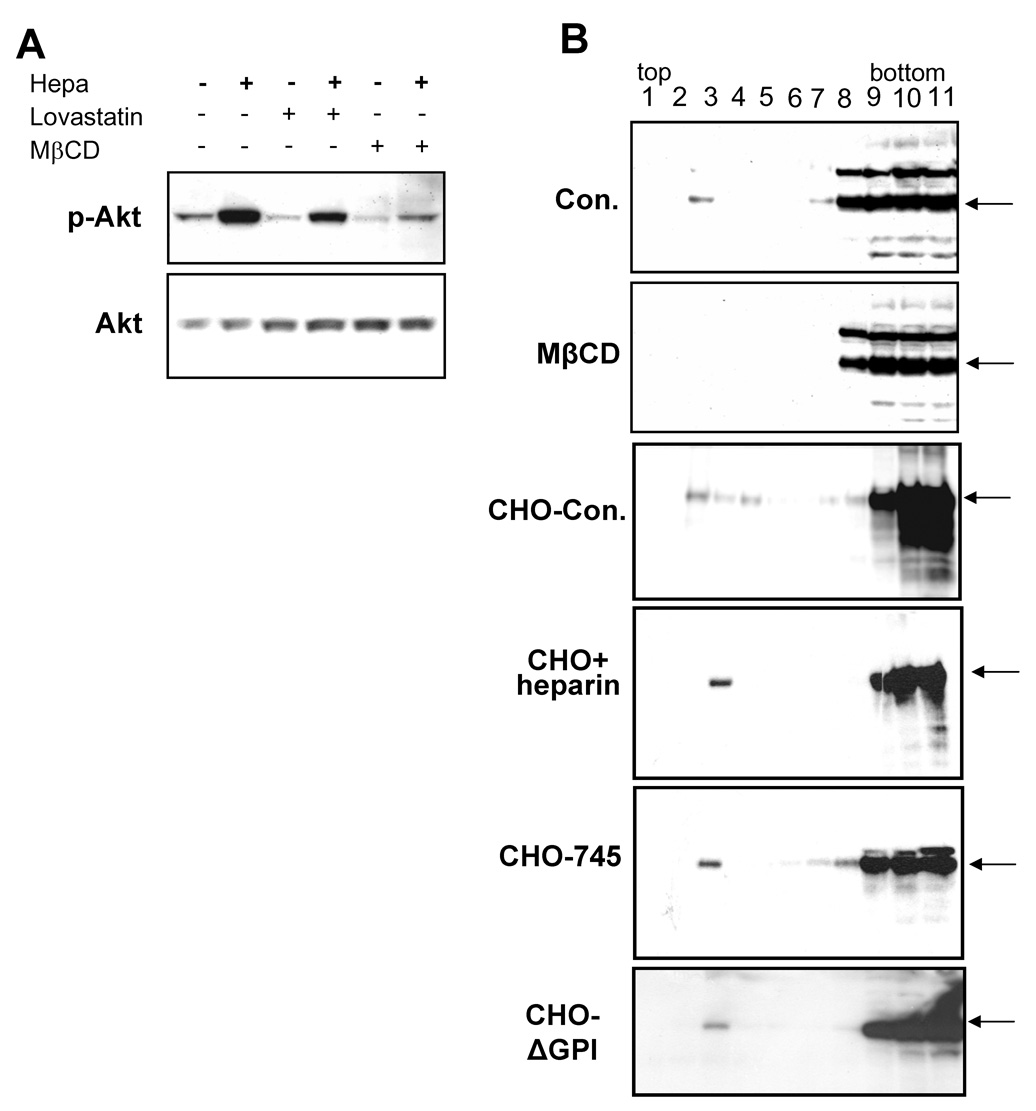
Akt activation following heparanase addition is mediated by lipid rafts component(s). A. Rafts disruption. HeLa cells were left untreated or incubated with Lovastatin (2 µM) for 18 h, or MβCD (2.5 mM) for 30 min. Heparanase (1 µg/ml) was then added for 30 min, and cell lysates were subjected for immunoblotting with anti-phospho Akt (upper panel) and anti-Akt (lower panel) antibodies. B. Heparanase binding. HeLa cells were left untreated as control (Con) or preincubated with MβCD (2.5 mM) for 30 min. Heparanase (1 µg/ml) was then added and cells were incubated at 4°C for 2h. Cells were then washed and lysate samples were loaded on Nycodenz flotation gradient, as described under "Materials and Methods". Fractions were collected from the top (rafts) to the bottom (non-rafts) of the gradient, pre-absorbed on Con A-Sepharose beads, and subjected to immunoblotting with anti-heparanase antibody. Note low but detectable levels of heparanase at rafts fractions (fractions 2–4) in control (upper panel) but not in MβCD treated cells (second panel). CHO K1 cells were incubated with heparanase (1µg/ml) without (Con, third panel) or with heparin (50 µg/ml, fourth panel) for 2 h at 4°C. Cell lysates were then loaded onto flotation gradient and analyzed as above. Note that heparin does not interfere with heparanase binding to lipid rafts. Interaction of heparanase with lipid rafts of CHO-745 cells deficient of HS (fifth panel), and CHO cells deficient of GPI-anchoring proteins and thus lacking cell surface glypicans (ΔGPI, lower panel), was similarly evaluated.
In order to ascertain binding of heparanase to lipid raft molecules, HeLa cells were left untreated as control, or treated with MβCD to disrupt rafts integrity. Cells were then incubated with heparanase for 2 h, solubilized with Triton X-100 at 4°C, and cell lysates were loaded on a Nycodenz gradient [18]. Fractions were collected and analyzed for the presence of heparanase by immunoblotting (Fig. 2B). Most of the heparanase reactivity appeared in non-raft fractions (Fig. 2B, upper panel, fractions 8–11), as expected. In addition, small, yet detectable amount of heparanase was found in lipid raft fractions of control, untreated cells (Con, Fig. 2B, fractions 2–4). Moreover, pretreatment with MβCD significantly reduced heparanase levels in the raft fractions (MβCD, Fig. 2B, second panel), and this effect appears specific since heparanase levels at the non-raft fractions were not affected (Fig. 2B, fractions 8–11). The major heparanase binding protein responsible for its presence in non-raft fractions is most likely HSPG. Syndecans are transmembrane HSPG which are not localized in rafts, but were shown to associate with these membrane microdomains following ligand binding [22; 23]. This possibility is excluded, however, by the following experiments. Heparanase appeared in the raft fractions of CHO K1 cells (Fig. 2B, third panel), and a similar reactivity was observed when binding was carried out in the presence of heparin (Fig. 2B, fourth panel). Moreover, heparanase appeared in rafts fractions upon its binding to HS-deficient CHO-745 cells (Fig. 2B, fifth panel), and to CHOΔGPI cells deficient of rafts glypicans (Fig. 2B, lower panel), collectively suggesting that the rafts component responsible for heparanase binding is not HSPG.
Heparanase induces Akt/PKB activation in lipid rafts
In order to further combine Akt/PKB activation and the presence of heparanase in lipid rafts, CHO K1 cells were left untreated as control, or incubated with heparanase at 0.03 or 1 µg/ml for 30 min on ice. Cell lysates were loaded on Nycodenz gradient and aliquot (5 µl) from each gradient fraction was applied onto nitrocellulose membrane as dots, and exposed to cholera toxin β-subunit. This protein binds with high affinity to ganglioside GM1 that localizes exclusively in rafts, thus enabling specific identification of raft fractions. As shown in figure 3A, fractions 3 and 4 exhibited the strongest reactivity with the toxin and are thus considered as the principal raft fractions, while fractions 9–11 are considered as non-rafts. Subsequently, rafts (#2–4) and non-rafts (#9–11) fractions were subjected to immunoblotting with anti-phospho-Akt antibody (Fig. 3B). Only low levels of phospho-Akt were detected in raft fractions prior to heparanase stimulation (Fig. 3B, rafts, 0). In contrast, Akt phosphorylation was markedly induced in raft fractions collected from cells that were incubated with a low concentration of heparanase (Fig. 3B, rafts, 0.03), and Akt phosphorylation was further stimulated upon incubation with higher concentrations (1µg/ml) of heparanase (Fig. 3B, rafts, 1). No significant Akt phosphorylation was observed in non-raft fractions (Fig. 3B, non-rafts), suggesting that heparanase binding to its raft-resident component facilitates Akt/PKB phosphorylation at the lipid rafts microdomain.
Figure 3.
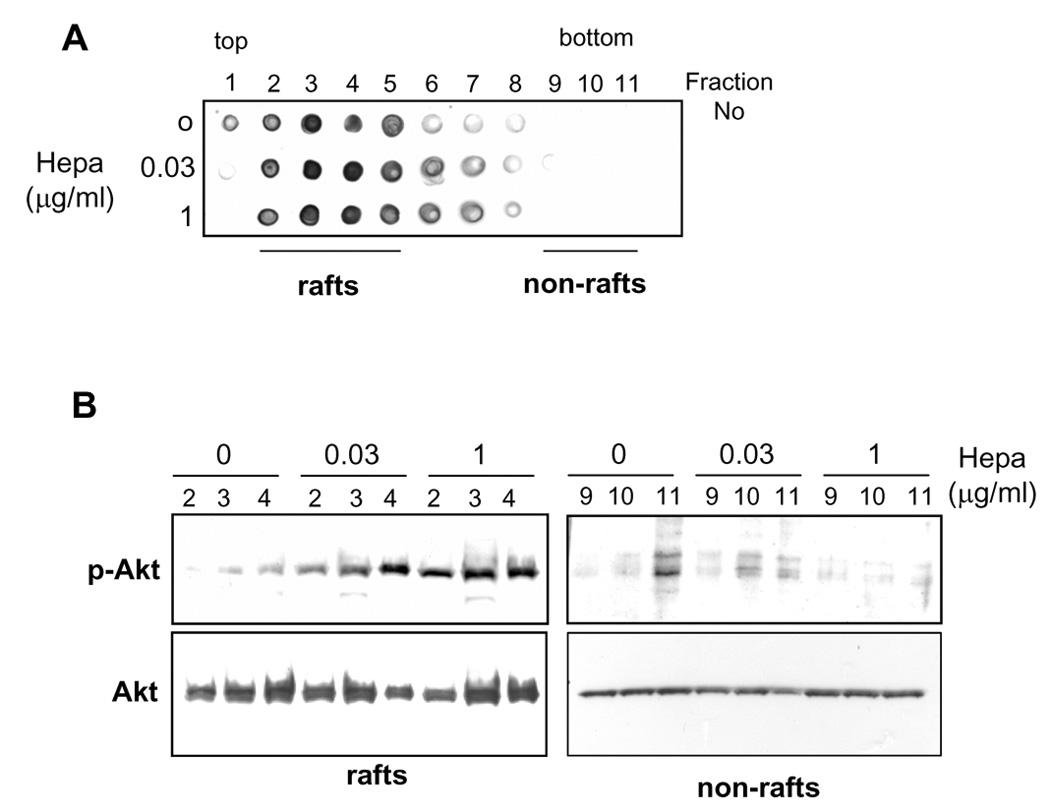
Heparanase stimulates Akt phosphorylation in lipid rafts. A. Identification of rafts fractions. HeLa cells were left untreated as control (0) or incubated with 0.03 and 1 µg/ml of heparanase for 1 h on ice. Cell lysates were loaded on flotation gradient and fractions were collected from the top (rafts) to the bottom (non-rafts) of the gradient. Aliquot (5 µl) from each fraction was applied to nitrocellulose membrane as dots, and the membrane was reacted with cholera toxin β-subunit-HRP conjugate to label rafts fractions. B. Akt activation. Fractions 2–4 and 9–11, representing rafts and non-rafts fractions, respectively, from control (0) and heparanase treated cells (0.03, 1) were subjected to immunoblotting with anti-phospho-Akt (upper panels) and anti-Akt (lower panels) antibodies. Note enhanced Akt phosphorylation in rafts following heparanase addition.
Discussion
Lipid rafts are membrane microdomains enriched in sphingolipids, cholesterol and protein complexes that initiate signal transduction. In the case of tyrosine kinase signaling, adaptors, scaffolds and enzymes are recruited to the cytoplasmic side of the plasma membrane following ligand activation, thus protecting the signaling complex from non-rafts enzymes (i.e., phosphatases) [24; 25]. Our findings suggest that the heparanase binding protein/receptor is similarly localized at lipid rafts, mediating Akt activation. The heparanase-binding rafts component seems not to be HSPG, since Akt activation was noted in HS-deficient cells [9]. Moreover, heparanase binding to the lipid rafts component was not affected by the presence of heparin, and a comparable binding was noted in wild type, HS-deficient (CHO-745), and GPI-deficient (CHO-ΔGPI) cells (Fig. 2B). In contrast, heparanase binding to the rafts component was significantly reduced following MβCD treatment, while binding to non-rafts proteins (i.e., syndecans, MPR, LRP) was not impaired (Fig. 2B). Significantly, Akt was activated by heparanase to a comparable extent in wild type, MPR-deficient, and LRP-deficient cells (Fig. 1), suggesting interaction of heparanase with a novel, raft-resident partner.
The involvement of lipid rafts in Akt/PKB activation following heparanase addition is evident by reduced Akt/PKB phosphorylation levels following Lovastatin, and even more so MβCD treatments (Fig. 2), both well established means to disrupt lipid rafts [25]. Lipid rafts disruption in HeLa cells also reduced basal Akt/PKB phosphorylation levels (Fig. 2A), in agreement with decreased Akt/PKB phosphorylation reported for prostate LNCaP [20; 21], epidermoid A431 [26] and small-cell lung carcinoma [27] cells following rafts disruption, correlating with increased apoptotic index [20; 21; 26]. The anti apoptotic effect of Akt/PKB is well established and is executed through phosphorylation and inhibition of pro-apoptotic mediators such as Bad, FOXO family members, and IκB kinase-β [28; 29]. Full activation of Akt/PKB requires phosphorylation of threonine308 and serine473 [30]. The kinase 3-phosphoinositide-dependent kinase-1 (PDK1), responsible for Akt phosphorylation of threonine308 is activated through interaction with phosphatydilinositol (3,4,5) triphosphate at the plasma membrane in a PI 3-K-dependent manner [31]. The kinase responsible for Akt/PKB phosphorylation of serine473, however, was only partially characterized. This activity is composed of a constitutively active complex of about 550 kDa, which resides in lipid rafts [31], thus rationalizing the observed decrease in Akt/PKB phosphorylation upon rafts disruption. Importantly, reduced Akt/PKB phosphorylation levels were noted upon heparanase down regulation by means of siRNA (Fig. 1A). This result clearly indicates that Akt/PKB phosphorylation levels are regulated by the relatively low levels of endogenous cellular heparanase, thus providing a strong clinical relevance for heparanase up regulation by primary human carcinomas [4; 5], and urge the development of heparanase inhibitors.
Attempts to inhibit heparanase enzymatic activity were initiated in parallel with the emerging clinical relevance of this activity. With the availability of recombinant heparanase and the establishment of high-throughput screen methods, a variety of inhibitory molecules were developed, including neutralizing antibodies, peptides, small molecules, chemically modified non-anticoagulant species of heparin, as well as several other polyanionic molecules, such as laminaran sulfate, suramin and PI-88 [5; 32; 33]. PI-88 is undergoing Phase III clinical trail in patients with advanced solid tumors [34]. Protein domains of heparanase responsible for non-enzymatic functions of the protein are yet to be characterized. The localization of heparanase binding component(s) to lipid rafts is a first step towards elucidation of the molecular machinery utilized by heparanase to elicit signal transduction, and will be employed for the development of inhibitors directed against non enzymatic functions of the heparanase molecule. These, combined with inhibitors of heparanase enzymatic activity, are expected to neutralize heparanase functions and reveal its significance in promoting tumor growth, angiogenesis and metastasis.
Acknowledgments
This work was supported by grants from the Israel Science Foundation (grant 549/06); National Cancer Institute, NIH (grant RO1-CA106456); the Israel Cancer Research Fund; and the Rappaport Family Institute Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dempsey LA, Brunn GJ, Platt JL. Heparanase, a potential regulator of cell-matrix interactions. Trends Biochem. Sci. 2000;25:349–351. doi: 10.1016/s0968-0004(00)01619-4. [DOI] [PubMed] [Google Scholar]
- 2.Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim. Biophys. Acta. 2001;1471:M99–M108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- 3.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J. Clin. Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Vlodavsky I, Abboud-Jarrous G, Elkin M, Naggi A, Casu B, Sasisekharan R, Ilan N. The impact of heparanese and heparin on cancer metastasis and angiogenesis. Pathophysiol. Haemost. Thromb. 2006;35:116–127. doi: 10.1159/000093553. [DOI] [PubMed] [Google Scholar]
- 6.Zetser A, Bashenko Y, Miao H-Q, Vlodavsky I, Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741. [PubMed] [Google Scholar]
- 7.Goldshmidt O, Zcharia E, Cohen M, Aingorn H, Cohen I, Nadav L, Katz B-Z, Geiger B, Vlodavsky I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003;17:1015–1025. doi: 10.1096/fj.02-0773com. [DOI] [PubMed] [Google Scholar]
- 8.Sotnikov I, Hershkoviz R, Grabovsky V, Ilan N, Cahalon L, Vlodavsky I, Alon R, Lider O. Enzymatically quiescent heparanase augments T cell interactions with VCAM-1 and extracellular matrix components under versatile dynamic contexts. J. Immunol. 2004;172:5185–5193. doi: 10.4049/jimmunol.172.9.5185. [DOI] [PubMed] [Google Scholar]
- 9.Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J. Biol. Chem. 2004;279:23536–23541. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- 10.Cohen I, Pappo O, Elkin M, San T, Bar-Shavit R, Hazan R, Peretz T, Vlodavsky I, Abramovitch R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int. J. Cancer. 2005;118:1609–1617. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 11.Vreys V, Delande N, Zhang Z, Coomans C, Roebroek A, Durr J, David G. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. J. Biol. Chem. 2005;280:33141–33148. doi: 10.1074/jbc.M503007200. [DOI] [PubMed] [Google Scholar]
- 12.Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY, Vlodavsky I, Ilan N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J. Biol.Chem. 2004;279:44084–44092. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 13.Zetser A, Levy-Adam F, Kaplan V, Gingis-Velitski S, Bashenko Y, Schubert S, Flugelman MY, Vlodavsky I, Ilan N. Processing and activation of latent heparanase occurs in lysosomes. J. Cell Sci. 2004;117:2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 14.Pohlmann R, Boeker MWC, von Figura K. The Two Mannose 6-Phosphate Receptors Transport Distinct Complements of Lysosomal Proteins. J. Biol. Chem. 1995;270:27311–27318. doi: 10.1074/jbc.270.45.27311. [DOI] [PubMed] [Google Scholar]
- 15.Willnow T, Herz J. Genetic deficiency in low density lipoprotein receptor-related protein confers cellular resistance to Pseudomonas exotoxin A. Evidence that this protein is required for uptake and degradation of multiple ligands. J. Cell Sci. 1994;107:719–726. [PubMed] [Google Scholar]
- 16.Abrami L, Fivaz M, Kobayashi T, Kinoshita T, Parton RG, van der Goot FG. Cross-talk between caveolae and glycosylphosphatidylinositol-rich domains. J. Biol. Chem. 2001;276:30729–30736. doi: 10.1074/jbc.M102039200. [DOI] [PubMed] [Google Scholar]
- 17.Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I. Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J. Natl. Cancer Inst. 2004;96:1219–1230. doi: 10.1093/jnci/djh230. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Zaken O, Tzaban S, Tal Y, Horonchik L, Esko JD, Vlodavsky I, Taraboulos A. Cellular heparan sulfate participates in the metabolism of prions. J. Biol. Chem. 2003;278:40041–40049. doi: 10.1074/jbc.M301152200. [DOI] [PubMed] [Google Scholar]
- 19.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate Akt-regulated survival in prostate cancer cells. Cancer Res. 2002;62:2227–2231. [PubMed] [Google Scholar]
- 22.Chu CL, Buczek-Thomas JA, Nugent MA. Heparan sulphate proteoglycans modulate fibroblast growth factor-2 binding through a lipid raft-mediated mechanism. Biochem. J. 2004;379:331–341. doi: 10.1042/BJ20031082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tkachenko E, Simons M. Clustering induces redistribution of syndecan-4 core protein into raft membrane domains. J. Biol. Chem. 2002;277:19946–19951. doi: 10.1074/jbc.M200841200. [DOI] [PubMed] [Google Scholar]
- 24.Rajendran L, Simons K. Lipid rafts and membrane dynamics. J. Cell Sci. 2005;118:1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- 25.Simons K, Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 26.Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168:1107–1118. doi: 10.2353/ajpath.2006.050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khanzada UK, Pardo OE, Meier C, Downward J, Seckl MJ, Arcaro A. Potent inhibition of small-cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signalling. Oncogene. 2006;25:877–887. doi: 10.1038/sj.onc.1209117. [DOI] [PubMed] [Google Scholar]
- 28.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 29.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 30.Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem. Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 31.Hill MM, Feng J, Hemmings BA. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr. Biol. 2002;12:1251–1255. doi: 10.1016/s0960-9822(02)00973-9. [DOI] [PubMed] [Google Scholar]
- 32.Ferro V, Hammond E, Fairweather JK. The development of inhibitors of heparanase, a key enzyme involved in tumour metastasis, angiogenesis and inflammation. Mini Rev. Med. Chem. 2004;4:693–702. doi: 10.2174/1389557043403729. [DOI] [PubMed] [Google Scholar]
- 33.Miao HQ, Liu H, Navarro E, Kussie P, Zhu Z. Development of heparanase inhibitors for anti-cancer therapy. Curr. Med. Chem. 2006;13:2101–2111. doi: 10.2174/092986706777935230. [DOI] [PubMed] [Google Scholar]
- 34.Basche M, Gustafson DL, Holden SN, O'Bryant CL, Gore L, Witta S, Schultz MK, Morrow M, Levin A, Creese BR, Kangas M, Roberts K, Nguyen T, Davis K, Addison RS, Moore JC, Eckhardt SG. A phase I biological and pharmacologic study of the heparanase inhibitor PI-88 in patients with advanced solid tumors. Clin. Cancer Res. 2006;12:5471–5480. doi: 10.1158/1078-0432.CCR-05-2423. [DOI] [PubMed] [Google Scholar]